
A Virus Genetic System to Analyze the Fusogenicity of Human Cytomegalovirus Glycoprotein B Variants


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Plasmids and Reagents
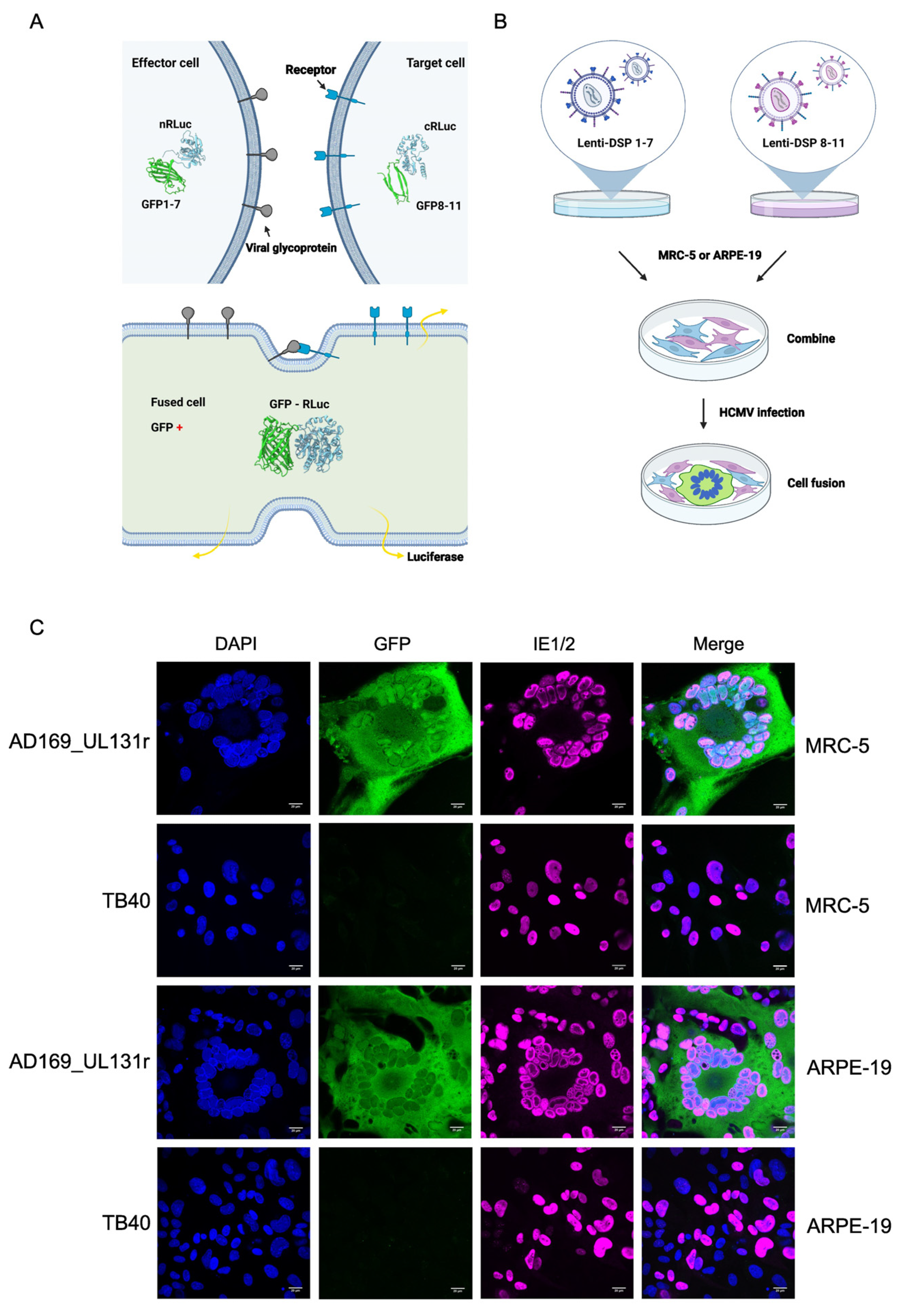
2.3. Cell–cell Fusion Assay
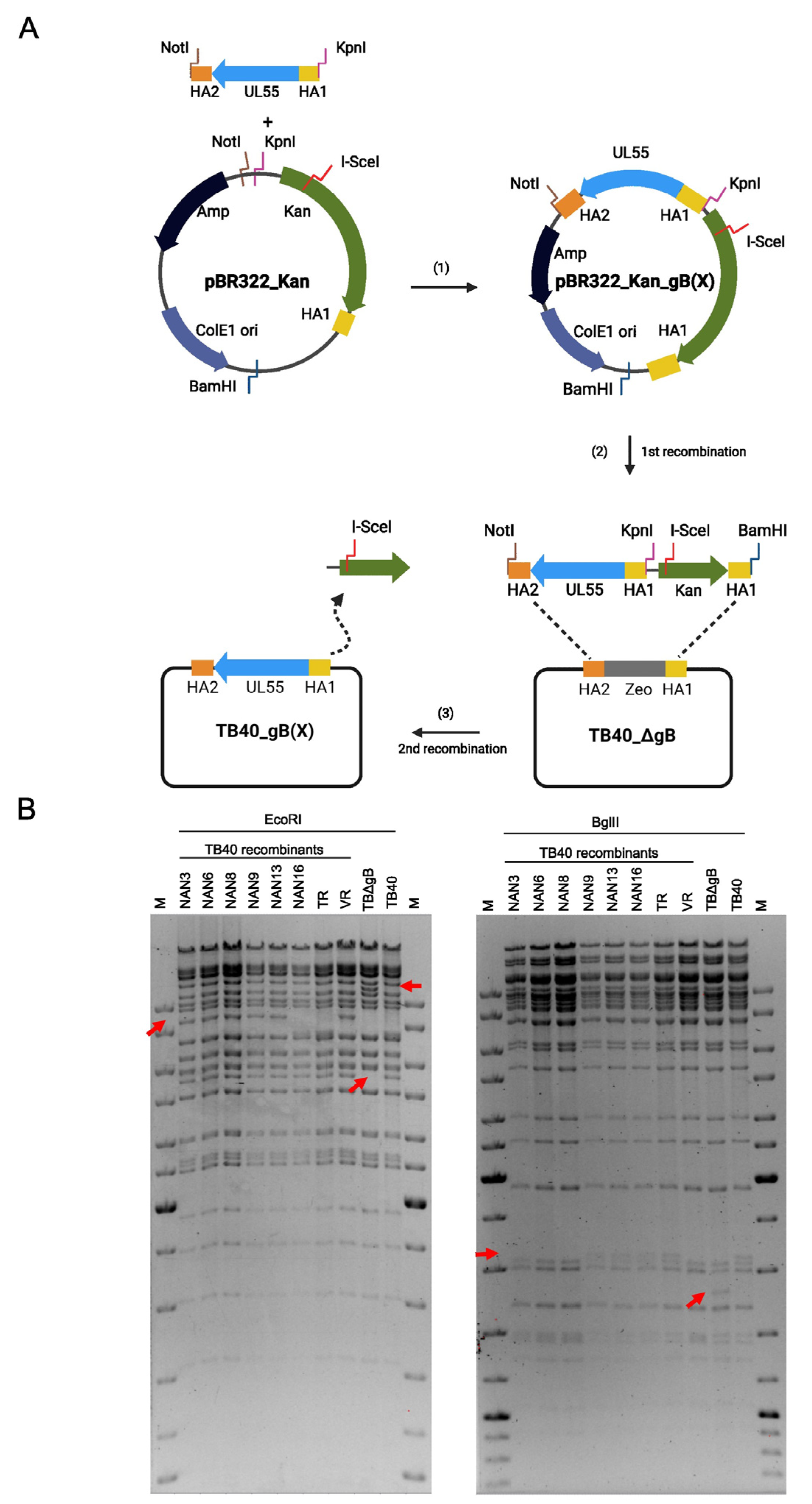
2.4. Mutagenesis of HCMV Genomes
