
In Silico and In Vitro Evaluation of Some Amidine Derivatives as Hit Compounds towards Development of Inhibitors against Coronavirus Diseases

 ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. In Silico Docking Study
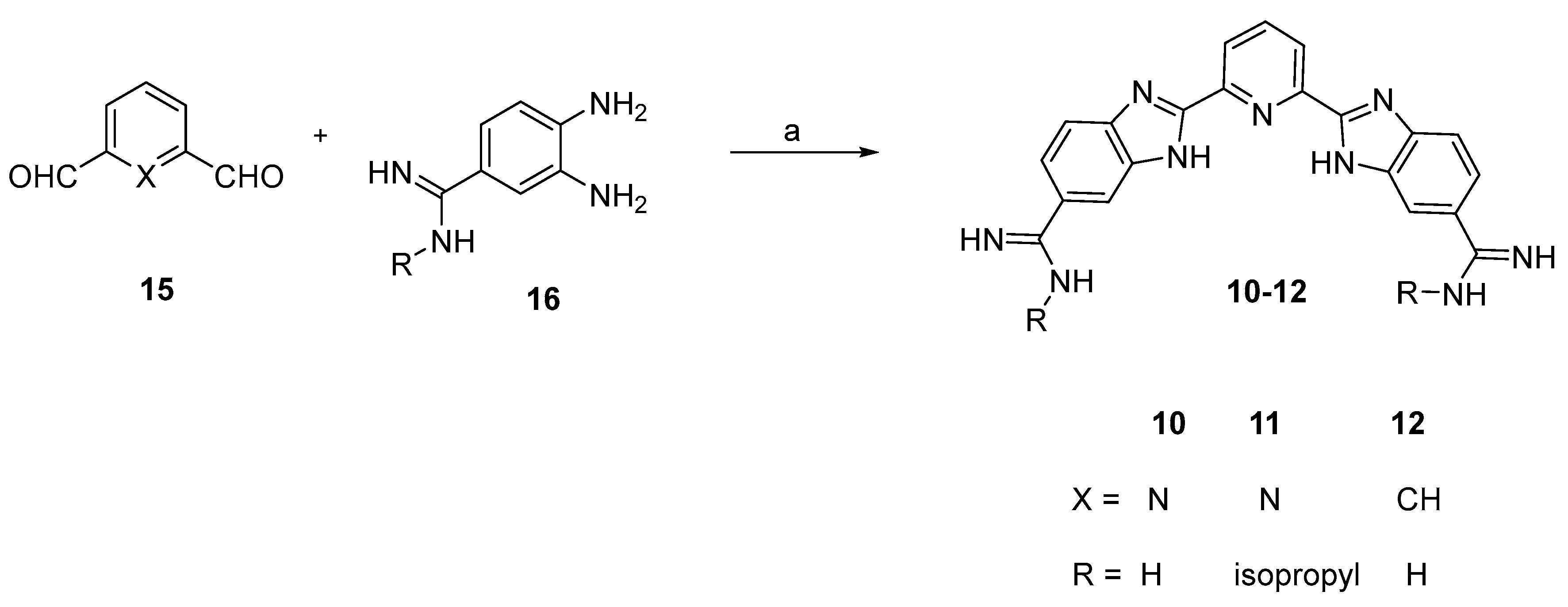
2.2. Chemistry
2.3. Cells and Materials
2.4. Cell Fusion Assay Using Dual Split Proteins (DSP)
2.5. Enzyme Assays
2.6. Pseudovirus Assay
2.7. Cell Toxicity Assay
3. Results
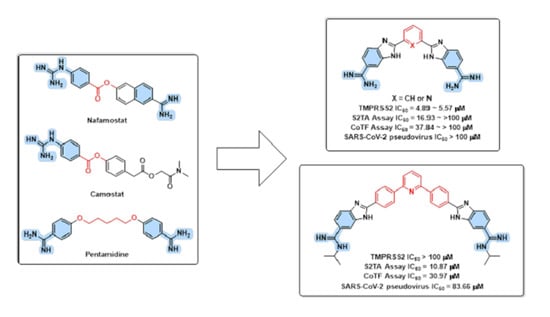
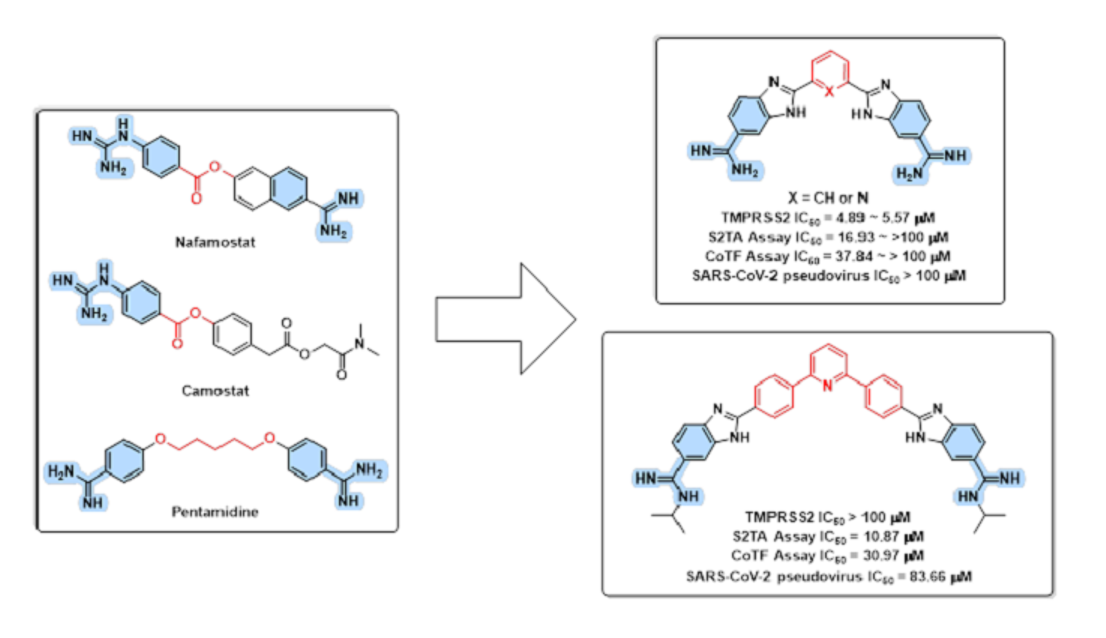
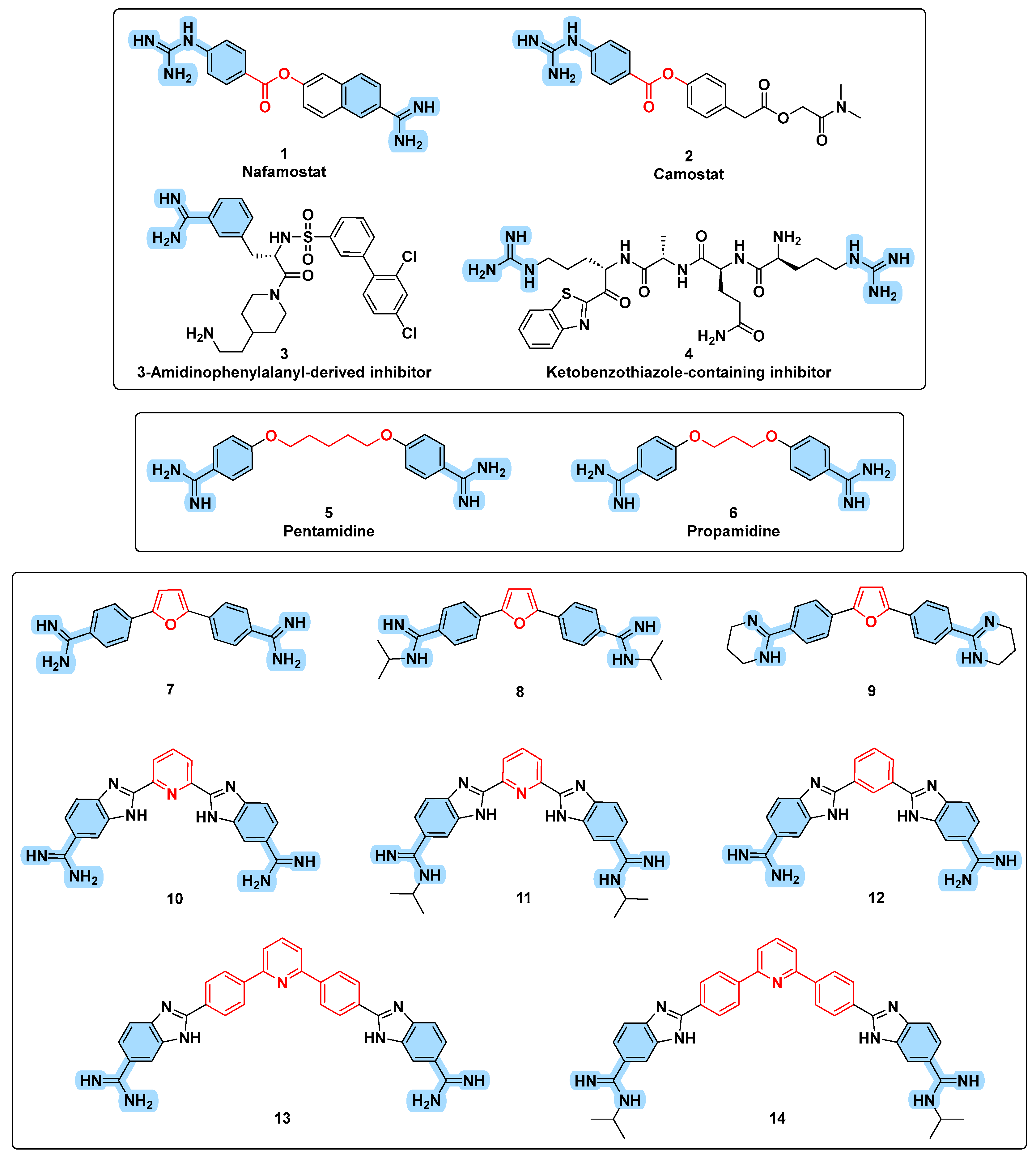
3.1. Design Rational
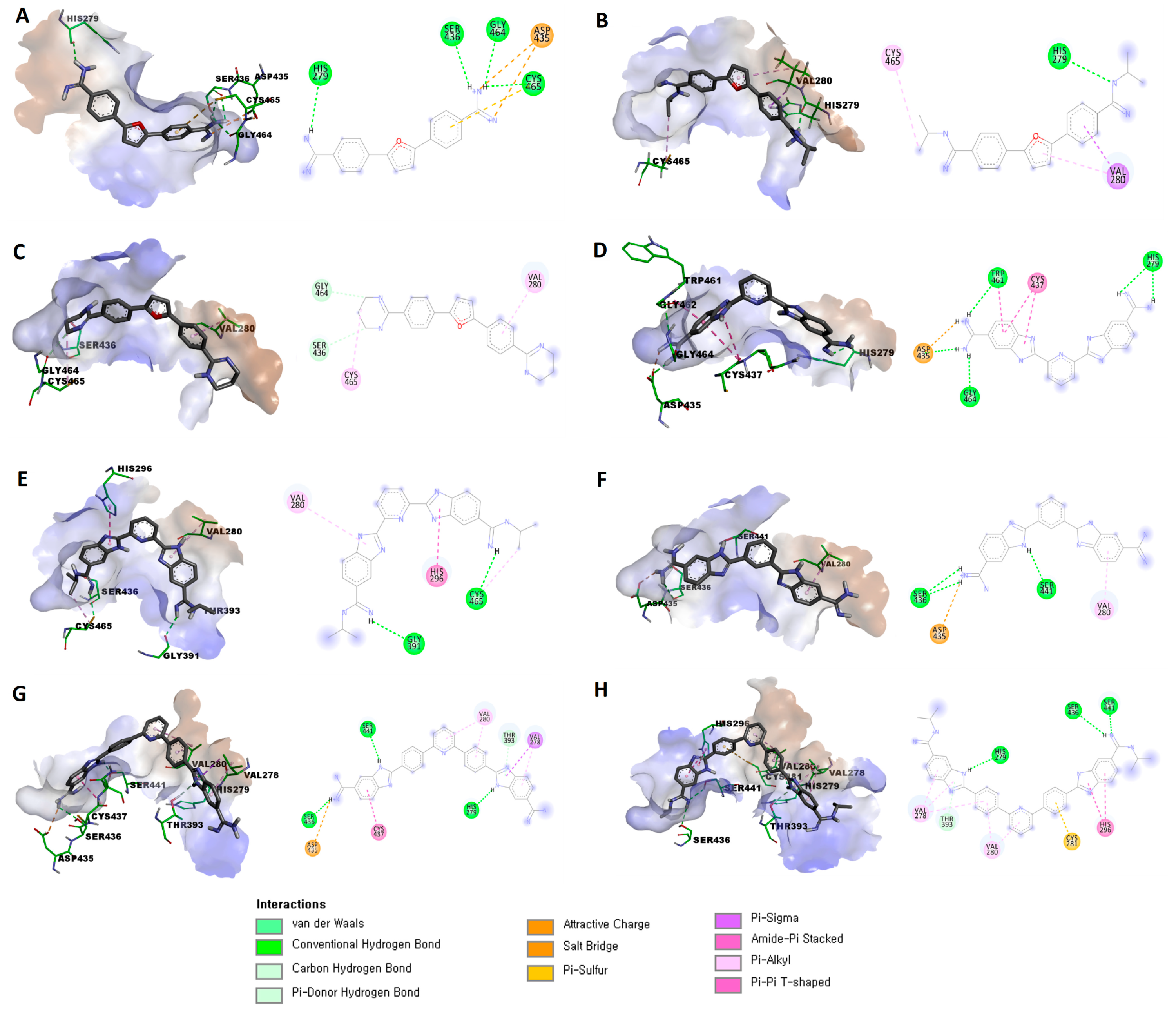
3.2. In Silico Evaluation
3.3. Chemistry
3.4. In Vitro Evaluations
3.4.1. TMPRSS2 Inhibition Assay
3.4.2. Cell Fusion Assays
3.4.3. Pseudovirus Entry and Cell Viability Assays
3.4.4. Thrombin and Factor Xa Enzyme Inhibition Assays
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chang, L.; Yan, Y.; Wang, L. Coronavirus Disease 2019: Coronaviruses and Blood Safety. Transfus. Med. Rev. 2020, 34, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Lian, X.; Su, X.; Wu, W.; Marraro, G.A.; Zeng, Y. From SARS and MERS to COVID-19: A brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir. Res. 2020, 21, 224. [Google Scholar] [CrossRef] [PubMed]
- Notarte, K.I.; Catahay, J.A.; Velasco, J.V.; Pastrana, A.; Ver, A.T.; Pangilinan, F.C.; Peligro, P.J.; Casimiro, M.; Guerrero, J.J.; Gellaco, M.M.L.; et al. Impact of COVID-19 vaccination on the risk of developing long-COVID and on existing long-COVID symptoms: A systematic review. EClinicalMedicine 2022, 53, 101624. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Shao, W.; Chen, X.; Zhang, B.; Wang, G.; Zhang, W. Real-world effectiveness of COVID-19 vaccines: A literature review and meta-analysis. Int. J. Infect. Dis. 2022, 114, 252–260. [Google Scholar] [CrossRef]
- Iacopetta, D.; Ceramella, J.; Catalano, A.; Saturnino, C.; Pellegrino, M.; Mariconda, A.; Longo, P.; Sinicropi, M.S.; Aquaro, S. COVID-19 at a Glance: An Up-to-Date Overview on Variants, Drug Design and Therapies. Viruses 2022, 14, 573. [Google Scholar] [CrossRef]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef]
- Fernández-de-las-Peñas, C. Long COVID: Current definition. Infection 2022, 50, 285–286. [Google Scholar] [CrossRef]
- Raveendran, A.V.; Jayadevan, R.; Sashidharan, S. Long COVID: An overview. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15, 869–875. [Google Scholar] [CrossRef]
- Parigger, L.; Krassnigg, A.; Schopper, T.; Singh, A.; Tappler, K.; Köchl, K.; Hetmann, M.; Gruber, K.; Steinkellner, G.; Gruber, C.C. Recent changes in the mutational dynamics of the SARS-CoV-2 main protease substantiate the danger of emerging resistance to antiviral drugs. Front. Med. 2022, 9, 1061142. [Google Scholar] [CrossRef]
- Jiao, Z.; Yan, Y.; Chen, Y.; Wang, G.; Wang, X.; Li, L.; Yang, M.; Hu, X.; Guo, Y.; Shi, Y.; et al. Adaptive Mutation in the Main Protease Cleavage Site of Feline Coronavirus Renders the Virus More Resistant to Main Protease Inhibitors. J. Virol. 2022, 96, e00907–e00922. [Google Scholar] [CrossRef]
- Kumari, M.; Lu, R.-M.; Li, M.-C.; Huang, J.-L.; Hsu, F.-F.; Ko, S.-H.; Ke, F.-Y.; Su, S.-C.; Liang, K.-H.; Yuan, J.P.-Y.; et al. A critical overview of current progress for COVID-19: Development of vaccines, antiviral drugs, and therapeutic antibodies. J. Biomed. Sci. 2022, 29, 68. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Chen, X.; Wu, J.; Duan, X.; Men, K. Small molecules in the treatment of COVID-19. Signal Transduct. Target. Ther. 2022, 7, 387. [Google Scholar] [CrossRef] [PubMed]
