A Repurposed Drug for Brain Cancer: Enhanced Atovaquone Amorphous Solid Dispersion by Combining a Spontaneously Emulsifying Component with a Polymer Carrier

 , , , ,
, , , , 
Abstract
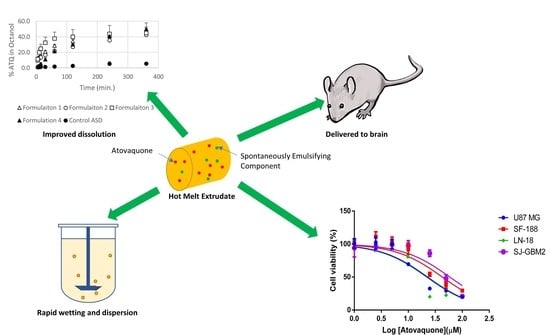
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Atovaquone Solubility in Pharmaceutical Excipients Used in the Formulations
2.3. Confirming the Composition of the Spontaneously Emulsifying Components
2.4. Atovaquone Solubility in Each of the Spontaneously Emulsifying Components
2.5. Flory–Huggins Theory and Miscibility of Compositions
2.6. Preparation of Atovaquone Formulations by Hot-Melt Extrusion
2.7. Characterization of Amorphous Solid Dispersions
2.7.1. X-Ray Diffractometry (XRD)
2.7.2. Differential Scanning Calorimetry (DSC)
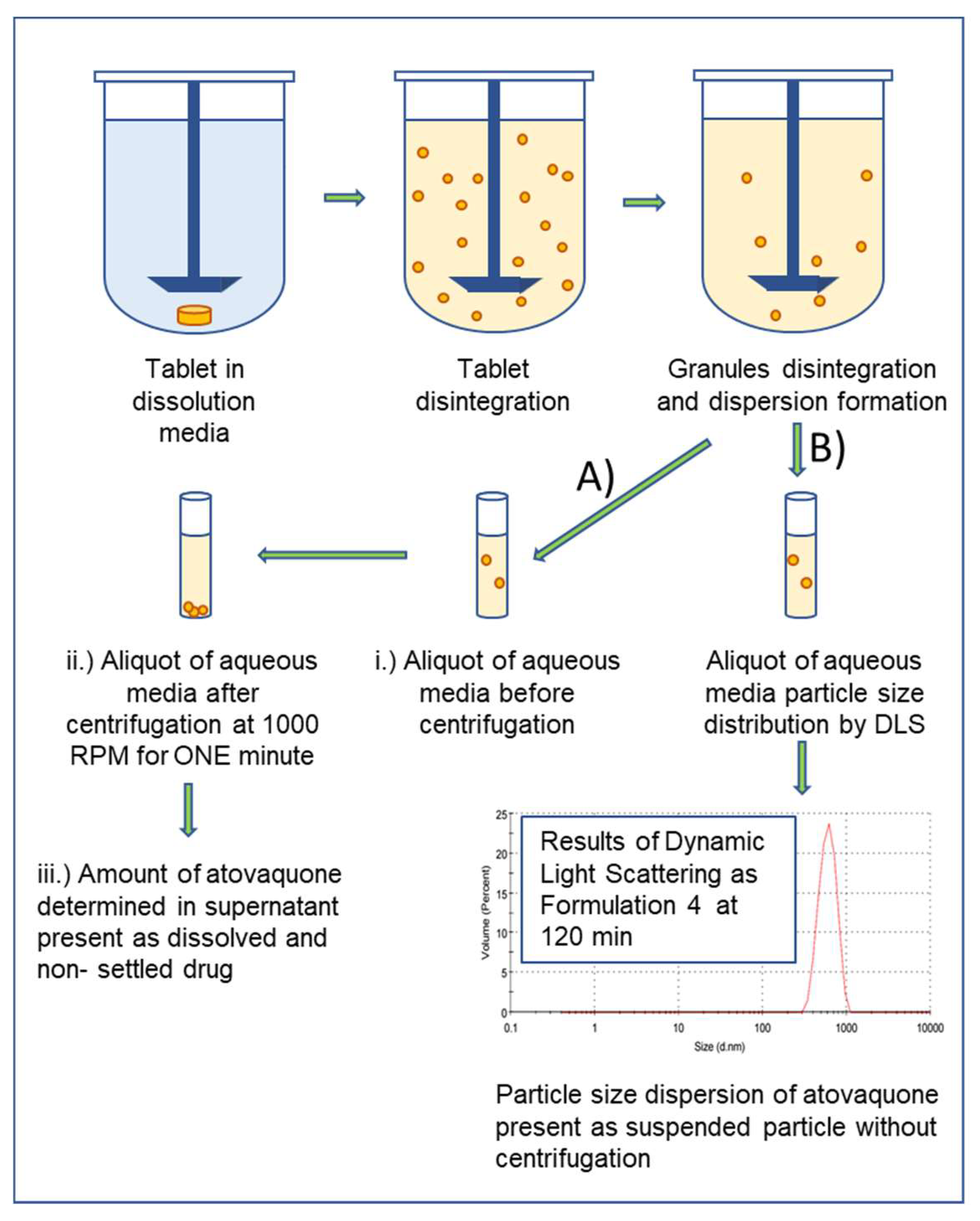
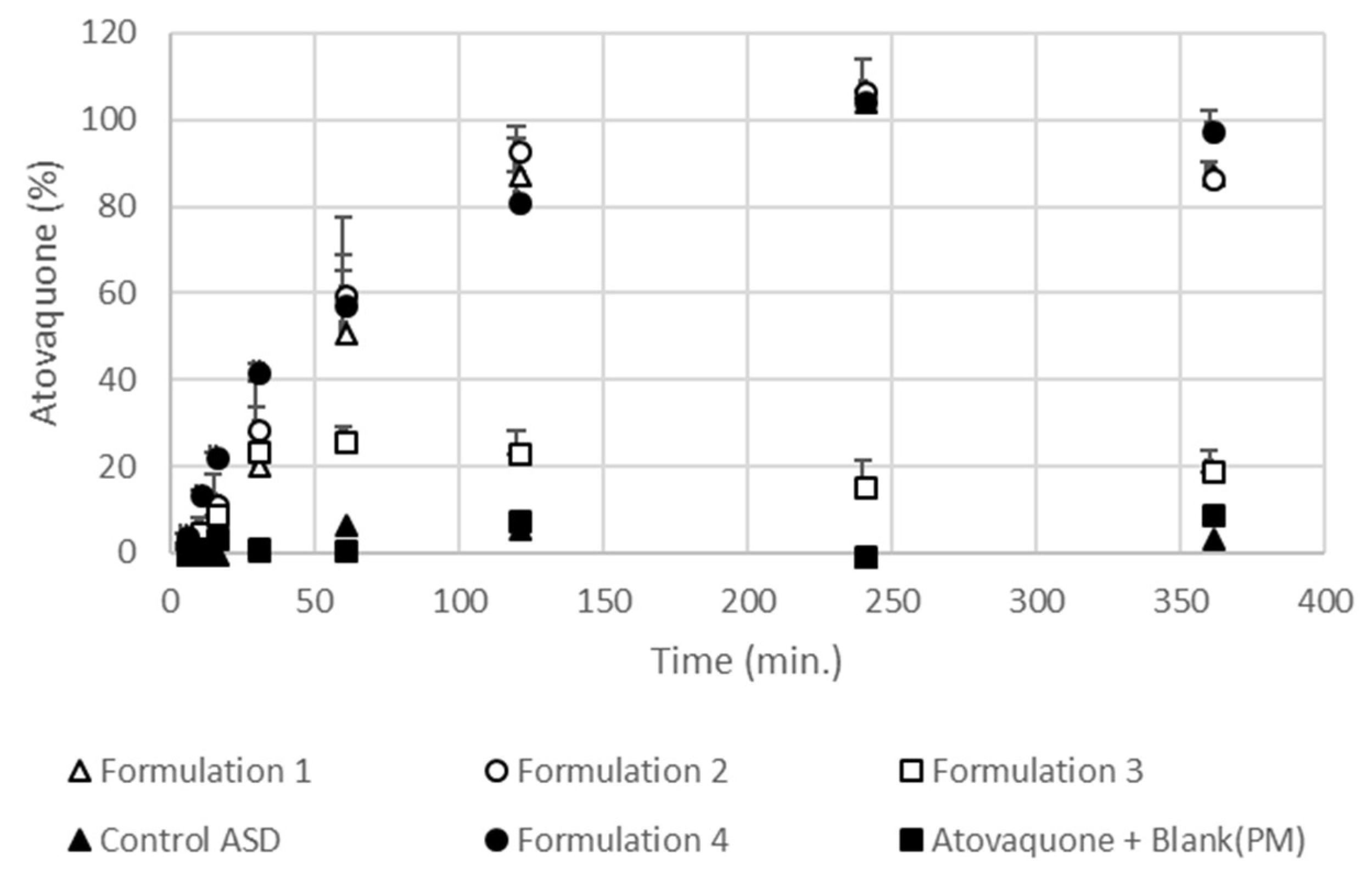
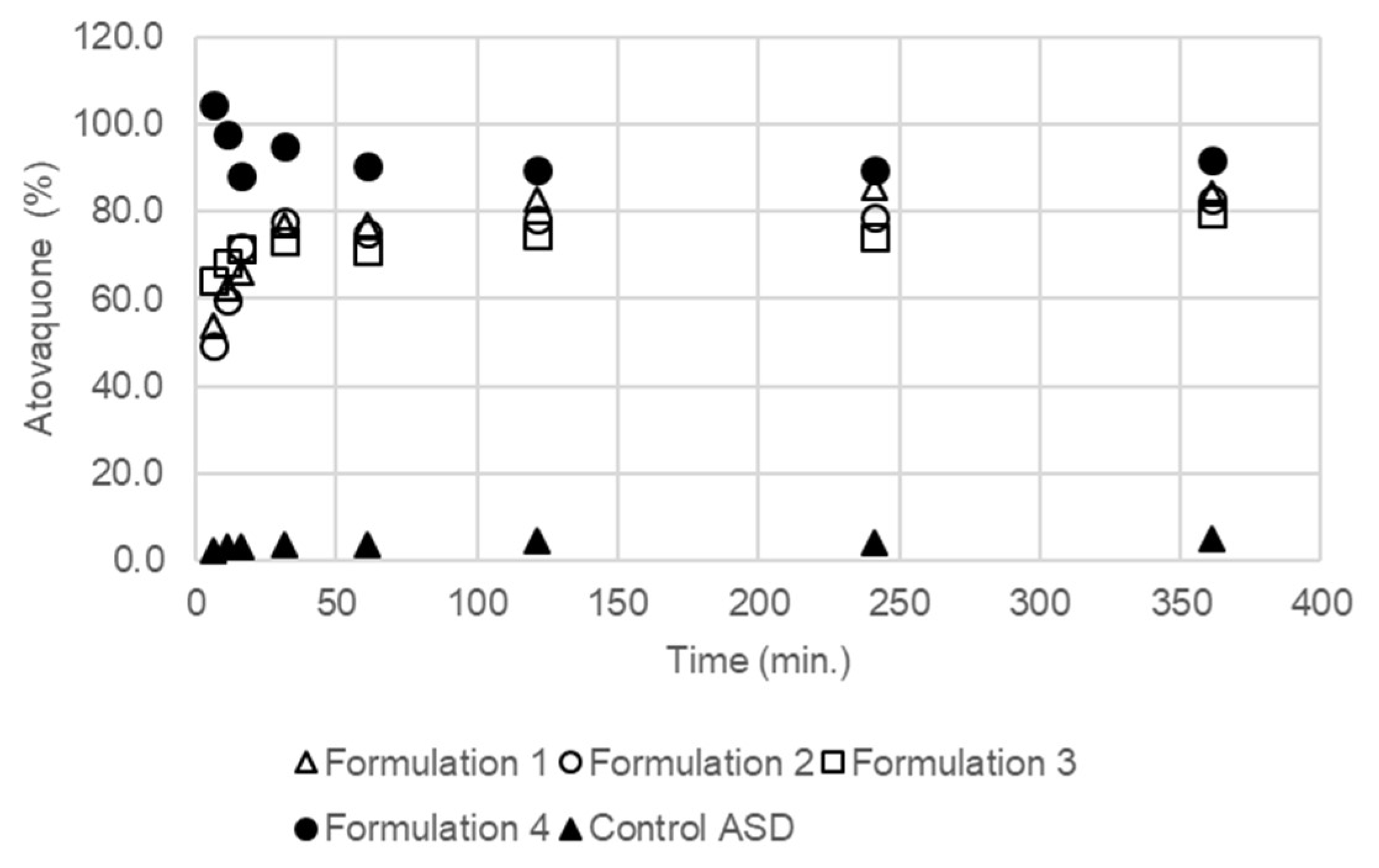
2.7.3. In Vitro Dispersion Test for Tablets
2.7.4. In Vitro Dispersion Test for Granules
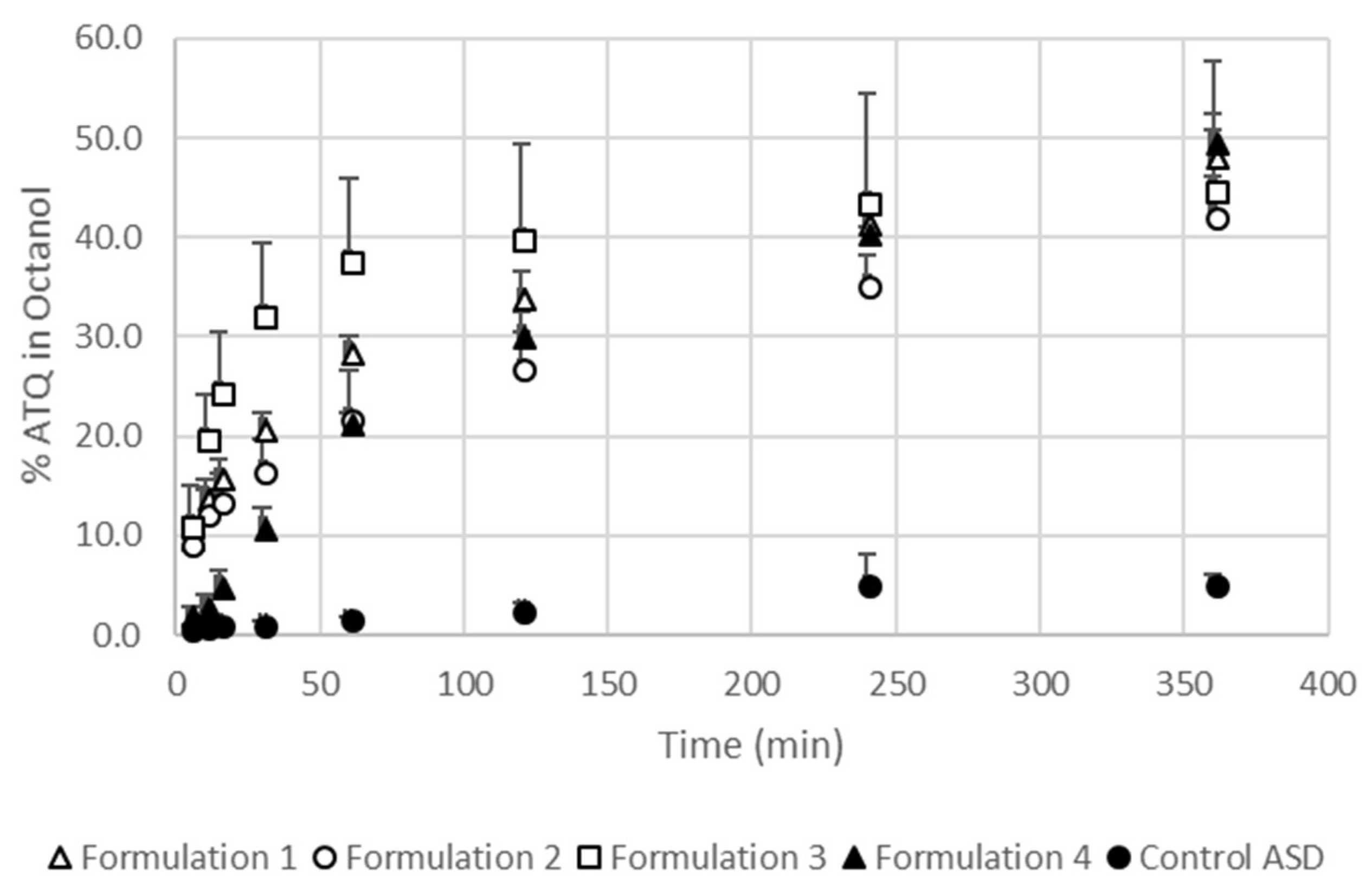
2.7.5. Biphasic Dissolution (Octanol/Water) Test
2.7.6. Cell Culture Conditions
2.7.7. Cell Viability Test in Glioblastoma Multiform Cell Lines
2.7.8. Proof-of-Concept Drug Exposure Study
2.7.9. Analysis of Atovaquone Concentrations in the Brain and Serum
3. Results and Discussion
3.1. Atovaquone Is Effective against Glioblastoma Multiforme Cell Lines
3.2. Developing the Atovaquone Formulations
3.2.1. Four Spontaneously Emulsifying Components Selected for the Formulations
3.2.2. PVP K30 Selected as the Polymer Carrier in the Thermally Processed Formulation
3.3. Solid State Characterization of the Thermally Processed Formulations
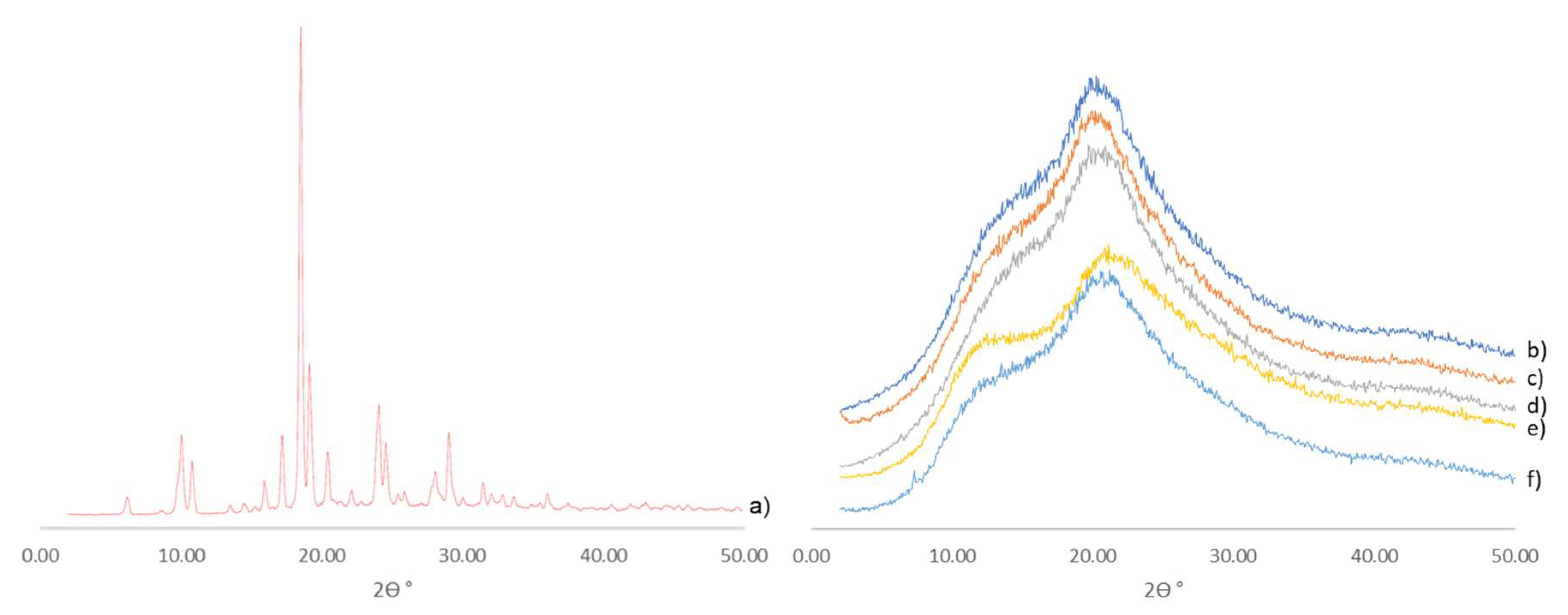
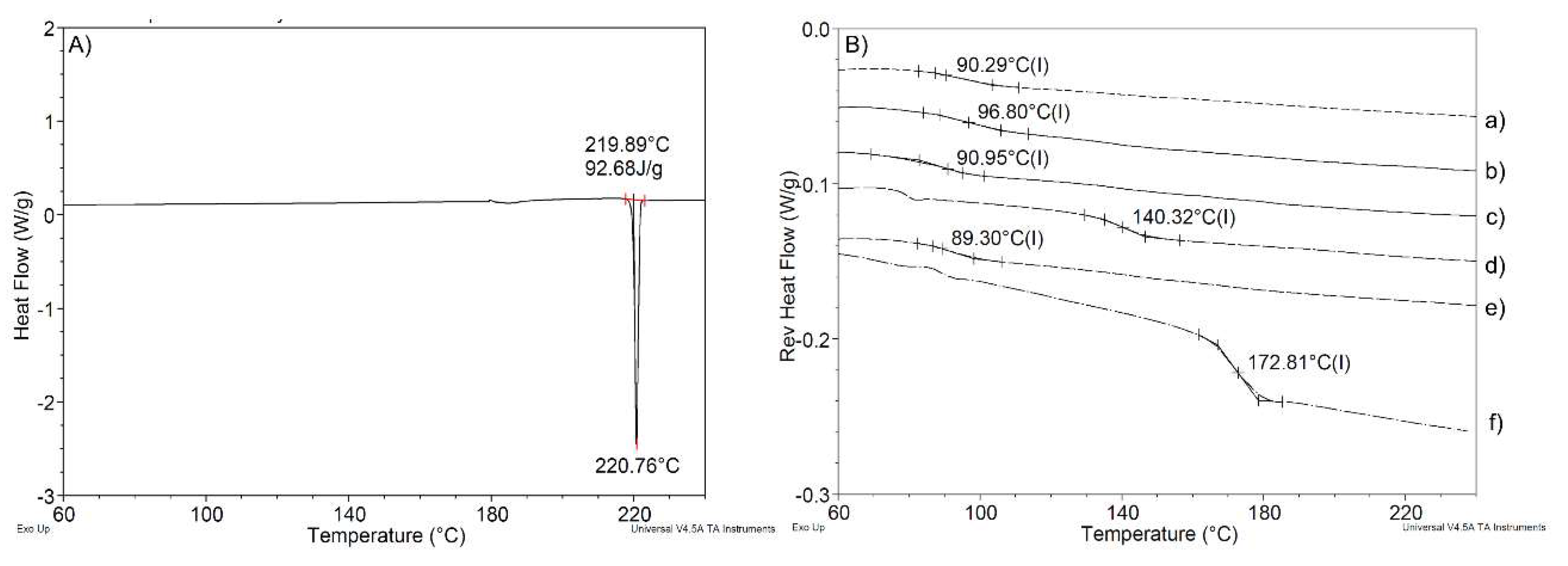
3.3.1. Hot-Melt-Extrusion-Processed Formulations Are Amorphous by Powder X-Ray Diffractometry (XRD)
3.3.2. The Glass Transition Temperature of Formulations Containing the SEC is Lower Than That of the Control Formulations
3.4. The Extent and Rate of Dissolution Is Greater When the SEC Is Present in the Formulation
3.5. Formulations Containing the SEC Rapidly Wet and Disperse into Fine Particles from Direct Compressed Tablets and Granules
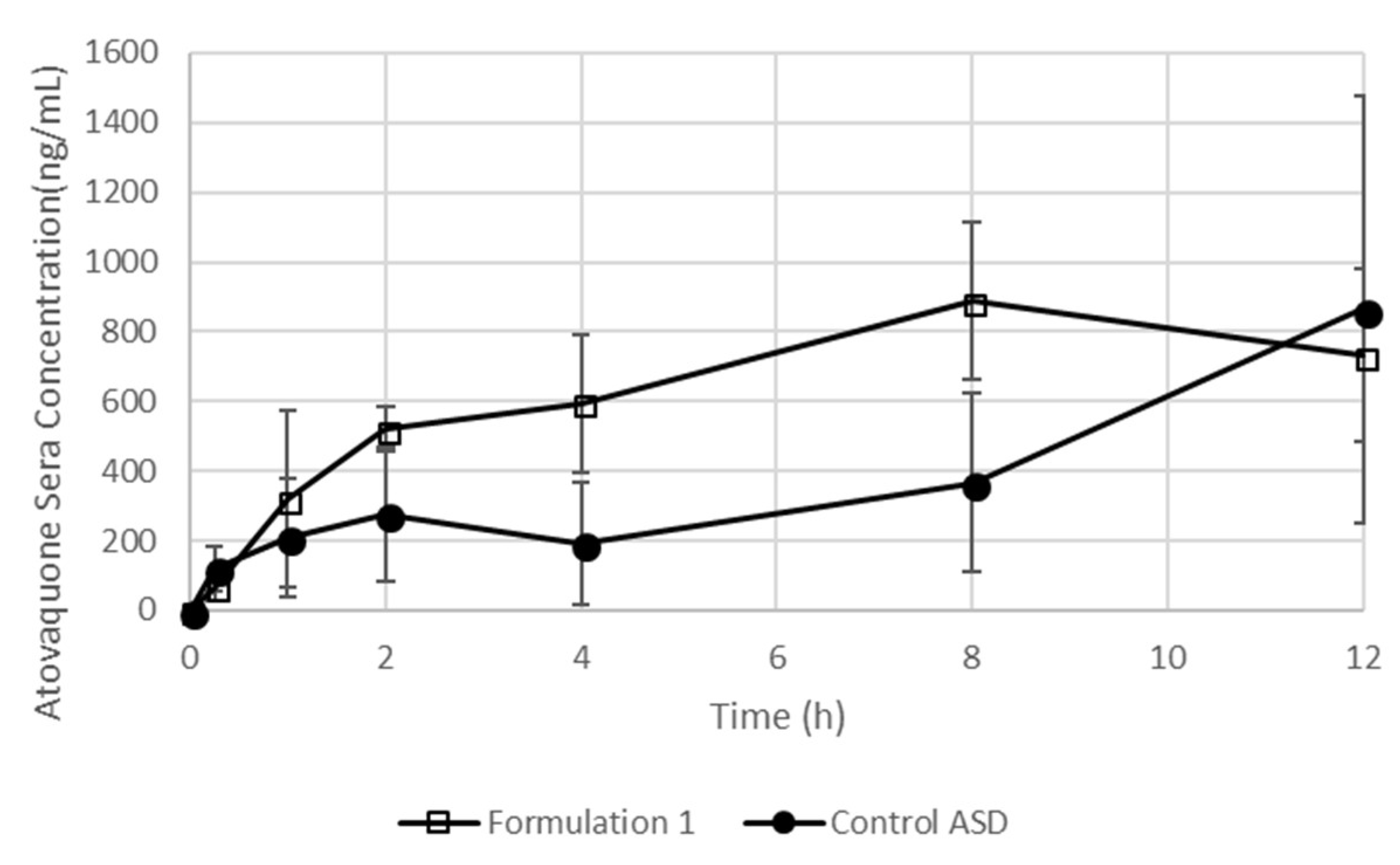
3.6. Preliminary Studies Confirm that SEC-Containing Formulations Lead to Effective Brain Levels Identified for Glioblastoma Multiforme Treatment
3.7. The Formulation Containing an SEC Leads To Increased Exposure after Dosing in Mice
4. Conclusions
Author Contributions
Declaration
Acknowledgments
Conflicts of Interest
References
