
Infectious Bursal Disease Virus Assembly Causes Endoplasmic Reticulum Stress and Lipid Droplet Accumulation
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell lines and Culture Conditions
2.2. Viral Stocks Production
2.3. Antibodies
2.4. Pharmacological Inhibitors
2.5. Oleic Acid Treatment and Viability Assay
2.6. Oil Red O Staining
2.7. Plasmids and Transfection Methods
2.8. Indirect Immunofluorescence
2.9. Western Blot
2.10. Viral Titration by Plaque Assay
2.11. Transmission Electron Microscopy
2.12. Cryo-Immunoelectron Microscopy
2.13. Statistical Analysis
3. Results
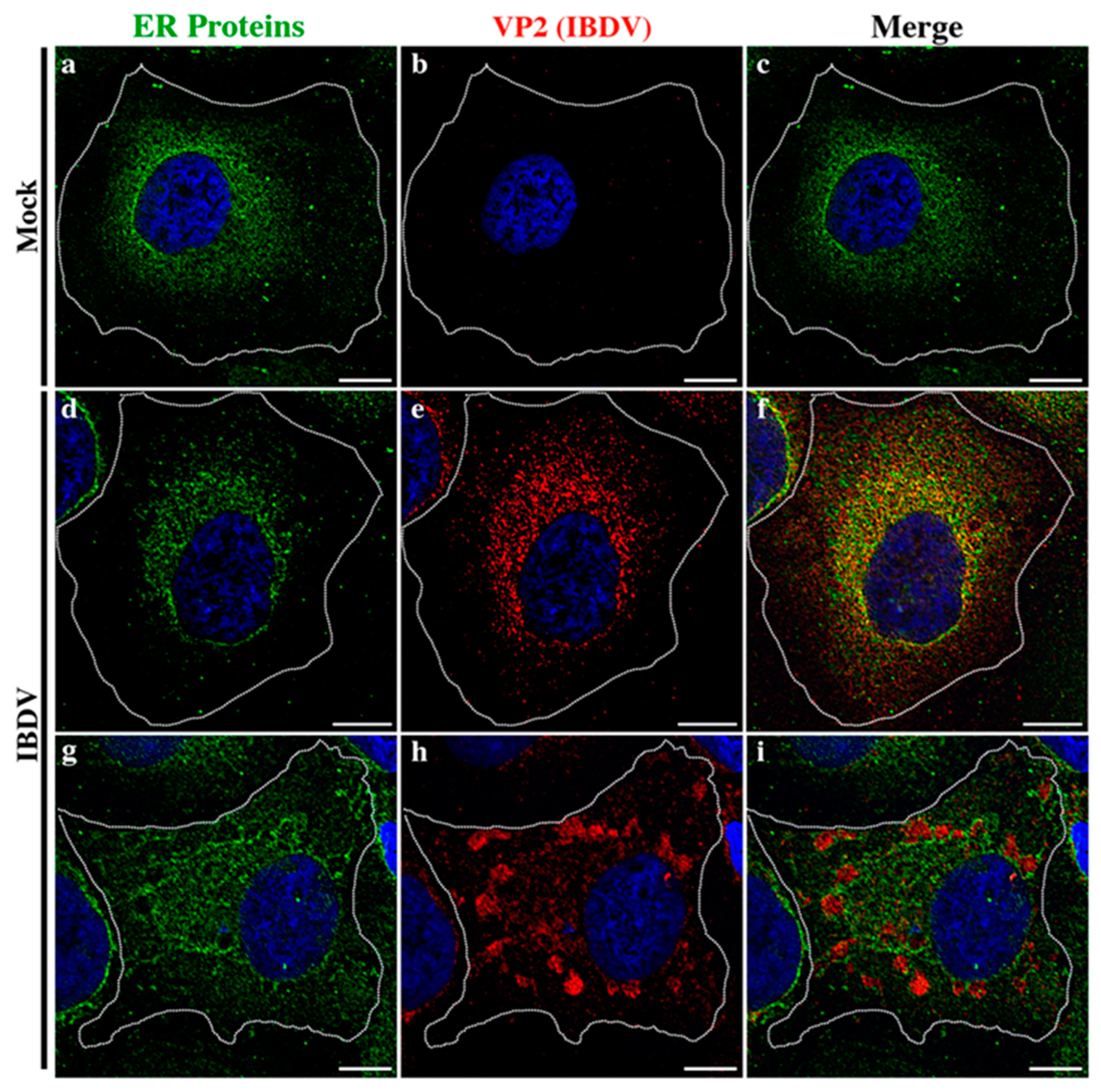
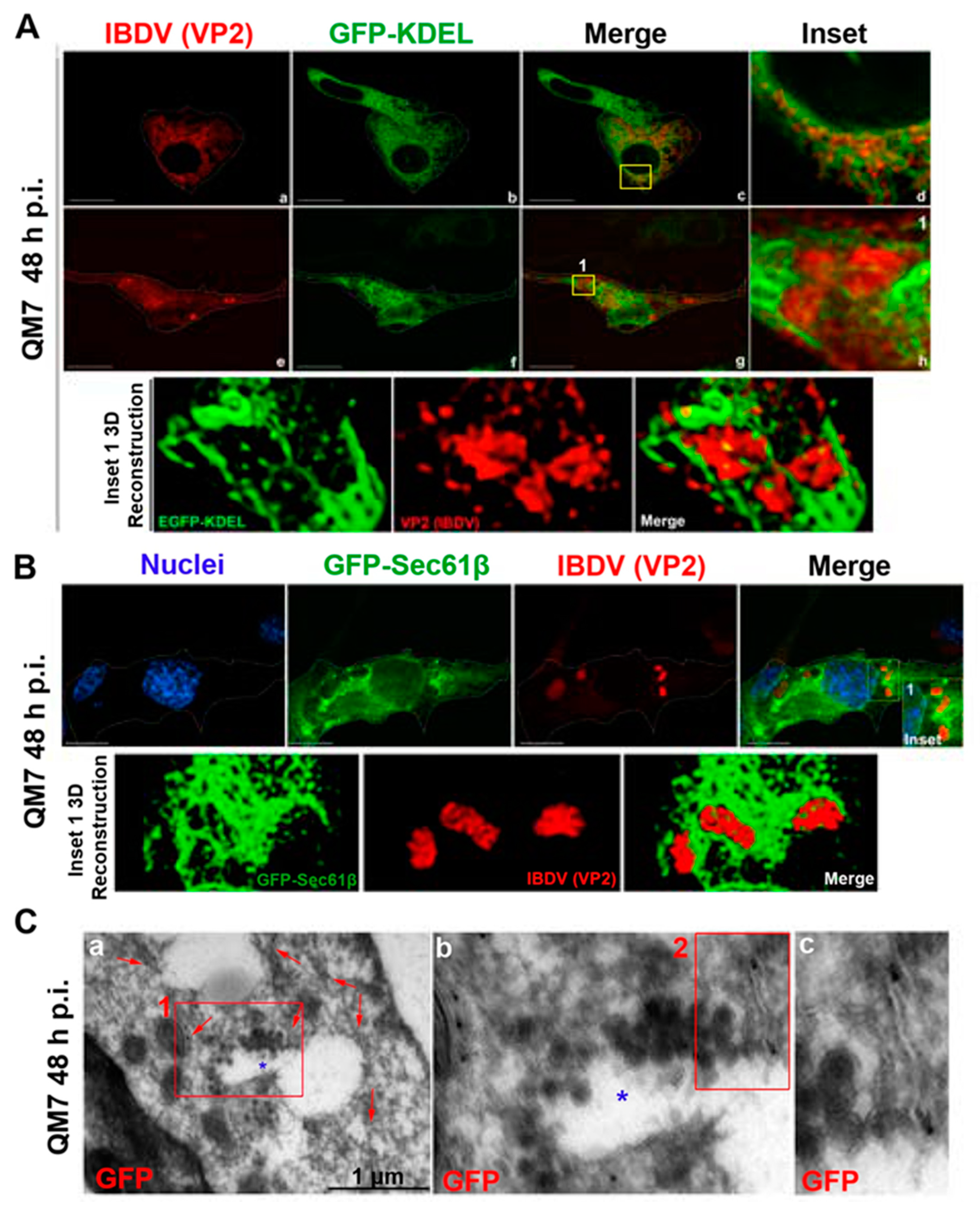
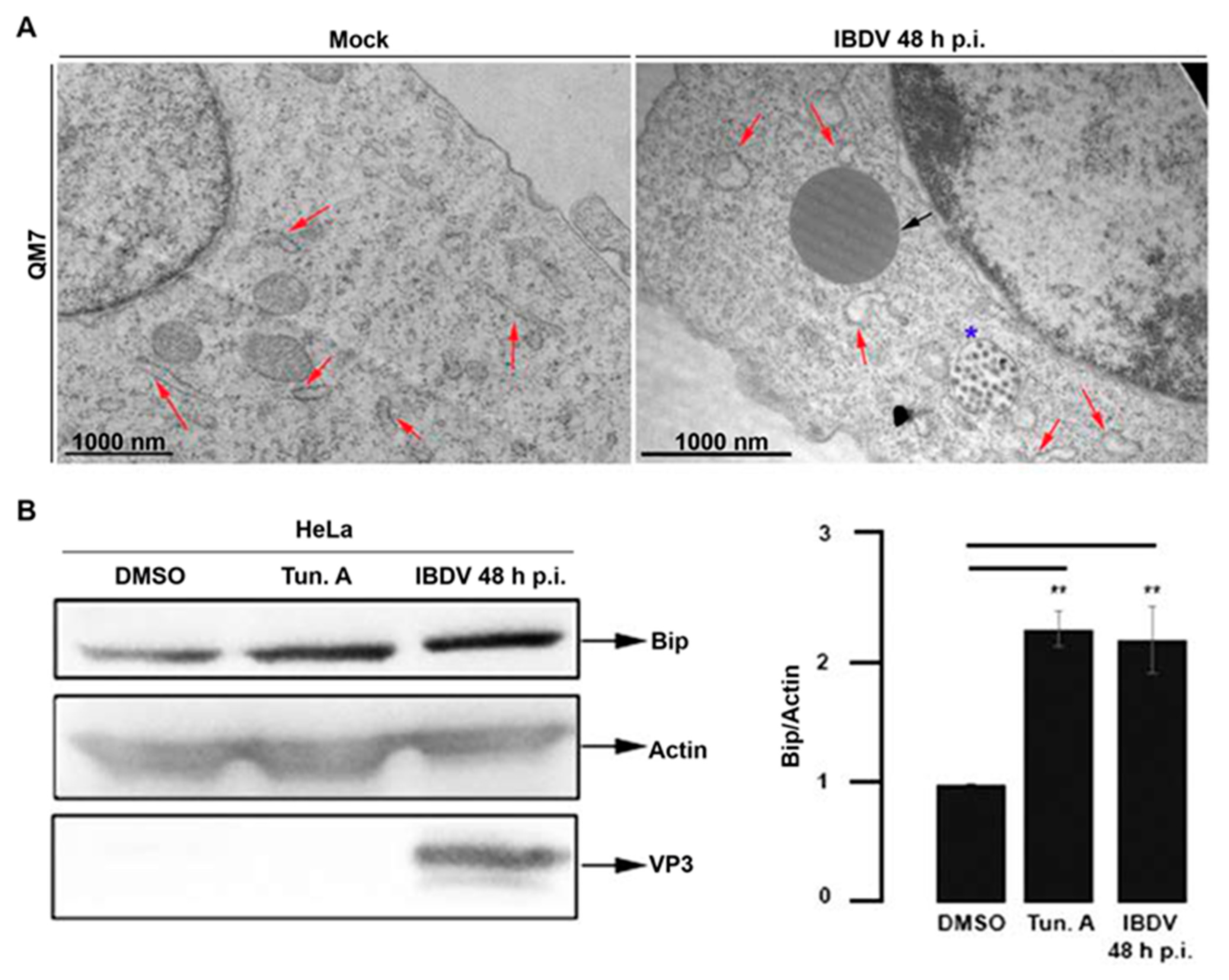
3.1. Newly Assembled IBDV Particles Are Enclosed within Single-Membrane Compartments Closely Associated with ER Membranes
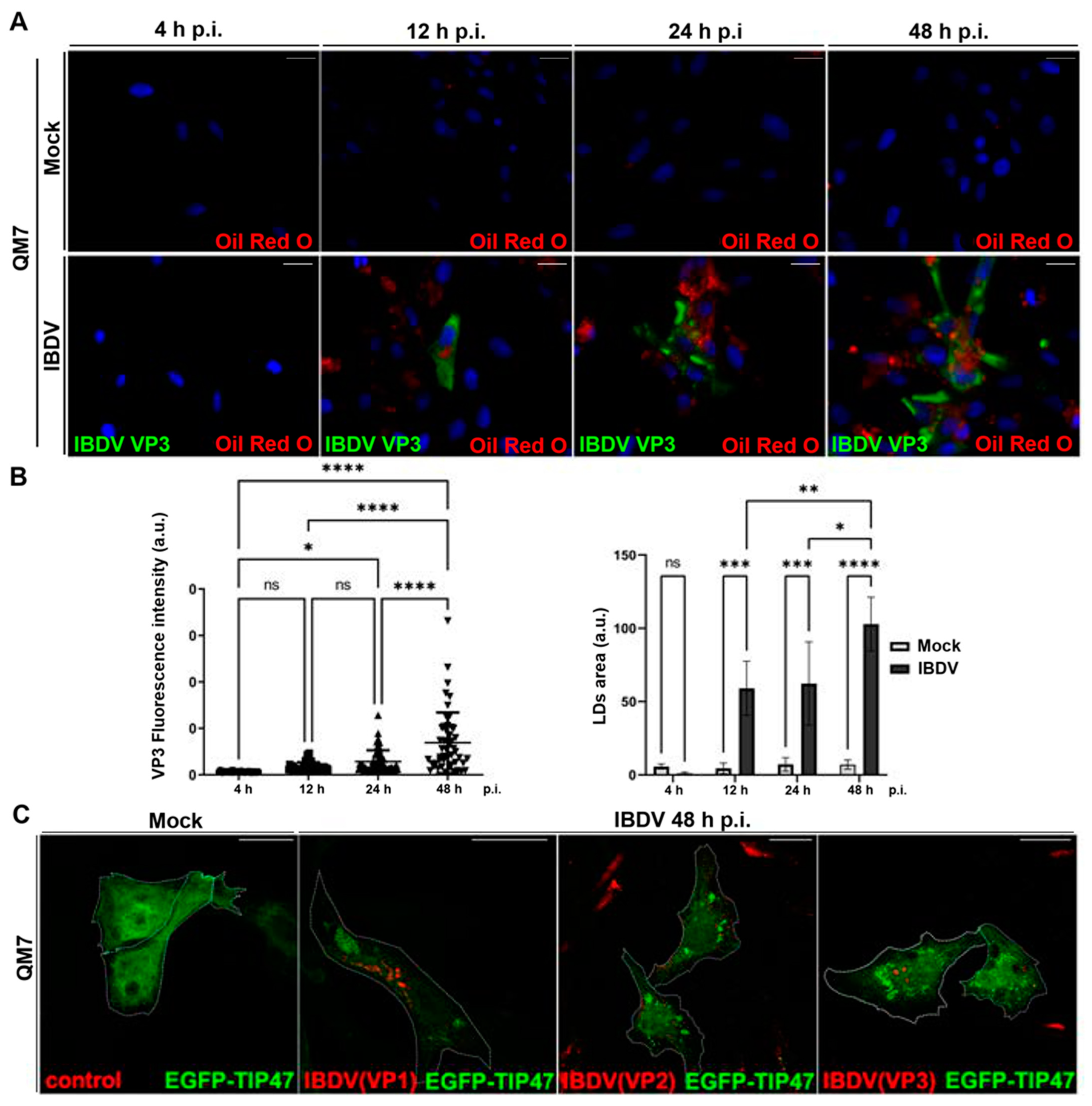
3.2. IBDV Infection Induces LDs and Chaperone BiP Accumulation in Host Cells
3.3. LDs Do Not Have a Role in IBDV Replication
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Payne, S. Family Birnaviridae. In Viruses; Payne, S., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 227–229. [Google Scholar]
- Coulibaly, F.; Chevalier, C.; Gutsche, I.; Pous, J.; Navaza, J.; Bressanelli, S.; Delmas, B.; Rey, F.A. The Birnavirus Crystal Structure Reveals Structural Relationships among Icosahedral Viruses. Cell 2005, 120, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, F.; Chevalier, C.; Delmas, B.; Rey, F.A. Crystal Structure of an Aquabirnavirus Particle: Insights into Antigenic Diversity and Virulence Determinism. J. Virol. 2010, 84, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Delmas, B.; Attoui, H.; Ghosh, S.; Malik, Y.S.; Mundt, E.; Vakharia, V.N. ICTV Virus Taxonomy Profile: Birnaviridae. J. Gen. Virol. 2019, 100, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Luque, D.; Rivas, G.; Alfonso, C.; Carrascosa, J.L.; Rodríguez, J.F.; Castón, J.R. Infectious Bursal Disease Virus Is an Icosahedral Polyploid DsRNA Virus. Proc. Natl. Acad. Sci. USA 2009, 106, 2148–2152. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Roy, P. The Molecular Biology of Bluetongue Virus Replication. Virus Res. 2014, 182, 5–20. [Google Scholar] [CrossRef]
- Pesavento, J.B.; Crawford, S.E.; Estes, M.K.; Prasad, B.V.V. Rotavirus Proteins: Structure and Assembly. Curr. Top. Microbiol. Immunol. 2006, 309, 189–219. [Google Scholar] [CrossRef]
- Lin, T.-W.; Lo, C.-W.; Lai, S.-Y.; Fan, R.-J.; Lo, C.-J.; Chou, Y.-M.; Thiruvengadam, R.; Wang, A.H.-J.; Wang, M.-Y. Chicken Heat Shock Protein 90 Is a Component of the Putative Cellular Receptor Complex of Infectious Bursal Disease Virus. J. Virol. 2007, 81, 8730–8741. [Google Scholar] [CrossRef]