- Mei, M.; Tan, X. Current Strategies of Antiviral Drug Discovery for COVID-19. Front. Mol. Biosci. 2021, 8, 671263. [Google Scholar] [CrossRef]
- Mahoney, M.; Damalanka, V.C.; Tartell, M.A.; Chung, D.h.; Lourenço, A.L.; Pwee, D.; Mayer Bridwell, A.E.; Hoffmann, M.; Voss, J.; Karmakar, P.; et al. A novel class of TMPRSS2 inhibitors potently block SARS-CoV-2 and MERS-CoV viral entry and protect human epithelial lung cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2108728118. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Du, W.; Song, M.; Liu, Q.; Herrmann, A.; Huang, Q. Spontaneous binding of potential COVID-19 drugs (Camostat and Nafamostat) to human serine protease TMPRSS2. Comput. Struct. Biotechnol. J. 2021, 19, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.Á.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 2020, 63, 12359–12386. [Google Scholar] [CrossRef]
- Fraser, B.J.; Beldar, S.; Seitova, A.; Hutchinson, A.; Mannar, D.; Li, Y.; Kwon, D.; Tan, R.; Wilson, R.P.; Leopold, K.; et al. Structure and activity of human TMPRSS2 protease implicated in SARS-CoV-2 activation. Nat. Chem. Biol. 2022, 18, 963–971. [Google Scholar] [CrossRef]
- Wettstein, L.; Kirchhoff, F.; Münch, J. The Transmembrane Protease TMPRSS2 as a Therapeutic Target for COVID-19 Treatment. Int. J. Mol. Sci. 2022, 23, 1351. [Google Scholar] [CrossRef]
- Hassan, A.H.E.; Kim, H.J.; Park, K.; Choi, Y.; Moon, S.; Lee, C.H.; Kim, Y.J.; Cho, S.B.; Gee, M.S.; Lee, D.; et al. Synthesis and Biological Evaluation of O6-Aminoalkyl-Hispidol Analogs as Multifunctional Monoamine Oxidase-B Inhibitors towards Management of Neurodegenerative Diseases. Antioxidants 2023, 12, 1033. [Google Scholar] [CrossRef]
- Hassan, A.H.E.; Wang, C.Y.; Lee, H.J.; Jung, S.J.; Kim, Y.J.; Cho, S.B.; Lee, C.H.; Ham, G.; Oh, T.; Lee, S.K.; et al. Scaffold hopping of N-benzyl-3,4,5-trimethoxyaniline: 5,6,7-Trimethoxyflavan derivatives as novel potential anticancer agents modulating hippo signaling pathway. Eur. J. Med. Chem. 2023, 256, 115421. [Google Scholar] [CrossRef]
- Gulia, K.; Hassan, A.H.E.; Lenhard, J.R.; Farahat, A.A. Escaping ESKAPE resistance: In vitro and in silico studies of multifunctional carbamimidoyl-tethered indoles against antibiotic-resistant bacteria. R. Soc. Open Sci. 2023, 10, 230020. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Paik, S.; Ali, E.M.H.; Hassan, A.H.E.; Kang, S.J.; Lee, K.; Roh, E.J. Identification of Novel Aryl Carboxamide Derivatives as Death-Associated Protein Kinase 1 (DAPK1) Inhibitors with Anti-Proliferative Activities: Design, Synthesis, In Vitro, and In Silico Biological Studies. Pharmaceuticals 2022, 15, 1050. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.H.E.; Kim, H.J.; Gee, M.S.; Park, J.-H.; Jeon, H.R.; Lee, C.J.; Choi, Y.; Moon, S.; Lee, D.; Lee, J.K.; et al. Positional scanning of natural product hispidol’s ring-B: Discovery of highly selective human monoamine oxidase-B inhibitor analogues downregulating neuroinflammation for management of neurodegenerative diseases. J. Enzyme Inhib. Med. Chem. 2022, 37, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-H.; Shin, J.-S.; Chung, K.-S.; Kim, J.-M.; Jung, S.-H.; Yoo, H.-S.; Hassan, A.H.E.; Lee, J.K.; Inn, K.-S.; Lee, S.; et al. 3ʹ,4ʹ-Dihydroxyflavone mitigates inflammatory responses by inhibiting LPS and TLR4/MD2 interaction. Phytomedicine 2023, 109, 154553. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.H.E.; Park, K.T.; Kim, H.J.; Lee, H.J.; Kwon, Y.H.; Hwang, J.Y.; Jang, C.-G.; Chung, J.H.; Park, K.D.; Lee, S.J.; et al. Fluorinated CRA13 analogues: Synthesis, in vitro evaluation, radiosynthesis, in silico and in vivo PET study. Bioorg. Chem. 2020, 99, 103834. [Google Scholar] [CrossRef]