- Zhang, X.; Zhang, W.E.I.; Cao, W.-D.; Cheng, G.; Zhang, Y.-Q. Glioblastoma multiforme: Molecular characterization and current treatment strategy (Review). Exp. Ther. Med. 2012, 3, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Holland, E.C. Glioblastoma multiforme: The terminator. Proc. Natl. Acad. Sci. USA 2000, 97, 6242–6244. [Google Scholar] [CrossRef] [PubMed]
- Luwor, R.B.; Stylli, S.S.; Kaye, A.H. The role of Stat3 in glioblastoma multiforme. J. Clin. Neurosci. 2013, 20, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Villalva, C.; Martin-Lannerée, S.; Cortes, U.; Dkhissi, F.; Wager, M.; Le Corf, A.; Tourani, J.-M.; Dusanter-Fourt, I.; Turhan, A.G.; Karayan-Tapon, L. STAT3 is essential for the maintenance of neurosphere-initiating tumor cells in patients with glioblastomas: A potential for targeted therapy? Int. J. Cancer 2011, 128, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.; Bakhshinyan, D.; Venugopal, C.; Mahendram, S.; Rosa, D.A.; Vijayakumar, T.; Manoranjan, B.; Hallett, R.; McFarlane, N.; Delaney, K.H.; et al. CD133+ brain tumor-initiating cells are dependent on STAT3 signaling to drive medulloblastoma recurrence. Oncogene 2016, 36, 606. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Patel, M.; Ruzevick, J.; Jackson, C.M.; Lim, M. STAT3 Activation in Glioblastoma: Biochemical and Therapeutic Implications. Cancers 2014, 6, 376–395. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.-S.; Chen, Y.-P.; Lin, Z.-X.; Wang, X.-F.; Zheng, Z.-Q.; Chen, L. STAT3 serine 727 phosphorylation influences clinical outcome in glioblastoma. Int. J. Clin. Exp. Pathol. 2014, 7, 3141–3149. [Google Scholar] [PubMed]
- Birner, P.; Toumangelova-Uzeir, K.; Natchev, S.; Guentchev, M. STAT3 tyrosine phosphorylation influences survival in glioblastoma. J. Neurooncol. 2010, 100, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Miyata, H.; Ashizawa, T.; Iizuka, A.; Kondou, R.; Nonomura, C.; Sugino, T.; Urakami, K.; Asai, A.; Hayashi, N.; Mitsuya, K.; et al. Combination of a STAT3 Inhibitor and an mTOR Inhibitor Against a Temozolomide-resistant Glioblastoma Cell Line. Cancer Genom. Proteom. 2017, 14, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Ashizawa, T.; Akiyama, Y.; Miyata, H.; Iizuka, A.; Komiyama, M.; Kume, A.; Omiya, M.; Sugino, T.; Asai, A.; Hayashi, N.; et al. Effect of the STAT3 inhibitor STX-0119 on the proliferation of a temozolomide-resistant glioblastoma cell line. Int. J. Oncol. 2014, 45, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, S.O.; Harbor, P.C.; Chernova, O.; Barnett, G.H.; Vogelbaum, M.A.; Haque, S.J. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene 2002, 21, 8404. [Google Scholar] [CrossRef] [PubMed]
- SHEN, J.; LI, R.; LI, G. Inhibitory Effects of Decoy-ODN Targeting Activated STAT3 on Human Glioma Growth In Vivo. In Vivo 2009, 23, 237–243. [Google Scholar] [PubMed]