- Chi, J.; You, L.; Li, P.; Teng, M.; Zhang, G.; Luo, J.; Wang, A. Surface IgM λ Light Chain Is Involved in the Binding and Infection of Infectious Bursal Disease Virus (IBDV) to DT40 Cells. Virus Genes 2018, 54, 236–245. [Google Scholar] [CrossRef]
- Liu, A.; Pan, Q.; Li, Y.; Yan, N.; Wang, J.; Yang, B.; Chen, Z.; Qi, X.; Gao, Y.; Gao, L.; et al. Identification of Chicken CD74 as a Novel Cellular Attachment Receptor for Infectious Bursal Disease Virus in Bursa B Lymphocytes. J. Virol. 2020, 94, e01712-19. [Google Scholar] [CrossRef]
- Liu, A.; Pan, Q.; Wang, S.; Zhang, Y.; Li, Y.; Wang, Y.; Qi, X.; Gao, L.; Liu, C.; Zhang, Y.; et al. Identification of Chicken CD44 as a Novel B Lymphocyte Receptor for Infectious Bursal Disease Virus. J. Virol. 2022, 96, e0011322. [Google Scholar] [CrossRef]
- Delgui, L.; Oña, A.; Gutiérrez, S.; Luque, D.; Navarro, A.; Castón, J.R.; Rodríguez, J.F. The Capsid Protein of Infectious Bursal Disease Virus Contains a Functional A4β1 Integrin Ligand Motif. Virology 2009, 386, 360–372. [Google Scholar] [CrossRef]
- Gimenez, M.C.; Rodríguez Aguirre, J.F.; Colombo, M.I.; Delgui, L.R. Infectious Bursal Disease Virus Uptake Involves Macropinocytosis and Trafficking to Early Endosomes in a Rab5-Dependent Manner. Cell. Microbiol. 2015, 17, 988–1007. [Google Scholar] [CrossRef]
- Galloux, M.; Libersou, S.; Morellet, N.; Bouaziz, S.; Da Costa, B.; Ouldali, M.; Lepault, J.; Delmas, B. Infectious Bursal Disease Virus, a Non-Enveloped Virus, Possesses a Capsid-Associated Peptide That Deforms and Perforates Biological Membranes. J. Biol. Chem. 2007, 282, 20774–20784. [Google Scholar] [CrossRef]
- Gimenez, M.C.; Zanetti, F.A.; Terebiznik, M.R.; Colombo, M.I.; Delgui, L.R. Infectious Bursal Disease Virus Hijacks Endosomal Membranes as the Scaffolding Structure for Viral Replication. J. Virol. 2018, 92, e01964-17. [Google Scholar] [CrossRef]
- Delgui, L.R.; Rodriguez, J.F.; Colombo, M.I. The Endosomal Pathway and the Golgi Complex Are Involved in the Infectious Bursal Disease Virus Life Cycle. J. Virol. 2013, 87, 8993–9007. [Google Scholar] [CrossRef]
- Gimenez, M.C.; Issa, M.; Sheth, J.; Colombo, M.I.; Terebiznik, M.R.; Delgui, L.R. Phosphatidylinositol 3-Phosphate Mediates the Establishment of Infectious Bursal Disease Virus Replication Complexes in Association with Early Endosomes. J. Virol. 2020, 95, e02313-20. [Google Scholar] [CrossRef]
- Campbell, E.A.; Reddy, V.R.A.P.; Gray, A.G.; Wells, J.; Simpson, J.; Skinner, M.A.; Hawes, P.C.; Broadbent, A.J. Discrete Virus Factories Form in the Cytoplasm of Cells Coinfected with Two Replication-Competent Tagged Reporter Birnaviruses That Subsequently Coalesce over Time. J. Virol. 2020, 94, e02107-19. [Google Scholar] [CrossRef]