- El-Demerdash, A.; Al-Karmalawy, A.A.; Abdel-Aziz, T.M.; Elhady, S.S.; Darwish, K.M.; Hassan, A.H.E. Investigating the structure–activity relationship of marine natural polyketides as promising SARS-CoV-2 main protease inhibitors. RSC Adv. 2021, 11, 31339–31363. [Google Scholar] [CrossRef]
- Hassan, A.H.E.; Lee, K.-T.; Lee, Y.S. Flavone-based arylamides as potential anticancers: Design, synthesis and in vitro cell-based/cell-free evaluations. Eur. J. Med. Chem. 2020, 187, 111965. [Google Scholar] [CrossRef]
- Paul, A.; Nanjunda, R.; Kumar, A.; Laughlin, S.; Nhili, R.; Depauw, S.; Deuser, S.S.; Chai, Y.; Chaudhary, A.S.; David-Cordonnier, M.-H.; et al. Mixed up minor groove binders: Convincing A·T specific compounds to recognize a G·C base pair. Bioorg. Med. Chem. Lett. 2015, 25, 4927–4932. [Google Scholar] [CrossRef]
- Anbazhagan, M.; Boykin, D.W.; Stephens, C.E. Direct Conversion of Amidoximes to Amidines via Transfer Hydrogenation. Synthesis 2003, 2003, 2467–2469. [Google Scholar] [CrossRef]
- Yamamoto, M.; Gohda, J.; Kobayashi, A.; Tomita, K.; Hirayama, Y.; Koshikawa, N.; Seiki, M.; Semba, K.; Akiyama, T.; Kawaguchi, Y.; et al. Metalloproteinase-Dependent and TMPRSS2-Independent Cell Surface Entry Pathway of SARS-CoV-2 Requires the Furin Cleavage Site and the S2 Domain of Spike Protein. mBio 2022, 13, e00519–e00522. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kiso, M.; Sakai-Tagawa, Y.; Iwatsuki-Horimoto, K.; Imai, M.; Takeda, M.; Kinoshita, N.; Ohmagari, N.; Gohda, J.; Semba, K.; et al. The Anticoagulant Nafamostat Potently Inhibits SARS-CoV-2 S Protein-Mediated Fusion in a Cell Fusion Assay System and Viral Infection In Vitro in a Cell-Type-Dependent Manner. Viruses 2020, 12, 629. [Google Scholar] [CrossRef] [PubMed]
- Tani, H.; Komoda, Y.; Matsuo, E.; Suzuki, K.; Hamamoto, I.; Yamashita, T.; Moriishi, K.; Fujiyama, K.; Kanto, T.; Hayashi, N.; et al. Replication-Competent Recombinant Vesicular Stomatitis Virus Encoding Hepatitis C Virus Envelope Proteins. J. Virol. 2007, 81, 8601–8612. [Google Scholar] [CrossRef] [PubMed]
- Tani, H.; Shiokawa, M.; Kaname, Y.; Kambara, H.; Mori, Y.; Abe, T.; Moriishi, K.; Matsuura, Y. Involvement of Ceramide in the Propagation of Japanese Encephalitis Virus. J. Virol. 2010, 84, 2798–2807. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.H.E.; Mahmoud, K.; Phan, T.-N.; Shaldam, M.A.; Lee, C.H.; Kim, Y.J.; Cho, S.B.; Bayoumi, W.A.; El-Sayed, S.M.; Choi, Y.; et al. Bestatin analogs-4-quinolinone hybrids as antileishmanial hits: Design, repurposing rational, synthesis, in vitro and in silico studies. Eur. J. Med. Chem. 2023, 250, 115211. [Google Scholar] [CrossRef]
- Hassan, A.H.E.; Phan, T.-N.; Choi, Y.; Moon, S.; No, J.H.; Lee, Y.S. Design, Rational Repurposing, Synthesis, In Vitro Evaluation, Homology Modeling and In Silico Study of Sulfuretin Analogs as Potential Antileishmanial Hit Compounds. Pharmaceuticals 2022, 15, 1058. [Google Scholar] [CrossRef]