- Kohsaka, S.; Wang, L.; Yachi, K.; Mahabir, R.; Narita, T.; Itoh, T.; Tanino, M.; Kimura, T.; Nishihara, H.; Tanaka, S. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol. Cancer Ther. 2012, 11, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.V.; Cseh, O.; Aman, A.; Weiss, S.; Luchman, H.A. The JAK2/STAT3 inhibitor pacritinib effectively inhibits patient-derived GBM brain tumor initiating cells in vitro and when used in combination with temozolomide increases survival in an orthotopic xenograft model. PLoS ONE 2017, 12, e0189670. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Asawa, R.; Zhang, Y.Q.; Cunningham, E.; Sun, H.; Tropsha, A.; Janzen, W.P.; Muratov, E.N.; Capuzzi, S.J.; Farag, S.; et al. Quantitative high-throughput phenotypic screening of pediatric cancer cell lines identifies multiple opportunities for drug repurposing. Oncotarget 2018, 9, 4758–4772. [Google Scholar] [CrossRef] [PubMed]
- Radloff, P.D.; Philipps, J.; Nkeyi, M.; Hutchinson, D.; Kremsner, P.G. Atovaquone and proguanil for Plasmodium falciparum malaria. Lancet 1996, 347, 1511–1514. [Google Scholar] [CrossRef]
- Douzinas, E.E.; Flevari, K.; Andrianakis, I.; Betrosian, A.P. Oral atovaquone for the treatment of severe Pneumocystis jirovecii pneumonia in a patient with glucose-6-phosphate dehydrogenase deficiency. Scand. J. Infect. Dis. 2010, 42, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Gabardi, S.; Millen, P.; Hurwitz, S.; Martin, S.; Roberts, K.; Chandraker, A. Atovaquone versus trimethoprim-sulfamethoxazole as Pneumocystis jirovecii pneumonia prophylaxis following renal transplantation. Clin. Transpl. 2012, 26, E184–E190. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Kim, H.; Ho, V.T.; Walker, S.R.; Bar-Natan, M.; Anahtar, M.; Liu, S.; Toniolo, P.A.; Kroll, Y.; Jones, N.; et al. Gene expression–based discovery of atovaquone as a STAT3 inhibitor and anticancer agent. Blood 2016, 128, 1845–1853. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Mutti, L.; Krstic-Demonacos, M.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 2016, 7, 34084–34099. [Google Scholar] [CrossRef] [PubMed]
- Ashton, T.M.; Fokas, E.; Kunz-Schughart, L.A.; Folkes, L.K.; Anbalagan, S.; Huether, M.; Kelly, C.J.; Pirovano, G.; Buffa, F.M.; Hammond, E.M.; et al. The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat. Commun. 2016, 7, 12308. [Google Scholar] [CrossRef] [PubMed]
- Sek, L.; Boyd, B.J.; Charman, W.N.; Porter, C.J. Examination of the impact of a range of Pluronic surfactants on the in-vitro solubilisation behaviour and oral bioavailability of lipidic formulations of atovaquone. J. Pharm. Pharmacol. 2006, 58, 809–820. [Google Scholar] [CrossRef] [PubMed]