- Reddy, V.R.A.P.; Campbell, E.A.; Wells, J.; Simpson, J.; Nazki, S.; Hawes, P.C.; Broadbent, A.J. Birnaviridae Virus Factories Show Features of Liquid-Liquid Phase Separation and Are Distinct from Paracrystalline Arrays of Virions Observed by Electron Microscopy. J. Virol. 2022, 96, e0202421. [Google Scholar] [CrossRef]
- Méndez, F.; Romero, N.; Cubas, L.L.; Delgui, L.R.; Rodríguez, D.; Rodríguez, J.F. Non-Lytic Egression of Infectious Bursal Disease Virus (IBDV) Particles from Infected Cells. PLoS ONE 2017, 12, e0170080. [Google Scholar] [CrossRef]
- Zhang, L.; Ren, X.; Chen, Y.; Gao, Y.; Wang, N.; Lu, Z.; Gao, L.; Qin, L.; Wang, Y.; Gao, H.; et al. Chondroitin Sulfate N-Acetylgalactosaminyltransferase-2 Contributes to the Replication of Infectious Bursal Disease Virus via Interaction with the Capsid Protein VP2. Viruses 2015, 7, 1474–1491. [Google Scholar] [CrossRef]
- Huang, H.L.; Wu, J.L.; Chen, M.H.C.; Hong, J.R. Aquatic Birnavirus-Induced ER Stress-Mediated Death Signaling Contribute to Downregulation of Bcl-2 Family Proteins in Salmon Embryo Cells. PLoS ONE 2011, 6, e22935. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, M.C.; Frontini-Lopez, Y.R.; Pocognoni, C.A.; Roldán, J.S.; García Samartino, C.; Uhart, M.; Colombo, M.I.; Terebiznik, M.R.; Delgui, L.R. Rab1b-GBF1-ARF1 Secretory Pathway Axis Is Required for Birnavirus Replication. J. Virol. 2022, 96, e0200521. [Google Scholar] [CrossRef] [PubMed]
- Farhan, H.; Rabouille, C. Signalling to and from the Secretory Pathway. J. Cell Sci. 2011, 124, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Tsai, B. How Viruses Use the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013250. [Google Scholar] [CrossRef]
- Samsa, M.M.; Mondotte, J.A.; Iglesias, N.G.; Assunção-Miranda, I.; Barbosa-Lima, G.; Da Poian, A.T.; Bozza, P.T.; Gamarnik, A.V. Dengue Virus Capsid Protein Usurps Lipid Droplets for Viral Particle Formation. PLoS Pathog. 2009, 5, e1000632. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, A. Virus-Induced ER Stress and the Unfolded Protein Response. Front. Plant Sci. 2012, 3, 293. [Google Scholar] [CrossRef]
- Grangeon, R.; Agbeci, M.; Chen, J.; Grondin, G.; Zheng, H.; Laliberté, J.-F. Impact on the Endoplasmic Reticulum and Golgi Apparatus of Turnip Mosaic Virus Infection. J. Virol. 2012, 86, 9255–9265. [Google Scholar] [CrossRef]
- Hansen, M.D.; Johnsen, I.B.; Stiberg, K.A.; Sherstova, T.; Wakita, T.; Richard, G.M.; Kandasamy, R.K.; Meurs, E.F.; Anthonsen, M.W. Hepatitis C Virus Triggers Golgi Fragmentation and Autophagy through the Immunity-Related GTPase M. Proc. Natl. Acad. Sci. USA 2017, 25, 3462–3471. [Google Scholar] [CrossRef]