2.5. Immunoblot and Immunofluorescence Analysis
2.6. Quantification of HCMV Genomes
3. Results
3.1. Polymorphism of HCMV Glycoprotein B
3.2. Development of a Genetic system to Analyze the Functional Differences of gB Variants
3.3. Adaptation of a DSP Reporter System for the Detection of HCMV-Induced Cell Fusion
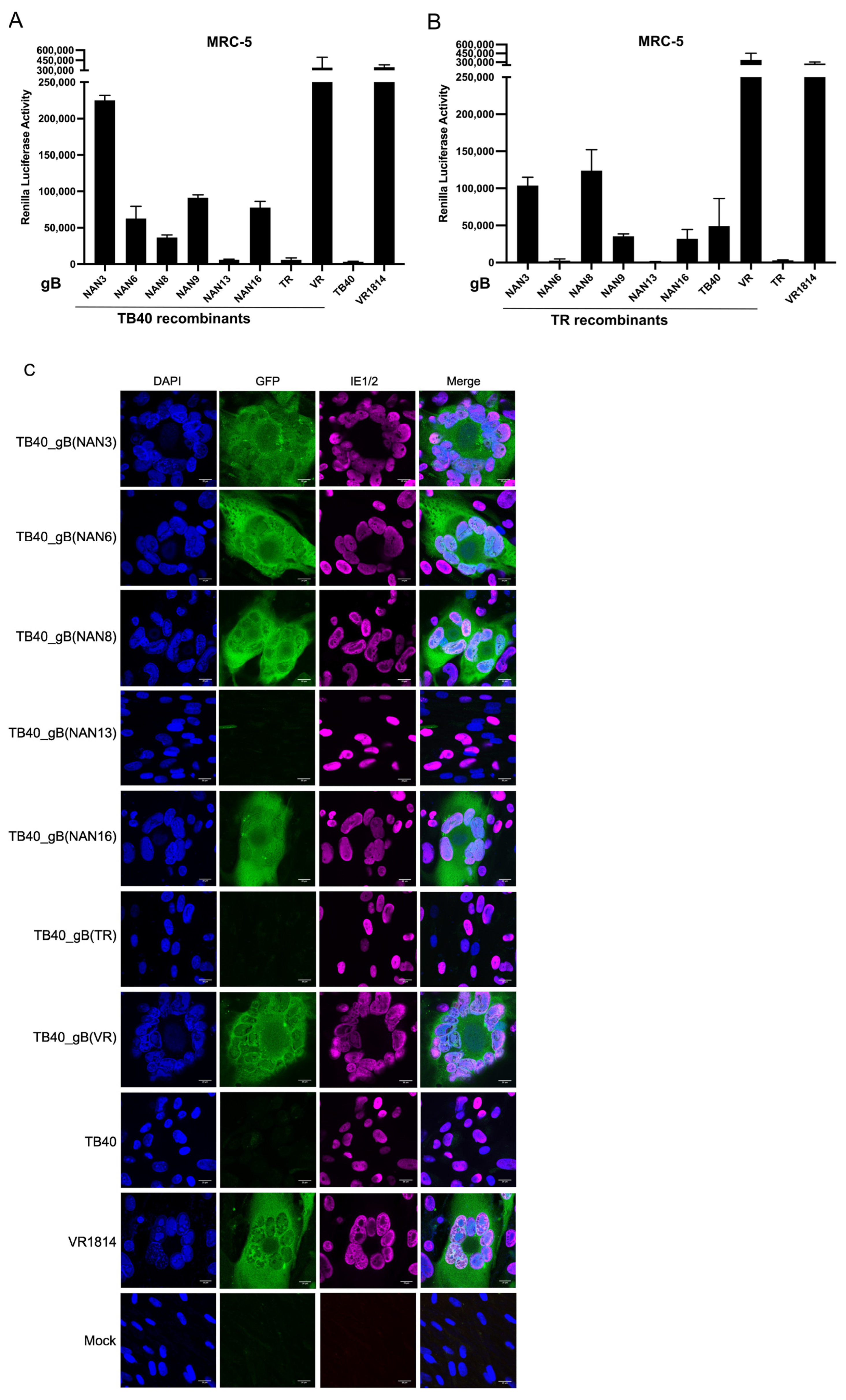