- GlaxoSmithKline. MALARONE (Atovaquone and Proguanil Hydrochloride) Tablets; GlaxoSmithKline: Brentford, UK, 2013. [Google Scholar]
- GlaxoSmithKline. Mepron (Atovaquone Oral Suspension) [Package Insert]; Research Triangle Park; GlaxoSmithKline: Brentford, UK, 2017. [Google Scholar]
- Ino, H.; Takahashi, N.; Hashimoto, H.f.; Doi, Y.; Hirama, T. Phase 1 study of atovquine and atovaquone/proguanil HCl. J. Infect. Chemother. 2013, 61, 335–342. [Google Scholar]
- Schöler, N.; Krause, K.; Kayser, O.; Müller, R.H.; Borner, K.; Hahn, H.; Liesenfeld, O. Atovaquone Nanosuspensions Show Excellent Therapeutic Effect in a New Murine Model of Reactivated Toxoplasmosis. Antimicrob. Agents Chemother. 2001, 45, 1771–1779. [Google Scholar] [CrossRef] [PubMed]
- Dunay, I.R.; Heimesaat, M.M.; Bushrab, F.N.; Müller, R.H.; Stocker, H.; Arasteh, K.; Kurowski, M.; Fitzner, R.; Borner, K.; Liesenfeld, O. Atovaquone Maintenance Therapy Prevents Reactivation of Toxoplasmic Encephalitis in a Murine Model of Reactivated Toxoplasmosis. Antimicrob. Agents Chemother. 2004, 48, 4848–4854. [Google Scholar] [CrossRef] [PubMed]
- Azami, S.J.; Amani, A.; Keshavarz, H.; Najafi-Taher, R.; Mohebali, M.; Faramarzi, M.A.; Mahmoudi, M.; Shojaee, S. Nanoemulsion of atovaquone as a promising approach for treatment of acute and chronic toxoplasmosis. Eur. J. Pharm. Sci. 2018, 117, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Goddeeris, C.; Willems, T.; Van den Mooter, G. Formulation of fast disintegrating tablets of ternary solid dispersions consisting of TPGS 1000 and HPMC 2910 or PVPVA 64 to improve the dissolution of the anti-HIV drug UC 781. Eur. J. Pharm. Sci. 2008, 34, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Hughey, J.R.; Keen, J.M.; Miller, D.A.; Kolter, K.; Langley, N.; McGinity, J.W. The use of inorganic salts to improve the dissolution characteristics of tablets containing Soluplus(R)-based solid dispersions. Eur. J. Pharm. Sci. 2013, 48, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhou, L.; Zhang, F. Reactive Melt Extrusion To Improve the Dissolution Performance and Physical Stability of Naproxen Amorphous Solid Dispersions. Mol. Pharm. 2017, 14, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Albadarin, A.B.; Potter, C.B.; Davis, M.T.; Iqbal, J.; Korde, S.; Pagire, S.; Paradkar, A.; Walker, G. Development of stability-enhanced ternary solid dispersions via combinations of HPMCP and Soluplus((R)) processed by hot melt extrusion. Int. J. Pharm. 2017, 532, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kang, W.S.; Piao, J.; Yoon, I.S.; Kim, D.D.; Cho, H.J. Soluplus(R)/TPGS-based solid dispersions prepared by hot-melt extrusion equipped with twin-screw systems for enhancing oral bioavailability of valsartan. Drug Des. Dev. Ther. 2015, 9, 2745–2756. [Google Scholar]