- Hassan, A.H.E.; Phan, T.-N.; Yoon, S.; Lee, C.J.; Jeon, H.R.; Kim, S.-H.; No, J.H.; Lee, Y.S. Pyrrolidine-based 3-deoxysphingosylphosphorylcholine analogs as possible candidates against neglected tropical diseases (NTDs): Identification of hit compounds towards development of potential treatment of Leishmania donovani. J. Enzyme Inhib. Med. Chem. 2021, 36, 1922–1930. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Paik, S.; Hassan, A.H.E.; Lee, Y.S.; Roh, E.J. Hit discovery of 4-amino-N-(4-(3-(trifluoromethyl)phenoxy)pyrimidin-5-yl)benzamide: A novel EGFR inhibitor from a designed small library. Bioorg. Chem. 2017, 75, 393–405. [Google Scholar] [CrossRef]
- Hassan, A.H.E.; Phan, T.-N.; Moon, S.; Lee, C.H.; Kim, Y.J.; Cho, S.B.; El-Sayed, S.M.; Choi, Y.; No, J.H.; Lee, Y.S. Design, synthesis, and repurposing of O6-aminoalkyl-sulfuretin analogs towards discovery of potential lead compounds as antileishmanial agents. Eur. J. Med. Chem. 2023, 251, 115256. [Google Scholar] [CrossRef]
- Farag, A.K.; Hassan, A.H.E.; Ahn, B.S.; Park, K.D.; Roh, E.J. Reprofiling of pyrimidine-based DAPK1/CSF1R dual inhibitors: Identification of 2,5-diamino-4-pyrimidinol derivatives as novel potential anticancer lead compounds. J. Enzyme Inhib. Med. Chem. 2020, 35, 311–324. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Hassan, A.H.E.; Paik, S.; Sup Lee, Y.; Lee, H.-H.; Shin, J.-S.; Lee, K.-T.; Roh, E.J. EGFR inhibitors from cancer to inflammation: Discovery of 4-fluoro-N-(4-(3-(trifluoromethyl)phenoxy)pyrimidin-5-yl)benzamide as a novel anti-inflammatory EGFR inhibitor. Bioorg. Chem. 2019, 86, 112–118. [Google Scholar] [CrossRef]
- Kang, S.; Lee, J.M.; Jeon, B.; Elkamhawy, A.; Paik, S.; Hong, J.; Oh, S.-J.; Paek, S.H.; Lee, C.J.; Hassan, A.H.E.; et al. Repositioning of the antipsychotic trifluoperazine: Synthesis, biological evaluation and in silico study of trifluoperazine analogs as anti-glioblastoma agents. Eur. J. Med. Chem. 2018, 151, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.H.E.; Yoo, S.Y.; Lee, K.W.; Yoon, Y.M.; Ryu, H.W.; Jeong, Y.; Shin, J.-S.; Kang, S.-Y.; Kim, S.-Y.; Lee, H.-H.; et al. Repurposing mosloflavone/5,6,7-trimethoxyflavone-resveratrol hybrids: Discovery of novel p38-α MAPK inhibitors as potent interceptors of macrophage-dependent production of proinflammatory mediators. Eur. J. Med. Chem. 2019, 180, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.J.; Hobbs, D.C.F.; Umeda, M.; Nagata, A.; Yamaguchi, R.; Sato, Y.; Sato, A.; Ohmatsu, K.; Ooi, T.; Yanai, T.; et al. In Silico Analysis and Synthesis of Nafamostat Derivatives and Evaluation of Their Anti-SARS-CoV-2 Activity. Viruses 2022, 14, 389. [Google Scholar] [CrossRef] [PubMed]
- Vardhan, S.; Sahoo, S.K. Virtual screening by targeting proteolytic sites of furin and TMPRSS2 to propose potential compounds obstructing the entry of SARS-CoV-2 virus into human host cells. J. Tradit. Complement. Med. 2022, 12, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Yang, J.; Xiao, D.; Yin, J.; Song, M.; Xu, Y.; Zhao, L.; Dai, Q.; Li, Y.; Wang, C.; et al. Nafamostat mesylate as a broad-spectrum candidate for the treatment of flavivirus infections by targeting envelope proteins. Antivir. Res. 2022, 202, 105325. [Google Scholar] [CrossRef]
- Midgley, I.; Hood, A.J.; Proctor, P.; Chasseaud, L.F.; Irons, S.R.; Cheng, K.N.; Brindley, C.J.; Bonn, R. Metabolic fate of 14C-camostat mesylate in man, rat and dog after intravenous administration. Xenobiotica 1994, 24, 79–92. [Google Scholar] [CrossRef]