- Fontana, J.; López-Montero, N.; Elliott, R.M.; Fernández, J.J.; Risco, C. The Unique Architecture of Bunyamwera Virus Factories around the Golgi Complex. Cell. Microbiol. 2008, 10, 2012–2028. [Google Scholar] [CrossRef]
- Jackson, W.T. Poliovirus-Induced Changes in Cellular Membranes throughout Infection. Curr. Opin. Virol. 2014, 9, 67–73. [Google Scholar] [CrossRef]
- Salonen, A.; Ahola, T.; Kaariainen, L. Viral RNA Replication in Association with Cellular Membranes. Curr. Top. Microbiol. Immunol. 2005, 285, 139–173. [Google Scholar] [CrossRef] [PubMed]
- Roldán, J.S.; Candurra, N.A.; Colombo, M.I.; Delgui, L.R. Junín Virus Promotes Autophagy To Facilitate the Virus Life Cycle. J. Virol. 2019, 93, e02307-18. [Google Scholar] [CrossRef]
- Gojanovich, A.D.; Gimenez, M.C.; Masone, D.; Rodriguez, T.M.; Dewey, R.A.; Delgui, L.R.; Bustos, D.M.; Uhart, M. Human Adipose-Derived Mesenchymal Stem/Stromal Cells Handling Protocols. Lipid Droplets and Proteins Double-Staining. Front. Cell Dev. Biol. 2018, 6, 33. [Google Scholar] [CrossRef]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harb. Protoc. 2018, 2018, 469–471. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Garriga, D.; Querol-Audí, J.; Abaitua, F.; Saugar, I.; Pous, J.; Verdaguer, N.; Castón, J.R.; Rodriguez, J.F. The 2.6-Angstrom Structure of Infectious Bursal Disease Virus-Derived T=1 Particles Reveals New Stabilizing Elements of the Virus Capsid. J. Virol. 2006, 80, 6895–6905. [Google Scholar] [CrossRef]
- Munafo, D.B.; Colombo, M.I. Induction of Autophagy Causes Dramatic Changes in the Subcellular Distribution of GFP-Rab24. Traffic 2002, 3, 472–482. [Google Scholar] [CrossRef]
- Terasaki, M.; Jaffe, L.A.; Hunnicutt, G.R.; Hammer, J.A., III. Structural Change of the Endoplasmic Reticulum during Fertilization: Evidence for Loss of Membrane Continuity Using the Green Fluorescent Protein. Dev. Biol. 1996, 179, 320–328. [Google Scholar] [CrossRef]
- Stornaiuolo, M.; Lotti, L.V.; Borgese, N.; Torrisi, M.-R.; Mottola, G.; Martire, G.; Bonatti, S. KDEL and KKXX Retrieval Signals Appended to the Same Reporter Protein Determine Different Trafficking between Endoplasmic Reticulum, Intermediate Compartment, and Golgi Complex. Mol. Biol. Cell 2003, 14, 889–902. [Google Scholar] [CrossRef]
- Raykhel, I.; Alanen, H.; Salo, K.; Jurvansuu, J.; Nguyen, V.D.; Latva-Ranta, M.; Ruddock, L. A Molecular Specificity Code for the Three Mammalian KDEL Receptors. J. Cell Biol. 2007, 179, 1193–1204. [Google Scholar] [CrossRef]
