
Evaluating Novel Quantification Methods for Infectious Baculoviruses

Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Baculovirus Stock
2.2. Quantification of Stock Titer
2.3. Quantitative Polymerase Chain Reaction
2.4. Flow Cytometric Titration
3. Results
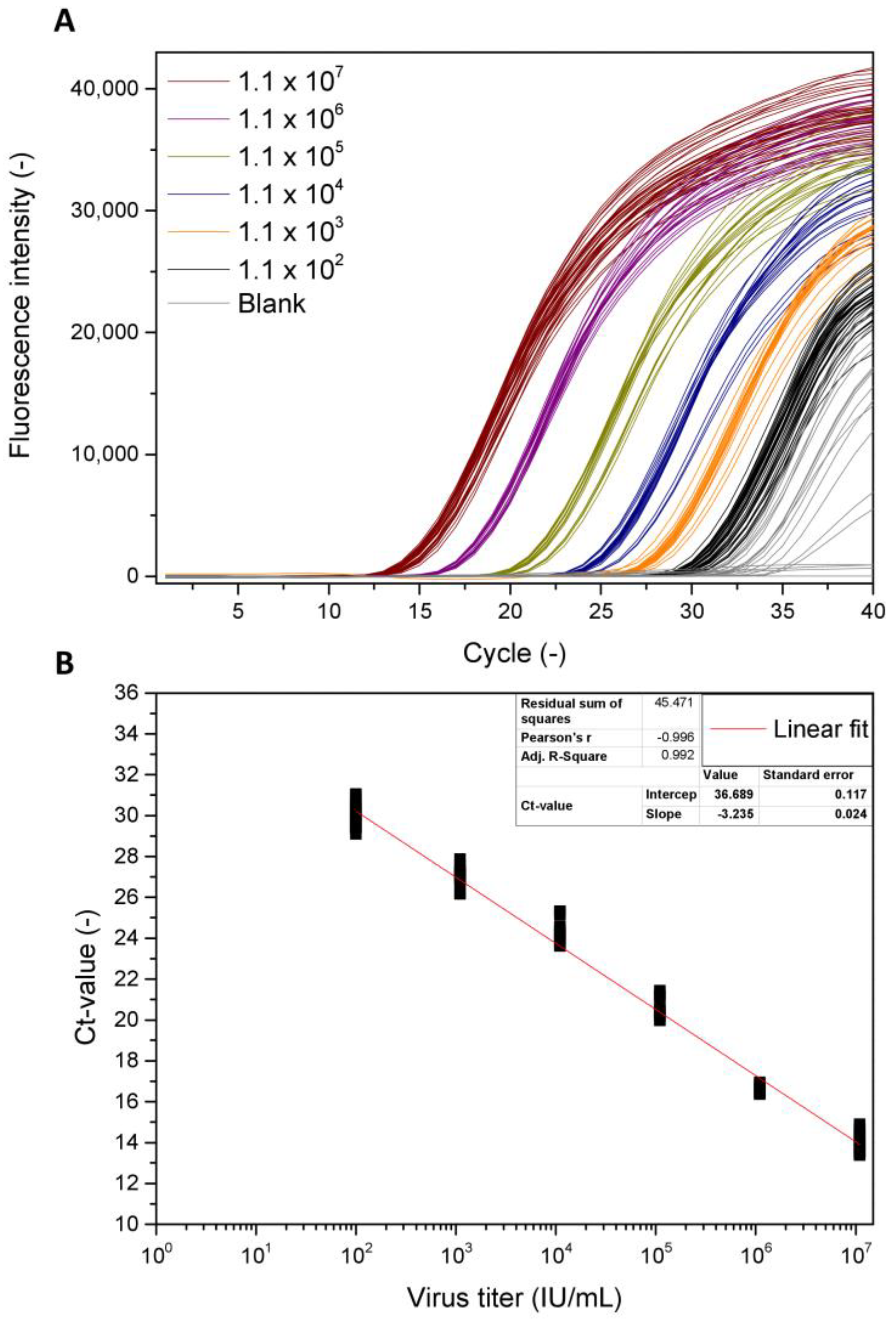
3.1. qPCR as a Referenrence Method for BV Quantification
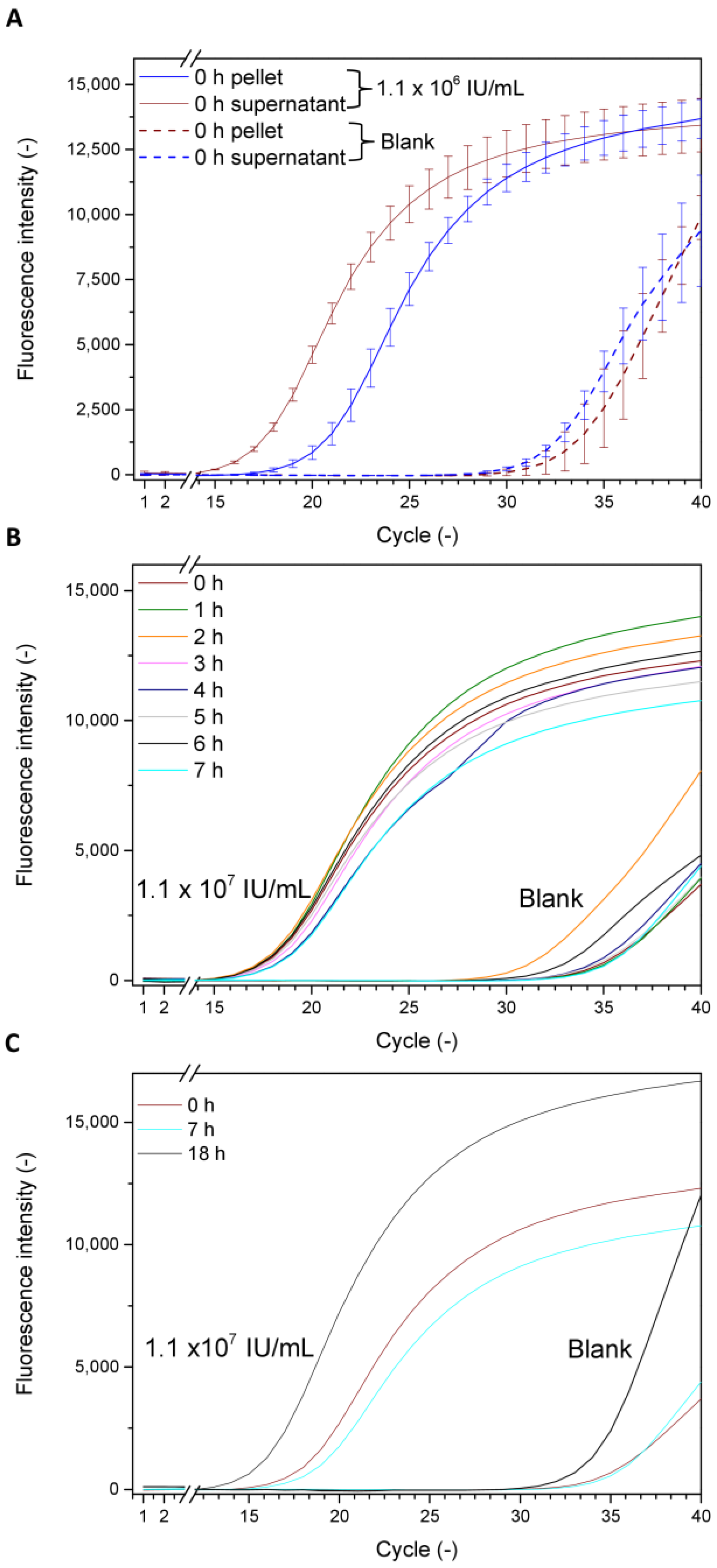
3.1.1. Estimating the Virus Infection Kinetic Based on qPCR Measurements
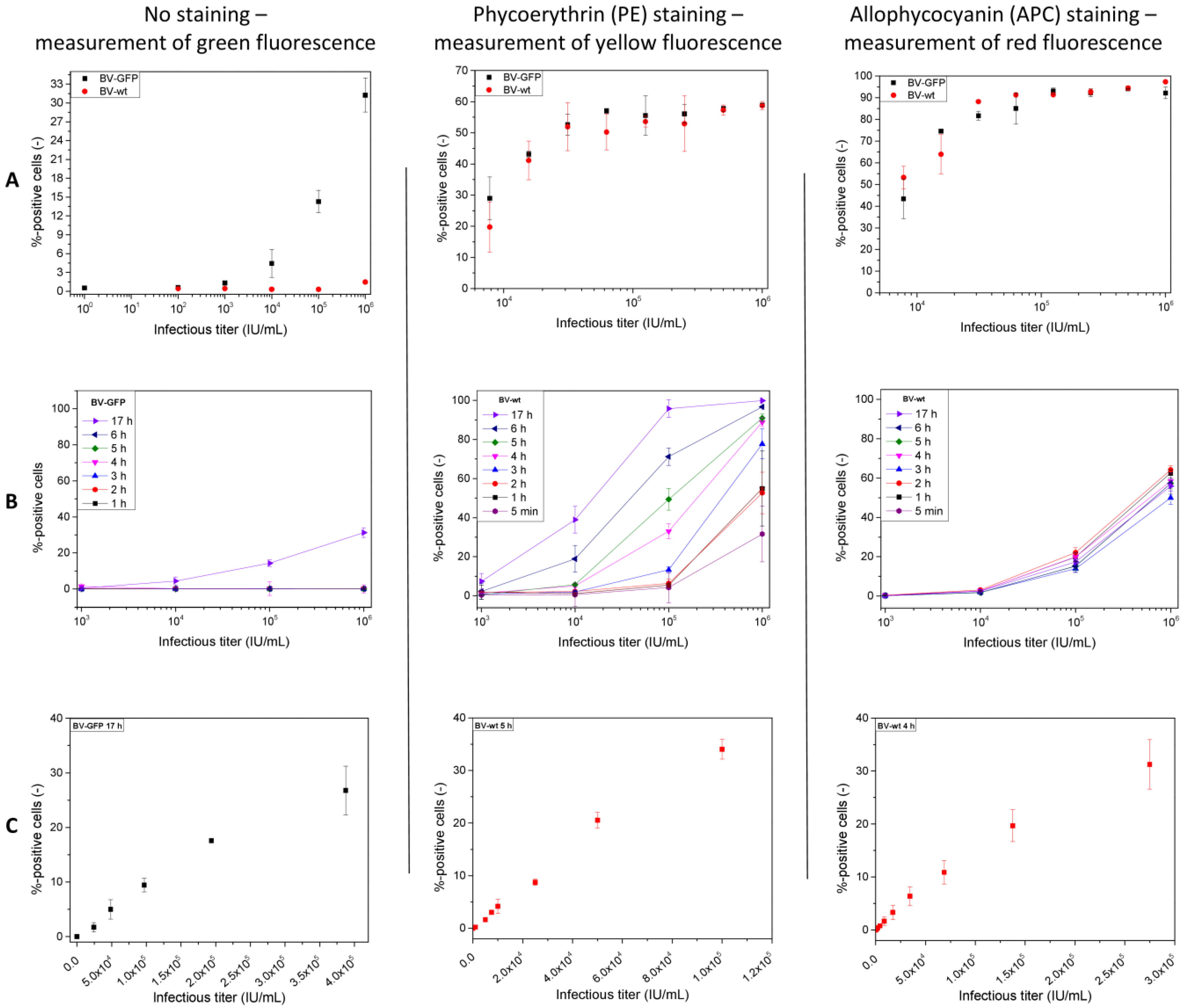
3.2. Flow Cytometric Quantification
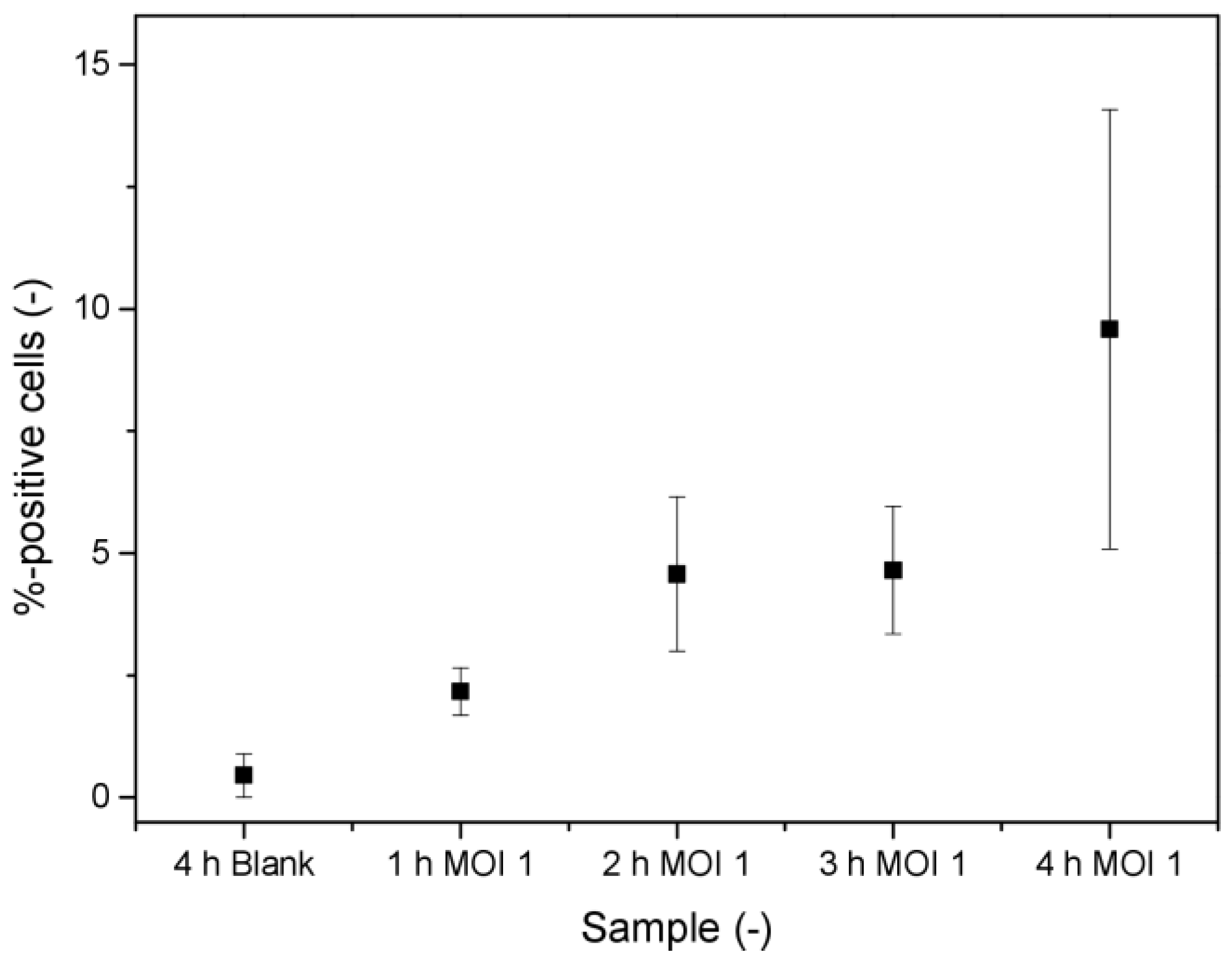
3.3. Labeling of Virus-Derived mRNA in Infected Cells for Flow Cytometric Detection
4. Discussion
4.1. qPCR as a Referenrence Method for BV Quantification
4.2. Flow Cytometric Quantification Based on Fluorophore Expression and Protein Labeling
4.3. Labeling of Virus-Derived mRNA in Infected Cells for Flow Cytometric Detection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heidbuechel, J.P.W.; Engeland, C.E. Paramyxoviruses for Tumor-targeted Immunomodulation: Design and Evaluation Ex Vivo. J. Vis. Exp. 2019, 143, 58651. [Google Scholar] [CrossRef]
- Wang, R.; Wang, Y.; Liu, F.; Luo, S. Orf virus: A promising new therapeutic agent. Rev. Med. Virol. 2019, 29, e2013. [Google Scholar] [CrossRef] [PubMed]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Keiser, M.S.; Davidson, B.L. Viral Vectors for Gene Transfer. Curr. Protoc. Mouse Biol. 2018, 8, e58. [Google Scholar] [CrossRef]
- Hemminki, O.; Dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, e84. [Google Scholar] [CrossRef]
- Lawler, S.E.; Speranza, M.-C.; Cho, C.-F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef]
- Genzel, Y. Designing cell lines for viral vaccine production: Where do we stand? Biotechnol. J. 2015, 10, 728–740. [Google Scholar] [CrossRef]