- Tsukagoshi, S. Pharmacokinetics studies of nafamostat mesilate (FUT), a synthetic protease inhibitor, which has been used for the treatments of DIC and acute pancreatitis, and as an anticoagulant in extracorporeal circulation. Gan Kagaku Ryoho 2000, 27, 767–774. [Google Scholar]
- González-Bello, C. Designing Irreversible Inhibitors—Worth the Effort? ChemMedChem 2016, 11, 22–30. [Google Scholar] [CrossRef]
- Hempel, T.; Raich, L.; Olsson, S.; Azouz, N.P.; Klingler, A.M.; Hoffmann, M.; Pöhlmann, S.; Rothenberg, M.E.; Noé, F. Molecular mechanism of inhibiting the SARS-CoV-2 cell entry facilitator TMPRSS2 with camostat and nafamostat. Chem. Sci. 2021, 12, 983–992. [Google Scholar] [CrossRef]
- Huang, X.; Pearce, R.; Omenn, G.S.; Zhang, Y. Identification of 13 Guanidinobenzoyl- or Aminidinobenzoyl-Containing Drugs to Potentially Inhibit TMPRSS2 for COVID-19 Treatment. Int. J. Mol. Sci. 2021, 22, 7060. [Google Scholar] [CrossRef]
- Peiffer, A.L.; Garlick, J.M.; Wu, Y.; Soellner, M.B.; Brooks, C.L.; Mapp, A.K. TMPRSS2 inhibitor discovery facilitated through an in silico and biochemical screening platform. bioRxiv 2021. [Google Scholar] [CrossRef]
- Meyer, D.; Sielaff, F.; Hammami, M.; Böttcher-Friebertshäuser, E.; Garten, W.; Steinmetzer, T. Identification of the first synthetic inhibitors of the type II transmembrane serine protease TMPRSS2 suitable for inhibition of influenza virus activation. Biochem. J. 2013, 452, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Pilgram, O.; Keils, A.; Benary, G.E.; Müller, J.; Merkl, S.; Ngaha, S.; Huber, S.; Chevillard, F.; Harbig, A.; Magdolen, V.; et al. Improving the selectivity of 3-amidinophenylalanine-derived matriptase inhibitors. Eur. J. Med. Chem. 2022, 238, 114437. [Google Scholar] [CrossRef] [PubMed]
- Colombo, É.; Désilets, A.; Duchêne, D.; Chagnon, F.; Najmanovich, R.; Leduc, R.; Marsault, E. Design and Synthesis of Potent, Selective Inhibitors of Matriptase. ACS Med. Chem. Lett. 2012, 3, 530–534. [Google Scholar] [CrossRef]
- Beaulieu, A.; Gravel, É.; Cloutier, A.; Marois, I.; Colombo, É.; Désilets, A.; Verreault, C.; Leduc, R.; Marsault, É.; Richter, M.V. Matriptase Proteolytically Activates Influenza Virus and Promotes Multicycle Replication in the Human Airway Epithelium. J. Virol. 2013, 87, 4237–4251. [Google Scholar] [CrossRef]
- Patrick, G.L. Plasmepsins as targets for antimalarial agents. In Antimalarial Agents; Patrick, G.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 217–270. [Google Scholar]
- Nanjappan, S.K.; Surendran, S.; Paul, D. Pharmacokinetics and pharmacodynamics of peptidomimetics. In Peptide and Peptidomimetic Therapeutics; Qvit, N., Rubin, S.J.S., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 195–211. [Google Scholar]
- Trabocchi, A. Principles and applications of small molecule peptidomimetics. In Small Molecule Drug Discovery; Trabocchi, A., Lenci, E., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 163–195. [Google Scholar]
- Pelay-Gimeno, M.; Glas, A.; Koch, O.; Grossmann, T.N. Structure-Based Design of Inhibitors of Protein–Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem. Int. Ed. 2015, 54, 8896–8927. [Google Scholar] [CrossRef]
- Li Petri, G.; Di Martino, S.; De Rosa, M. Peptidomimetics: An Overview of Recent Medicinal Chemistry Efforts toward the Discovery of Novel Small Molecule Inhibitors. J. Med. Chem. 2022, 65, 7438–7475. [Google Scholar] [CrossRef]