- Cui, X.A.; Zhang, H.; Ilan, L.; Liu, A.X.; Kharchuk, I.; Palazzo, A.F. MRNA Encoding Sec61β, a Tail-Anchored Protein, Is Localized on the Endoplasmic Reticulum. J. Cell Sci. 2015, 128, 3398–3410. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A. A New Paradigm: Innate Immune Sensing of Viruses via the Unfolded Protein Response. Front. Microbiol. 2014, 5, 222. [Google Scholar] [CrossRef]
- Ozcan, U. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Wu, J.; Back, S.-H.; Callaghan, M.U.; Ferris, S.P.; Iqbal, J.; Clark, R.; Miao, H.; Hassler, J.R.; Fornek, J.; et al. UPR Pathways Combine to Prevent Hepatic Steatosis Caused by ER Stress-Mediated Suppression of Transcriptional Master Regulators. Dev. Cell 2008, 15, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Fei, W.; Wang, H.; Fu, X.; Bielby, C.; Yang, H. Conditions of Endoplasmic Reticulum Stress Stimulate Lipid Droplet Formation in Saccharomyces Cerevisiae. Biochem. J. 2009, 424, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy Counterbalances Endoplasmic Reticulum Expansion during the Unfolded Protein Response. PLoS Biol. 2006, 4, e423. [Google Scholar] [CrossRef]
- Cao, X.; Jin, X.; Zhang, X.; Li, Y.; Wang, C.; Wang, X.; Hong, J.; Wang, X.; Li, D.; Zhang, Y. Morphogenesis of Endoplasmic Reticulum Membrane-Invaginated Vesicles during Beet Black Scorch Virus Infection: Role of Auxiliary Replication Protein and New Implications of Three-Dimensional Architecture. J. Virol. 2015, 89, 6184–6195. [Google Scholar] [CrossRef]
- Bamunusinghe, D.; Seo, J.-K.; Rao, A.L.N. Subcellular Localization and Rearrangement of Endoplasmic Reticulum by Brome Mosaic Virus Capsid Protein. J. Virol. 2011, 85, 2953–2963. [Google Scholar] [CrossRef]
- Cnop, M.; Igoillo-Esteve, M.; Cunha, D.A.; Ladrière, L.; Eizirik, D.L. An Update on Lipotoxic Endoplasmic Reticulum Stress in Pancreatic Beta-Cells. Biochem. Soc. Trans. 2008, 36, 909–915. [Google Scholar] [CrossRef]
- Guan, C.; Xu, W.; Hong, W.; Zhou, M.; Lin, F.; Fu, L.; Liu, D.; Xu, A. Quercetin Attenuates the Effects of H2O2 on Endoplasmic Reticulum Morphology and Tyrosinase Export from the Endoplasmic Reticulum in Melanocytes. Mol. Med. Rep. 2015, 11, 4285–4290. [Google Scholar] [CrossRef]
- Iinuma, T.; Aoki, T.; Arasaki, K.; Hirose, H.; Yamamoto, A.; Samata, R.; Hauri, H.-P.; Arimitsu, N.; Tagaya, M.; Tani, K. Role of Syntaxin 18 in the Organization of Endoplasmic Reticulum Subdomains. J. Cell Sci. 2009, 122, 1680–1690. [Google Scholar] [CrossRef]