- Grein, T.A.; Loewe, D.; Dieken, H.; Salzig, D.; Weidner, T.; Czermak, P. High titer oncolytic measles virus production process by integration of dielectric spectroscopy as online monitoring system. Biotechnol. Bioeng. 2018, 115, 1186–1194. [Google Scholar] [CrossRef]
- Bissinger, T.; Wu, Y.; Marichal-Gallardo, P.; Riedel, D.; Liu, X.; Genzel, Y.; Tan, W.S.; Reichl, U. Towards integrated production of an influenza A vaccine candidate with MDCK suspension cells. Biotechnol. Bioeng. 2021, 118, 3996–4013. [Google Scholar] [CrossRef]
- Wolff, M.W.; Reichl, U. Downstream processing of cell culture-derived virus particles. Expert Rev. Vaccines 2011, 10, 1451–1475. [Google Scholar] [CrossRef]
- Lothert, K.; Eilts, F.; Wolff, M.W. Quantification methods for viruses and virus-like particles applied in biopharmaceutical production processes. Expert Rev. Vaccines 2022, 21, 1029–1044. [Google Scholar] [CrossRef] [PubMed]
- Hohl, A.; Ramms, A.S.; Dohmen, C.; Mantwill, K.; Bielmeier, A.; Kolk, A.; Ruppert, A.; Nawroth, R.; Holm, P.S. Adenovirus Particle Quantification in Cell Lysates Using Light Scattering. Hum. Gene Ther. Methods 2017, 28, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, H.N.; Xu, P.; Servellita, V.; Miller, S.; Liu, L.; Gopez, A.; Chiu, C.Y.; Abate, A.R. Digital droplet PCR accurately quantifies SARS-CoV-2 viral load from crude lysate without nucleic acid purification. Sci. Rep. 2021, 11, 780. [Google Scholar] [CrossRef] [PubMed]
- Abachin, E.; Convers, S.; Falque, S.; Esson, R.; Mallet, L.; Nougarede, N. Comparison of reverse-transcriptase qPCR and droplet digital PCR for the quantification of dengue virus nucleic acid. Biologicals 2018, 52, 49–54. [Google Scholar] [CrossRef]
- Wang, Y.; Bergelson, S.; Feschenko, M. Determination of Lentiviral Infectious Titer by a Novel Droplet Digital PCR Method. Hum. Gene Ther. Methods 2018, 29, 96–103. [Google Scholar] [CrossRef]