- Thiry, J.; Kok, M.G.; Collard, L.; Frere, A.; Krier, F.; Fillet, M.; Evrard, B. Bioavailability enhancement of itraconazole-based solid dispersions produced by hot melt extrusion in the framework of the Three Rs rule. Eur. J. Pharm. Sci. 2017, 99, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.T.; Pervez, H.; Shehzad, M.T.; Mahmood, Z.; Razi, M.T.; Ranjha, N.M.; Khanum, N. Improved physicochemical characteristics of artemisinin-nicotinamide solid dispersions by solvent evaporation and freeze dried methods. Pak. J. Pharm. Sci. 2012, 25, 447–456. [Google Scholar] [PubMed]
- Tayyab Ansari, M.; Arshad, M.S.; Hussain, A.; Ahmad, Z. Improvement of solubility, dissolution and stability profile of artemether solid dispersions and self emulsified solid dispersions by solvent evaporation method. Pharm. Dev. Technol. 2016, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhou, L.L.; Wei, L.L.; Hong, G.; Nie, S.F.; Yang, X.G.; Tang, R.; Pan, W.S. Preparation of budesonide-poly (ethylene oxide) solid dispersions using supercritical fluid technology. Drug Dev. Ind. Pharm. 2007, 33, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Obaidat, R.M.; Tashtoush, B.M.; Awad, A.A.; Al Bustami, R.T. Using Supercritical Fluid Technology (SFT) in Preparation of Tacrolimus Solid Dispersions. AAPS PharmSciTech 2017, 18, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Van Nijlen, T.; Brennan, K.; Van den Mooter, G.; Blaton, N.; Kinget, R.; Augustijns, P. Improvement of the dissolution rate of artemisinin by means of supercritical fluid technology and solid dispersions. Int. J. Pharm. 2003, 254, 173–181. [Google Scholar] [CrossRef]
- Friesen, D.T.; Shanker, R.; Crew, M.; Smithey, D.T.; Curatolo, W.; Nightingale, J. Hydroxypropyl methylcellulose acetate succinate-based spray-dried dispersions: An overview. Mol. Pharm. 2008, 5, 1003–1019. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Shah, S.; Jo, S.; Majumdar, S.; Gryczke, A.; Kolter, K.; Langley, N.; Repka, M.A. Investigation of phase diagrams and physical stability of drug-polymer solid dispersions. Pharm. Dev. Technol. 2015, 20, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Huang, J.; Hussain, M.A. Drug-polymer solubility and miscibility: Stability consideration and practical challenges in amorphous solid dispersion development. J. Pharm. Sci. 2010, 99, 2941–2947. [Google Scholar] [CrossRef] [PubMed]