- Sachdeva, M.M.; Claiborn, K.C.; Khoo, C.; Yang, J.; Groff, D.N.; Mirmira, R.G.; Stoffers, D.A. Pdx1 (MODY4) Regulates Pancreatic Beta Cell Susceptibility to ER Stress. Proc. Natl. Acad. Sci. USA 2009, 106, 19090–19095. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the Unfolded Protein Response Regulator GRP78/BiP in Development, Cancer, and Neurological Disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Cervantes-Ortiz, S.L.; Zamorano Cuervo, N.; Grandvaux, N. Respiratory Syncytial Virus and Cellular Stress Responses: Impact on Replication and Physiopathology. Viruses 2016, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Egan, P.A.; Sobkowiak, M.; Chan, S.-W. Hepatitis C Virus Envelope Protein E1 Binds PERK and Represses the Unfolded Protein Response. Open Virol. J. 2013, 7, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Jordan, R.; Wang, L.; Graczyk, T.M.; Block, T.M.; Romano, P.R. Replication of a Cytopathic Strain of Bovine Viral Diarrhea Virus Activates PERK and Induces Endoplasmic Reticulum Stress-Mediated Apoptosis of MDBK Cells. J. Virol. 2002, 76, 9588–9599. [Google Scholar] [CrossRef] [PubMed]
- Su, H.-L.; Liao, C.-L.; Lin, Y.-L. Japanese Encephalitis Virus Infection Initiates Endoplasmic Reticulum Stress and an Unfolded Protein Response. J. Virol. 2002, 76, 4162–4171. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. Measuring ER Stress and the Unfolded Protein Response Using Mammalian Tissue Culture System. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2011; Volume 490, pp. 71–92. [Google Scholar]
- Crawford, S.E.; Desselberger, U. Lipid Droplets Form Complexes with Viroplasms and Are Crucial for Rotavirus Replication. Curr. Opin. Virol. 2016, 19, 11–15. [Google Scholar] [CrossRef]
- Meyers, A.; del Rio, Z.P.; Beaver, R.A.; Morris, R.M.; Weiskittel, T.M.; Alshibli, A.K.; Mannik, J.; Morrell-Falvey, J.; Dalhaimer, P. Lipid Droplets Form from Distinct Regions of the Cell in the Fission Yeast Schizosaccharomyces Pombe. Traffic 2016, 17, 657–669. [Google Scholar] [CrossRef]
- Wolins, N.E.; Rubin, B.; Brasaemle, D.L. TIP47 Associates with Lipid Droplets. J. Biol. Chem. 2001, 276, 5101–5108. [Google Scholar] [CrossRef]
- Ohsaki, Y.; Maeda, T.; Maeda, M.; Tauchi-Sato, K.; Fujimoto, T. Recruitment of TIP47 to Lipid Droplets Is Controlled by the Putative Hydrophobic Cleft. Biochem. Biophys. Res. Commun. 2006, 347, 279–287. [Google Scholar] [CrossRef]
- Roingeard, P.; Melo, R.C.N. Lipid Droplet Hijacking by Intracellular Pathogens. Cell. Microbiol. 2017, 19, e12688. [Google Scholar] [CrossRef]
- Fader Kaiser, C.M.; Romano, P.S.; Vanrell, M.C.; Pocognoni, C.A.; Jacob, J.; Caruso, B.; Delgui, L.R. Biogenesis and Breakdown of Lipid Droplets in Pathological Conditions. Front. Cell Dev. Biol. 2022, 9, 826248. [Google Scholar] [CrossRef]
- Farías, M.A.; Diethelm-Varela, B.; Navarro, A.J.; Kalergis, A.M.; González, P.A. Interplay between Lipid Metabolism, Lipid Droplets, and DNA Virus Infections. Cells 2022, 11, 2224. [Google Scholar] [CrossRef]