- Tamanini, E.; Buck, I.M.; Chessari, G.; Chiarparin, E.; Day, J.E.H.; Frederickson, M.; Griffiths-Jones, C.M.; Hearn, K.; Heightman, T.D.; Iqbal, A.; et al. Discovery of a Potent Nonpeptidomimetic, Small-Molecule Antagonist of Cellular Inhibitor of Apoptosis Protein 1 (cIAP1) and X-Linked Inhibitor of Apoptosis Protein (XIAP). J. Med. Chem. 2017, 60, 4611–4625. [Google Scholar] [CrossRef]
- Kumar, R.; Bavi, R.; Jo, M.G.; Arulalapperumal, V.; Baek, A.; Rampogu, S.; Kim, M.O.; Lee, K.W. New compounds identified through in silico approaches reduce the α-synuclein expression by inhibiting prolyl oligopeptidase in vitro. Sci. Rep. 2017, 7, 10827. [Google Scholar] [CrossRef]
- Trent, J.O.; Clark, G.R.; Kumar, A.; Wilson, W.D.; Boykin, D.W.; Hall, J.E.; Tidwell, R.R.; Blagburn, B.L.; Neidle, S. Targeting the Minor Groove of DNA: Crystal Structures of Two Complexes between Furan Derivatives of Berenil and the DNA Dodecamer d(CGCGAATTCGCG)2. J. Med. Chem. 1996, 39, 4554–4562. [Google Scholar] [CrossRef]
- Das, B.P.; Boykin, D.W. Synthesis and antiprotozoal activity of 2,5-bis(4-guanylphenyl)furans. J. Med. Chem. 1977, 20, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Moumbock, A.F.; Tran, H.T.; Lamy, E.; Günther, S. BC-11 is a covalent TMPRSS2 fragment inhibitor that impedes SARS-CoV-2 host cell entry. Arch. Pharm. 2022, 356, e2200371. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Kumar, A.; Nanjunda, R.; Farahat, A.A.; Boykin, D.W.; Wilson, W.D. Systematic synthetic and biophysical development of mixed sequence DNA binding agents. Org. Biomol. Chem. 2017, 15, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Depauw, S.; Lambert, M.; Jambon, S.; Paul, A.; Peixoto, P.; Nhili, R.; Marongiu, L.; Figeac, M.; Dassi, C.; Paul-Constant, C.; et al. Heterocyclic Diamidine DNA Ligands as HOXA9 Transcription Factor Inhibitors: Design, Molecular Evaluation, and Cellular Consequences in a HOXA9-Dependant Leukemia Cell Model. J. Med. Chem. 2019, 62, 1306–1329. [Google Scholar] [CrossRef]
- Fuentes-Prior, P. Priming of SARS-CoV-2 S protein by several membrane-bound serine proteinases could explain enhanced viral infectivity and systemic COVID-19 infection. J. Biol. Chem. 2021, 296, 100135. [Google Scholar] [CrossRef]
- Kandeel, M.; Yamamoto, M.; Tani, H.; Kobayashi, A.; Gohda, J.; Kawaguchi, Y.; Park, B.K.; Kwon, H.-J.; Inoue, J.-i.; Alkattan, A. Discovery of New Fusion Inhibitor Peptides against SARS-CoV-2 by Targeting the Spike S2 Subunit. Biomol. Ther. 2021, 29, 282–289. [Google Scholar] [CrossRef]
- Giannis, D.; Ziogas, I.A.; Gianni, P. Coagulation disorders in coronavirus infected patients: COVID-19, SARS-CoV-1, MERS-CoV and lessons from the past. J. Clin. Virol. 2020, 127, 104362. [Google Scholar] [CrossRef]
- Mackman, N.; Antoniak, S.; Wolberg, A.S.; Kasthuri, R.; Key, N.S. Coagulation Abnormalities and Thrombosis in Patients Infected with SARS-CoV-2 and Other Pandemic Viruses. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2033–2044. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Mercadante, M.; Nilsson-Payant, B.; Johnson, J.L.; Jaimes, J.A.; Muecksch, F.; Weisblum, Y.; Bram, Y.; Chandar, V.; Whittaker, G.R.; et al. Coagulation factors directly cleave SARS-CoV-2 spike and enhance viral entry. eLife 2022, 11, e77444. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Iwata-Yoshikawa, N.; Kakizaki, M.; Shiwa-Sudo, N.; Okura, T.; Tahara, M.; Fukushi, S.; Maeda, K.; Kawase, M.; Asanuma, H.; Tomita, Y.; et al. Essential role of TMPRSS2 in SARS-CoV-2 infection in murine airways. Nat. Commun. 2022, 13, 6100. [Google Scholar] [CrossRef] [PubMed]