- Bruce, E.A.; Mills, M.G.; Sampoleo, R.; Perchetti, G.A.; Huang, M.L.; Despres, H.W.; Schmidt, M.M.; Roychoudhury, P.; Shirley, D.J.; Jerome, K.R.; et al. Predicting infectivity: Comparing four PCR-based assays to detect culturable SARS-CoV-2 in clinical samples. EMBO Mol. Med. 2022, 14, e15290. [Google Scholar] [CrossRef]
- Li, D.; De Keuckelaere, A.; Uyttendaele, M. Application of long-range and binding reverse transcription-quantitative PCR to indicate the viral integrities of noroviruses. Appl. Environ. Microbiol. 2014, 80, 6473–6479. [Google Scholar] [CrossRef]
- Cresta, D.; Warren, D.C.; Quirouette, C.; Smith, A.P.; Lane, L.C.; Smith, A.M.; Beauchemin, C.A.A. Time to revisit the endpoint dilution assay and to replace the TCID50 as a measure of a virus sample’s infection concentration. PLoS Comput. Biol. 2021, 17, e1009480. [Google Scholar] [CrossRef]
- Hochdorfer, D.; Businger, R.; Hotter, D.; Seifried, C.; Solzin, J. Automated, label-free TCID50 assay to determine the infectious titer of virus-based therapeutics. J. Virol. Methods 2021, 299, e114318. [Google Scholar] [CrossRef]
- Lei, C.; Yang, J.; Hu, J.; Sun, X. On the Calculation of TCID50 for Quantitation of Virus Infectivity. Virol. Sin. 2021, 36, 141–144. [Google Scholar] [CrossRef]
- Smither, S.J.; Lear-Rooney, C.; Biggins, J.; Pettitt, J.; Lever, M.S.; Olinger, G.G., Jr. Comparison of the plaque assay and 50% tissue culture infectious dose assay as methods for measuring filovirus infectivity. J. Virol. Methods 2013, 193, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, E.J.; Manguiat, K.; Wood, H.; Drebot, M. Two Detailed Plaque Assay Protocols for the Quantification of Infectious SARS-CoV-2. Curr. Protoc. Microbiol. 2020, 57, ecpmc105. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ling, L.; Liu, X.; Laus, R.; Delcayre, A. A flow cytometry-based immuno-titration assay for rapid and accurate titer determination of modified vaccinia Ankara virus vectors. J. Virol. Methods 2010, 169, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Lothert, K.; Pagallies, F.; Feger, T.; Amann, R.; Wolff, M.W. Selection of chromatographic methods for the purification of cell culture-derived Orf virus for its application as a vaccine or viral vector. J. Biotechnol. 2020, 323, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Pidre, M.L.; Arrias, P.N.; Amoros Morales, L.C.; Romanowski, V. The Magic Staff: A Comprehensive Overview of Baculovirus-Based Technologies Applied to Human and Animal Health. Viruses 2022, 15, 80. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, C.M.; Marchi, S.; Montomoli, E. The baculovirus expression vector system: A modern technology for the future of influenza vaccine manufacturing. Expert Rev. Vaccines 2022, 21, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Sprick, G.; Weidner, T.; Salzig, D.; Czermak, P. Baculovirus-induced recombinant protein expression in human mesenchymal stromal stem cells: A promoter study. N. Biotechnol. 2017, 39, 161–166. [Google Scholar] [CrossRef]
- King, L.A.; Hitchman, R.; Possee, R.D. Recombinant Baculovirus Isolation. Methods Mol. Biol. 2016, 1350, 73–94. [Google Scholar] [CrossRef]
- Eilts, F.; Harnischfeger, J.; Loewe, D.; Wolff, M.W.; Salzig, D.; Czermak, P. Production of Baculovirus and Stem Cells for Baculovirus-Mediated Gene Transfer into Human Mesenchymal Stem Cells. Methods Mol. Biol. 2021, 2183, 367–390. [Google Scholar] [CrossRef]
- Zhai, Y.; Zhang, D.; Yu, L.; Sun, F.; Sun, F. SmartBac, a new baculovirus system for large protein complex production. J. Struct. Biol. X 2019, 1, 100003. [Google Scholar] [CrossRef]