- Bansal, K.; Baghel, U.S.; Thakral, S. Construction and Validation of Binary Phase Diagram for Amorphous Solid Dispersion Using Flory-Huggins Theory. AAPS PharmSciTech 2016, 17, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Marsac, P.J.; Shamblin, S.L.; Taylor, L.S. Theoretical and practical approaches for prediction of drug-polymer miscibility and solubility. Pharm. Res. 2006, 23, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; O’Donnell, K.P.; Keen, J.M.; Rickard, M.A.; McGinity, J.W.; Williams, R.O., 3rd. A New Extrudable Form of Hypromellose: AFFINISOL HPMC HME. AAPS PharmSciTech 2016, 17, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.J.; Pygall, S.R.; Cooper, V.B.; Mann, J.C. Overcoming sink limitations in dissolution testing: A review of traditional methods and the potential utility of biphasic systems. J. Pharm. Pharmacol. 2012, 64, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Heigoldt, U.; Sommer, F.; Daniels, R.; Wagner, K.-G. Predicting in vivo absorption behavior of oral modified release dosage forms containing pH-dependent poorly soluble drugs using a novel pH-adjusted biphasic in vitro dissolution test. Eur. J. Pharm. Biopharm. 2010, 76, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Vangani, S.; Li, X.; Zhou, P.; Del-Barrio, M.-A.; Chiu, R.; Cauchon, N.; Gao, P.; Medina, C.; Jasti, B. Dissolution of poorly water-soluble drugs in biphasic media using USP 4 and fiber optic system. Clin. Res. Regul. Aff. 2009, 26, 8–19. [Google Scholar] [CrossRef]
- Sarode, A.L.; Wang, P.; Obara, S.; Worthen, D.R. Supersaturation, nucleation, and crystal growth during single- and biphasic dissolution of amorphous solid dispersions: Polymer effects and implications for oral bioavailability enhancement of poorly water soluble drugs. Eur. J. Pharm. Biopharm. 2014, 86, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Perazzoli, G.; Prados, J.; Ortiz, R.; Caba, O.; Cabeza, L.; Berdasco, M.; Gonzalez, B.; Melguizo, C. Temozolomide Resistance in Glioblastoma Cell Lines: Implication of MGMT, MMR, P-Glycoprotein and CD133 Expression. PLoS ONE 2015, 10, e0140131. [Google Scholar] [CrossRef] [PubMed]
- Han, T.J.; Cho, B.J.; Choi, E.J.; Kim, D.H.; Song, S.H.; Paek, S.H.; Kim, I.A. Inhibition of STAT3 enhances the radiosensitizing effect of temozolomide in glioblastoma cells in vitro and in vivo. J. Neurooncol. 2016, 130, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Choi, E.; Cho, B. Inhibition of STAT3 Enhances the Radiosensitizing Effect of Temozolomide in Malignant Glioma Cells. Int. J. Radiat. Oncol. Biol. Phys. 2013, 87, S647. [Google Scholar] [CrossRef]
- Jermain, S.V.; Brough, C.; Williams, R.O. Amorphous solid dispersions and nanocrystal technologies for poorly water-soluble drug delivery—An update. Int. J. Pharm. 2018, 535, 379–392. [Google Scholar] [CrossRef] [PubMed]
- GlaxoSmithKline. Mepron Suspension [Saftey Data Sheet]; Research Triangle Park; GlaxoSmithKline: Brentford, UK, 2014. [Google Scholar]