- Quinn, T.M.; Gaughan, E.E.; Bruce, A.; Antonelli, J.; O’Connor, R.; Li, F.; McNamara, S.; Koch, O.; MacKintosh, C.; Dockrell, D.; et al. Randomised controlled trial of intravenous nafamostat mesylate in COVID pneumonitis: Phase 1b/2a experimental study to investigate safety, Pharmacokinetics and Pharmacodynamics. eBioMedicine 2022, 76, 103856. [Google Scholar] [CrossRef]
- Kinoshita, T.; Shinoda, M.; Nishizaki, Y.; Shiraki, K.; Hirai, Y.; Kichikawa, Y.; Tsushima, K.; Shinkai, M.; Komura, N.; Yoshida, K.; et al. A multicenter, double-blind, randomized, parallel-group, placebo-controlled study to evaluate the efficacy and safety of camostat mesilate in patients with COVID-19 (CANDLE study). BMC Med. 2022, 20, 342. [Google Scholar] [CrossRef]
- Gunst, J.D.; Staerke, N.B.; Pahus, M.H.; Kristensen, L.H.; Bodilsen, J.; Lohse, N.; Dalgaard, L.S.; Brønnum, D.; Fröbert, O.; Hønge, B.; et al. Efficacy of the TMPRSS2 inhibitor camostat mesilate in patients hospitalized with COVID-19-a double-blind randomized controlled trial. eClinicalMedicine 2021, 35, 100849. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Binding Score 1 | Established Interactions |
|---|---|---|
| 7 | −5.7172 | Three hydrogen bonds between (Gly464, Cys465, and Ser436) and the protons of one amidine group. |
| Hydrogen bond between His279 with a proton of the other amidine group. | ||
| Attractive charge between Asp435 and one amidine group. | ||
| π-sulfur interaction between Cys465 and the phenyl (linker) group. | ||
| 8 | −6.5412 | Hydrogen bond between His279 with protons of one amidine group. |
| π-σ interaction between Val280 and the phenyl (linker) group. | ||
| π-alkyl interaction between Val280 and the furan central ring. | ||
| Alkyl interaction between Cys465 and isopropyl substitution on one amidine group. | ||
| 9 | −6.0349 | C-H bond between Ser436 and protons of the tetrahydropyrimidine ring. |
| C-H bond between Gly464 and protons of the tetrahydropyrimidine ring. | ||
| π-alkyl interaction between Val280 and the phenyl (linker) group. | ||
| Alkyl interaction between Cys465 and the tetrahydropyrimidine ring. | ||
| 10 | −6.2871 | Salt bridge between Asp435 and one amidine group. |
| Three hydrogen bonds between (Asp435, Gly464, and Trp461) and the protons of one amidine group. | ||
| Two hydrogen bonds between His279 and the other amidine group. | ||
| Two amide-π stacked interactions between Cys437 and the benzimidazole ring. | ||
| Amide-π stacked interactions between Trp461 and the benzimidazole ring. | ||
| 11 | −7.4058 | Hydrogen bond between Cys465 with protons of one amidine group. |
| Hydrogen bond between Gly391 with protons of the other amidine group. | ||
| Alkyl interaction between Cys465 and isopropyl substitution on one amidine group. | ||
| π-π T-shaped interaction between His296 and the benzimidazole ring. | ||
| π -alkyl interaction between Val280 and the benzimidazole ring. | ||
| 12 | −6.4987 | Salt bridge between Asp435 and one amidine group. |
| Two hydrogen bonds between Ser436 and (the same) amidine group. | ||
| Hydrogen bond between Ser441 with a proton of the benzimidazole ring. | ||
| π-alkyl interaction between Val280 and the benzimidazole ring. | ||
| 13 | −7.6965 | Salt bridge between Asp435 and one amidine group. |
| Hydrogen bond between Ser436 and (the same) amidine group. | ||
| Hydrogen bond between Ser441 with a proton of the benzimidazole ring. | ||