- Neuhold, J.; Radakovics, K.; Lehner, A.; Weissmann, F.; Garcia, M.Q.; Romero, M.C.; Berrow, N.S.; Stolt-Bergner, P. GoldenBac: A simple, highly efficient, and widely applicable system for construction of multi-gene expression vectors for use with the baculovirus expression vector system. BMC Biotechnol. 2020, 20, 26. [Google Scholar] [CrossRef] [PubMed]
- Aucoin, M.G.; Mena, J.A.; Kamen, A.A. Bioprocessing of baculovirus vectors: A review. Curr. Gene Ther. 2010, 10, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Lothert, K.; Sprick, G.; Beyer, F.; Lauria, G.; Czermak, P.; Wolff, M.W. Membrane-based steric exclusion chromatography for the purification of a recombinant baculovirus and its application for cell therapy. J. Virol. Methods 2020, 275, e113756. [Google Scholar] [CrossRef]
- Nasimuzzaman, M.; Lynn, D.; van der Loo, J.C.; Malik, P. Purification of baculovirus vectors using heparin affinity chromatography. Mol. Ther. Methods Clin. Dev. 2016, 3, 16071. [Google Scholar] [CrossRef] [PubMed]
- Vicente, T.; Peixoto, C.; Carrondo, M.J.T.; Alves, P.M. Purification of recombinant baculoviruses for gene therapy using membrane processes. Gene Ther. 2009, 16, 766–775. [Google Scholar] [CrossRef]
- Basak, S.; Kang, H.J.; Chu, K.B.; Oh, J.; Quan, F.S. Simple and rapid plaque assay for recombinant baculoviruses expressing influenza hemagglutinin. Sci. Prog. 2021, 104, e368504211004261. [Google Scholar] [CrossRef]
- Shen, C.F.; Meghrous, J.; Kamen, A. Quantitation of baculovirus particles by flow cytometry. J. Virol. Methods 2002, 105, 321–330. [Google Scholar] [CrossRef]
- Qingsheng, W.; Yuanyuan, L. Establishment, verification and application of rapid detection of baculovirus infectious titer by flow cytometry. J. Virol. Methods 2022, 303, 114495. [Google Scholar] [CrossRef] [PubMed]
- Merkling, S.H.; van Rij, R.P. Analysis of resistance and tolerance to virus infection in Drosophila. Nat. Protoc. 2015, 10, 1084–1097. [Google Scholar] [CrossRef]