- Zhao, G.; Huang, J.; Xue, K.; Si, L.; Li, G. Enhanced intestinal absorption of etoposide by self-microemulsifying drug delivery systems: Roles of P-glycoprotein and cytochrome P450 3A inhibition. Eur. J. Pharm. Sci. 2013, 50, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Cerpnjak, K.; Zvonar Pobirk, A.; Vrecer, F.; Gasperlin, M. Tablets and minitablets prepared from spray-dried SMEDDS containing naproxen. Int. J. Pharm. 2015, 495, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Guo, X.; Hu, X.; Abbasi, A.M.; Zhou, L.; Li, T.; Fu, X.; Liu, R.H. Fabrication and Optimization of Self-Microemulsions to Improve the Oral Bioavailability of Total Flavones of Hippophae rhamnoides L. J. Food Sci. 2017, 82, 2901–2909. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, L.; Yue, S.; Huang, Q.; Zhang, Y.; Yang, J. Characterization and evaluation of a self-microemulsifying drug delivery system containing tectorigenin, an isoflavone with low aqueous solubility and poor permeability. Drug Deliv. 2017, 24, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Mahlin, D.; Bergström, C.A.S. Early drug development predictions of glass-forming ability and physical stability of drugs. Eur. J. Pharm. Sci. 2013, 49, 323–332. [Google Scholar] [CrossRef] [PubMed]
- DiNunzio, J.C.; Miller, D.A.; Yang, W.; McGinity, J.W.; Williams, R.O. Amorphous Compositions Using Concentration Enhancing Polymers for Improved Bioavailability of Itraconazole. Mol. Pharm. 2008, 5, 968–980. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Cheng, G.; Gu, J.C.; Xu, C.H. Development of solid self-emulsifying drug delivery systems: Preparation techniques and dosage forms. Drug Discov. Today 2008, 13, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Devraj, R.; Williams, H.D.; Warren, D.B.; Mohsin, K.; Porter, C.J.H.; Pouton, C.W. In vitro assessment of drug-free and fenofibrate-containing lipid formulations using dispersion and digestion testing gives detailed insights into the likely fate of formulations in the intestine. Eur. J. Pharm. Sci. 2013, 49, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Mohsin, K.; Alanazi, F. The fate of paclitaxel during in vitro dispersion testing of different lipid-based formulations. J. Drug Deliv. Sci. Technol. 2012, 22, 197–204. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thermally Processed Formulations by Hot-Melt Extrusion (% w/w) | ||||||
|---|---|---|---|---|---|---|
| Formulation 1 | Formulation 2 | Formulation 3 | Formulation 4 | Control ASD (No SEC) | Blank (No Drug) | |
| Atovaquone | 20 | 20 | 20 | 20 | 20 | |
| PVP K30 | 70 | 70 | 70 | 70 | 80 | 90 |
| Spontaneously Emulsifying Component Composition | ||||||
| Benzyl benzoate | - | - | - | 3 | - | 3 |