- Brink, J.T.R.; Fourie, R.; Sebolai, O.; Albertyn, J.; Pohl, C.H. The Role of Lipid Droplets in Microbial Pathogenesis. J. Med. Microbiol. 2021, 70, 001383. [Google Scholar] [CrossRef]
- Lai, M.; De Carli, A.; Filipponi, C.; Iacono, E.; La Rocca, V.; Lottini, G.; Piazza, C.R.; Quaranta, P.; Sidoti, M.; Pistello, M.; et al. Lipid Balance Remodelling by Human Positive-Strand RNA Viruses and the Contribution of Lysosomes. Antivir. Res. 2022, 206, 105398. [Google Scholar] [CrossRef]
- Den Boon, J.A.; Diaz, A.; Ahlquist, P. Cytoplasmic Viral Replication Complexes. Cell Host Microbe 2010, 8, 77–85. [Google Scholar] [CrossRef]
- Novoa, R.R.; Calderita, G.; Arranz, R.; Fontana, J.; Granzow, H.; Risco, C. Virus Factories: Associations of Cell Organelles for Viral Replication and Morphogenesis. Biol. Cell 2005, 2, 147–172. [Google Scholar] [CrossRef]
- De Castro, I.F.; Volonté, L.; Risco, C. Virus Factories: Biogenesis and Structural Design. Cell. Microbiol. 2013, 1, 24–34. [Google Scholar] [CrossRef]
- Cosgrove, A.S. An Apparently New Disease of Chickens: Avian Nephrosis. Avian Dis. 1962, 6, 385. [Google Scholar] [CrossRef]
- Romero-Brey, I.; Bartenschlager, R. Endoplasmic Reticulum: The Favorite Intracellular Niche for Viral Replication and Assembly. Viruses 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Romero-Brey, I.; Bartenschlager, R. Membranous Replication Factories Induced by Plus-Strand RNA Viruses. Viruses 2014, 6, 2826–2857. [Google Scholar] [CrossRef] [PubMed]
- Harak, C.; Lohmann, V. Ultrastructure of the Replication Sites of Positive-Strand RNA Viruses. Virology 2015, 479–480, 418–433. [Google Scholar] [CrossRef] [PubMed]
- Wolff, G.; Melia, C.E.; Snijder, E.J.; Bárcena, M. Double-Membrane Vesicles as Platforms for Viral Replication. Trends Microbiol. 2020, 28, 1022–1033. [Google Scholar] [CrossRef]
- Belov, G.A.; Nair, V.; Hansen, B.T.; Hoyt, F.H.; Fischer, E.R.; Ehrenfeld, E. Complex Dynamic Development of Poliovirus Membranous Replication Complexes. J. Virol. 2012, 86, 302–312. [Google Scholar] [CrossRef]
- Limpens, R.W.A.L.; van der Schaar, H.M.; Kumar, D.; Koster, A.J.; Snijder, E.J.; van Kuppeveld, F.J.M.; Bárcena, M. The Transformation of Enterovirus Replication Structures: A Three-Dimensional Study of Single- and Double-Membrane Compartments. mBio 2011, 2, e00166-11. [Google Scholar] [CrossRef]
- Melia, C.E.; van der Schaar, H.M.; de Jong, A.W.M.; Lyoo, H.R.; Snijder, E.J.; Koster, A.J.; van Kuppeveld, F.J.M.; Bárcena, M. The Origin, Dynamic Morphology, and PI4P-Independent Formation of Encephalomyocarditis Virus Replication Organelles. mBio 2018, 9, e00420-18. [Google Scholar] [CrossRef]