- Stevanovic, A.L.; Johnson, K.N. Infectivity of Drosophila C virus following oral delivery in Drosophila larvae. J. Gen. Virol. 2015, 96, 1490–1496. [Google Scholar] [CrossRef]
- Hamza, I.A.; Jurzik, L.; Uberla, K.; Wilhelm, M. Methods to detect infectious human enteric viruses in environmental water samples. Int. J. Hyg. Environ. Health 2011, 214, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Rosinski, M.; Reid, S.; Nielsen, L.K. Kinetics of baculovirus replication and release using real-time quantitative polymerase chain reaction. Biotechnol. Bioeng. 2002, 77, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, M.; Hemberg, M.; Rorsman, P.; Stahlberg, A. Quantification of mRNA in single cells and modelling of RT-qPCR induced noise. BMC Mol. Biol. 2008, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Shirima, R.R.; Maeda, D.G.; Kanju, E.; Ceasar, G.; Tibazarwa, F.I.; Legg, J.P. Absolute quantification of cassava brown streak virus mRNA by real-time qPCR. J. Virol. Methods 2017, 245, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Lothert, K.; Pagallies, F.; Eilts, F.; Sivanesapillai, A.; Hardt, M.; Moebus, A.; Feger, T.; Amann, R.; Wolff, M.W. A scalable downstream process for the purification of the cell culture-derived Orf virus for human or veterinary applications. J. Biotechnol. 2020, 323, 221–230. [Google Scholar] [CrossRef]
- Eckhardt, D.; Dieken, H.; Loewe, D.; Grein, T.A.; Salzig, D.; Czermak, P. Purification of oncolytic measles virus by cation-exchange chromatography using resin-based stationary phases. Sep. Sci. Technol. 2021, 57, 886–896. [Google Scholar] [CrossRef]
- Tang, V.A.; Renner, T.M.; Varette, O.; Le Boeuf, F.; Wang, J.; Diallo, J.S.; Bell, J.C.; Langlois, M.A. Single-particle characterization of oncolytic vaccinia virus by flow virometry. Vaccine 2016, 34, 5082–5089. [Google Scholar] [CrossRef]