| PEG 400 | - | - | - | 3 | - | 3 |
| Capmul MCM NF | 2.5 | 2.5 | 0.5 | - | - | - |
| Captex 300 NF | 2.5 | 2.5 | 0.5 | - | - | - |
| Tween 20 | - | - | - | 4 | - | 4 |
| Tween 80 | 5 | - | - | - | - | - |
| Cremophor RH 40 | - | 5 | - | - | - | - |
| Labrasol | - | - | 9 | - | - | - |
| Excipient and Excipient Combinations | Solubility of Atovaquone ± Standard Deviation (mg/mL) | Droplet Size Distribution (dv,50; nm) for SEC Diluted in Water (10 μL of SEC Diluted into 10 mL of Water) |
|---|---|---|
| Benzyl benzoate | 14.3 ± 0.4 | - |
| PEG 400 | 7.2 ± 0.7 | - |
| Capmul MCM NF | 3.5 ± 0.3 | - |
| Captex 300 NF | 4.6 ± 0.6 | - |
| Tween 20 | 9.7 ± 0.7 | - |
| Tween 80 | 10.2 ± 1.2 | - |
| Labrasol | 10.2 ± 0.5 | - |
| SEC for formulation 1 | 8.7 ± 0.5 | 122.4 |
| SEC for formulation 2 | 8.6 ± 0.4 | 64.1 |
| SEC for formulation 3 | 12.5 ± 0.1 | 186.1 |
| SEC for formulation 4 | 13.5 ± 0.6 | 51.0 |
| Dosage Form | Z = Average Particle Size (μm) | ||||||
|---|---|---|---|---|---|---|---|
| Time (min) | 30 | 60 | 120 | 240 | 360 | ||
| Tablet | Formulation | 1 | 2.7 | 2.6 | 2.1 | 1.7 | 1.6 |
| 2 | 2.3 | 1.6 | 1.5 | 1.2 | 1.2 | ||
| 3 | 1.4 | 1.6 | - 2 | - 2 | - 2 | ||
| 4 | 0.86 | 0.72 | 0.70 | 0.59 | 0.65 | ||
| Control ASD | - 2 | - 2 | - 2 | - 2 | - 2 | ||
| 5 1 | - 2 | - 2 | - 2 | - 2 | - 2 | ||
| Granule | 1 | 0.87 | 0.79 | 0.74 | 0.68 | 0.73 | |
| 2 | 0.81 | 0.61 | 0.59 | 0.56 | 0.54 | ||
| 3 | 0.39 | 0.40 | 0.39 | 0.38 | 0.36 | ||
| 4 | 0.49 | 0.49 | 0.48 | 0.46 | 0.47 | ||
| Control ASD | - 2 | - 2 | - 2 | - 2 | - 2 | ||
| 5 1 | - 2 | - 2 | - 2 | - 2 | - 2 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takabe, H.; Warnken, Z.N.; Zhang, Y.; Davis, D.A.; Smyth, H.D.C.; Kuhn, J.G.; Weitman, S.; Williams III, R.O. A Repurposed Drug for Brain Cancer: Enhanced Atovaquone Amorphous Solid Dispersion by Combining a Spontaneously Emulsifying Component with a Polymer Carrier. Pharmaceutics 2018, 10, 60. https://doi.org/10.3390/pharmaceutics10020060
Takabe H, Warnken ZN, Zhang Y, Davis DA, Smyth HDC, Kuhn JG, Weitman S, Williams III RO. A Repurposed Drug for Brain Cancer: Enhanced Atovaquone Amorphous Solid Dispersion by Combining a Spontaneously Emulsifying Component with a Polymer Carrier. Pharmaceutics. 2018; 10(2):60. https://doi.org/10.3390/pharmaceutics10020060
Chicago/Turabian StyleTakabe, Hiroyuki, Zachary N. Warnken, Yajie Zhang, Daniel A. Davis, Hugh D. C. Smyth, John G. Kuhn, Steve Weitman, and Robert O. Williams III. 2018. "A Repurposed Drug for Brain Cancer: Enhanced Atovaquone Amorphous Solid Dispersion by Combining a Spontaneously Emulsifying Component with a Polymer Carrier" Pharmaceutics 10, no. 2: 60. https://doi.org/10.3390/pharmaceutics10020060