| Hydrogen bond between His279 with a proton of the other benzimidazole ring. | ||
| π-donor hydrogen bond between Thr393 with a proton of the benzimidazole ring. | ||
| π-alkyl interaction between Val278 with the (same) benzimidazole ring. | ||
| π-σ interaction between Val278 with the (same) benzimidazole ring. | ||
| π-alkyl interaction between Val280 and the phenyl (linker) group. | ||
| π-alkyl interaction between Val280 and the pyridine central ring. | ||
| Amide-π stacked interaction between Cys437 and the benzimidazole ring. | ||
| 14 | −8.4438 | Two hydrogen bonds between Ser441 and Ser436 with a proton of one amidine group. |
| Hydrogen bond between His279 with a proton of the benzimidazole ring. | ||
| Two π-alkyl interaction between Val278 with the (same) benzimidazole ring. | ||
| π-π interaction between His296 with the (other) benzimidazole ring. | ||
| π-alkyl interaction between Val278 with the phenyl (linker) group. | ||
| π-alkyl interaction between Val280 with the phenyl (linker) group. | ||
| π-alkyl interaction between Val280 with the pyridine central ring. | ||
| π-donor hydrogen bond between Thr393 with a proton of the benzimidazole ring. | ||
| π-sulfur interaction between Cys281 with the phenyl (linker) group. |
| Compound | TMPRSS2 IC50 (µM) | S2TA Assay | CoTF Assay IC50 (µM) | |
|---|---|---|---|---|
| % Inhibition 1 | IC50 (µM) | |||
| 7 | 82.7 | 39.91 | >100 | >100 |
| 10 | 5.57 | 89.53 | 16.93 | 37.84 |
| 11 | >10 2 | 44.17 | >100 | >100 |
| 12 | 4.89 | 55.42 | >100 | >100 |
| 13 | 72.4 | 93.98 | 13.70 | 37.00 |
| 14 | >100 | 97.33 | 10.87 | 30.97 |
| Pentamidine | 3.98 | 71.30 | 32.27 | >100 |
| Nafamostat | <0.001 | 89.32 | 0.007115 | >100 |
| Compound | SARS-CoV-2 Pseudovirus | Cell Viability Inhibition IC50 (µM) | |
|---|---|---|---|
| % Inhibition 1 | IC50 (µM) | ||
| 7 | 2.66 | >100 | >100 |
| 10 | 19.57 | >100 | >100 |
| 11 | 27.02 | >100 | >100 |
| 12 | 17.07 | >100 | >100 |
| 13 | 20.53 | >100 | >100 |
| 14 | 77.44 | 83.66 | >100 |
| Pentamidine | 69.48 | 45 | >100 |
| Nafamostat | 87.34 | 0.06 | >100 |
| Compound | IC50 (µM) | |
|---|---|---|
| Thrombin | Factor Xa | |
| 7 | 79.1 | 30.9 |
| 10 | 0.921 | 5.93 |
| 11 | >1 1 | >1 1 |
| 12 | 0.862 | 34.1 |
| 13 | 0.929 | 4.89 |
| 14 | 2.25 | >10 1 |
| Pentamidine | 1.51 | 6.22 |
| Nafamostat | 0.0341 | 1.74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, A.H.E.; El-Sayed, S.M.; Yamamoto, M.; Gohda, J.; Matsumoto, T.; Shirouzu, M.; Inoue, J.-i.; Kawaguchi, Y.; Mansour, R.M.A.; Anvari, A.; et al. In Silico and In Vitro Evaluation of Some Amidine Derivatives as Hit Compounds towards Development of Inhibitors against Coronavirus Diseases. Viruses 2023, 15, 1171. https://doi.org/10.3390/v15051171
Hassan AHE, El-Sayed SM, Yamamoto M, Gohda J, Matsumoto T, Shirouzu M, Inoue J-i, Kawaguchi Y, Mansour RMA, Anvari A, et al. In Silico and In Vitro Evaluation of Some Amidine Derivatives as Hit Compounds towards Development of Inhibitors against Coronavirus Diseases. Viruses. 2023; 15(5):1171. https://doi.org/10.3390/v15051171
Chicago/Turabian StyleHassan, Ahmed H. E., Selwan M. El-Sayed, Mizuki Yamamoto, Jin Gohda, Takehisa Matsumoto, Mikako Shirouzu, Jun-ichiro Inoue, Yasushi Kawaguchi, Reem M. A. Mansour, Abtin Anvari, and et al. 2023. "In Silico and In Vitro Evaluation of Some Amidine Derivatives as Hit Compounds towards Development of Inhibitors against Coronavirus Diseases" Viruses 15, no. 5: 1171. https://doi.org/10.3390/v15051171