- Doerflinger, S.Y.; Cortese, M.; Romero-Brey, I.; Menne, Z.; Tubiana, T.; Schenk, C.; White, P.A.; Bartenschlager, R.; Bressanelli, S.; Hansman, G.S.; et al. Membrane Alterations Induced by Nonstructural Proteins of Human Norovirus. PLOS Pathog. 2017, 13, e1006705. [Google Scholar] [CrossRef]
- Ferraris, P.; Beaumont, E.; Uzbekov, R.; Brand, D.; Gaillard, J.; Blanchard, E.; Roingeard, P. Sequential Biogenesis of Host Cell Membrane Rearrangements Induced by Hepatitis C Virus Infection. Cell. Mol. Life Sci. 2013, 70, 1297–1306. [Google Scholar] [CrossRef]
- Paul, D.; Madan, V.; Ramirez, O.; Bencun, M.; Stoeck, I.K.; Jirasko, V.; Bartenschlager, R. Glycine Zipper Motifs in Hepatitis C Virus Nonstructural Protein 4B Are Required for the Establishment of Viral Replication Organelles. J. Virol. 2018, 92, e01890-17. [Google Scholar] [CrossRef]
- van der Hoeven, B.; Oudshoorn, D.; Koster, A.J.; Snijder, E.J.; Kikkert, M.; Bárcena, M. Biogenesis and Architecture of Arterivirus Replication Organelles. Virus Res. 2016, 220, 70–90. [Google Scholar] [CrossRef]
- Pelham, H.R.; Hardwick, K.G.; Lewis, M.J. Sorting of Soluble ER Proteins in Yeast. EMBO J. 1988, 7, 1757–1762. [Google Scholar] [CrossRef]
- Wang, Y.; Duan, Y.; Han, C.; Yao, S.; Qi, X.; Gao, Y.; Maier, H.J.; Britton, P.; Chen, L.; Zhang, L.; et al. Infectious Bursal Disease Virus Subverts Autophagic Vacuoles To Promote Viral Maturation and Release. J. Virol. 2017, 91, e01883-16. [Google Scholar] [CrossRef]
- Gamil, A.; Mutoloki, S.; Evensen, Ø. A Piscine Birnavirus Induces Inhibition of Protein Synthesis in CHSE-214 Cells Primarily through the Induction of EIF2α Phosphorylation. Viruses 2015, 7, 1987–2005. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frontini-López, Y.R.; Rivera, L.; Pocognoni, C.A.; Roldán, J.S.; Colombo, M.I.; Uhart, M.; Delgui, L.R. Infectious Bursal Disease Virus Assembly Causes Endoplasmic Reticulum Stress and Lipid Droplet Accumulation. Viruses 2023, 15, 1295. https://doi.org/10.3390/v15061295
Frontini-López YR, Rivera L, Pocognoni CA, Roldán JS, Colombo MI, Uhart M, Delgui LR. Infectious Bursal Disease Virus Assembly Causes Endoplasmic Reticulum Stress and Lipid Droplet Accumulation. Viruses. 2023; 15(6):1295. https://doi.org/10.3390/v15061295
Chicago/Turabian StyleFrontini-López, Yesica R., Lautaro Rivera, Cristian A. Pocognoni, Julieta S. Roldán, María I. Colombo, Marina Uhart, and Laura R. Delgui. 2023. "Infectious Bursal Disease Virus Assembly Causes Endoplasmic Reticulum Stress and Lipid Droplet Accumulation" Viruses 15, no. 6: 1295. https://doi.org/10.3390/v15061295