- Labisch, J.J.; Wiese, G.P.; Barnes, K.; Bollmann, F.; Pflanz, K. Infectious titer determination of lentiviral vectors using a temporal immunological real-time imaging approach. PLoS ONE 2021, 16, e0254739. [Google Scholar] [CrossRef]
- Laasfeld, T.; Kopanchuk, S.; Rinken, A. Image-based cell-size estimation for baculovirus quantification. Biotechniques 2017, 63, 161–168. [Google Scholar] [CrossRef]
- Blissard, G.W.; Wenz, J.R. Baculovirus gp64 envelope glycoprotein is sufficient to mediate pH-dependent membrane fusion. J. Virol. 1992, 66, 6829–6835. [Google Scholar] [CrossRef]
- Chen, Y.R.; Zhong, S.; Fei, Z.; Hashimoto, Y.; Xiang, J.Z.; Zhang, S.; Blissard, G.W. The transcriptome of the baculovirus Autographa californica multiple nucleopolyhedrovirus in Trichoplusia ni cells. J. Virol. 2013, 87, 6391–6405. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter [Unit] | No Staining | PE Staining | APC Staining |
|---|---|---|---|
| Seeding cell concentration [cells/mL] | 0.8 × 106 | 1.0 × 106 | 1.0 × 106 |
| Volume per well [µL] | 150 | 150 | 150 |
| Incubation time of virus infection [h] | 17 | 5 | 4 |
| Concentration of fluorescent antibody [mg/mL] | Not applicable | 0.001 | 0.00025 |
| Volume per well [µL] | 50 | 50 | |
| Incubation time of antibody staining [min] | Not applicable | 30 | 15 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lothert, K.; Bagrin, E.; Wolff, M.W. Evaluating Novel Quantification Methods for Infectious Baculoviruses. Viruses 2023, 15, 998. https://doi.org/10.3390/v15040998
Lothert K, Bagrin E, Wolff MW. Evaluating Novel Quantification Methods for Infectious Baculoviruses. Viruses. 2023; 15(4):998. https://doi.org/10.3390/v15040998
Chicago/Turabian StyleLothert, Keven, Elena Bagrin, and Michael W. Wolff. 2023. "Evaluating Novel Quantification Methods for Infectious Baculoviruses" Viruses 15, no. 4: 998. https://doi.org/10.3390/v15040998