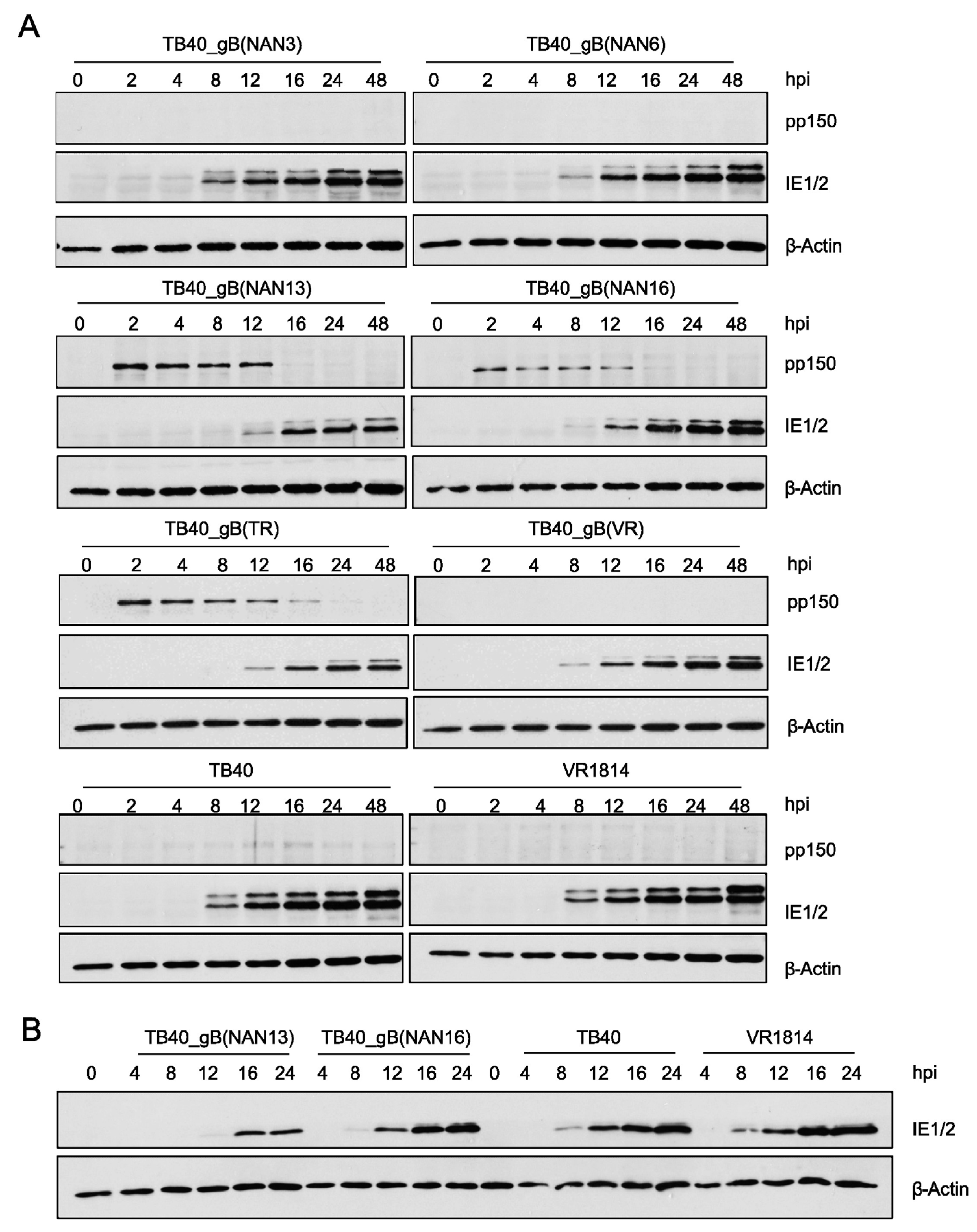
3.4. Analysis of the Fusogenicity of gB Variants from Congenital HCMV Strains
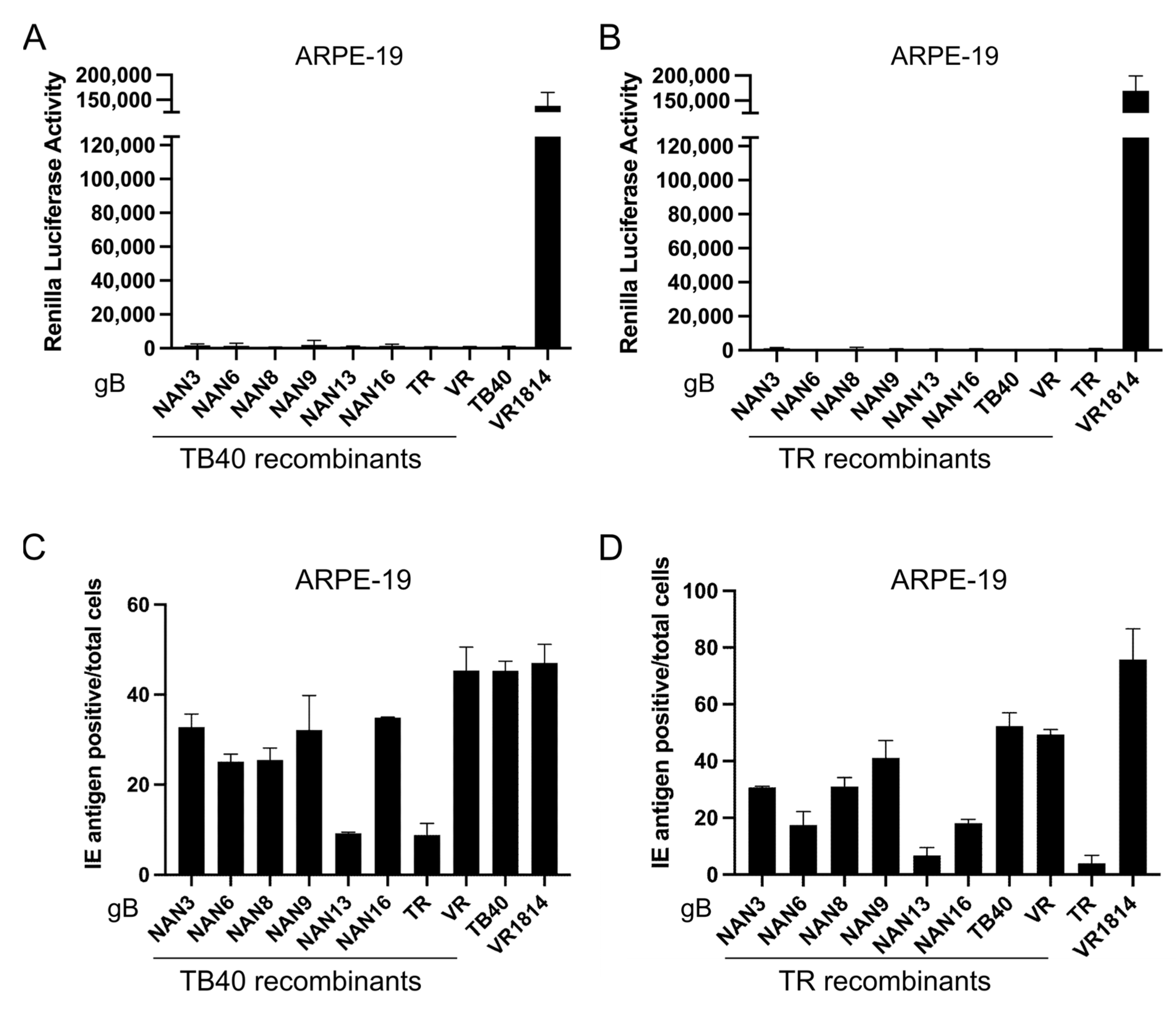
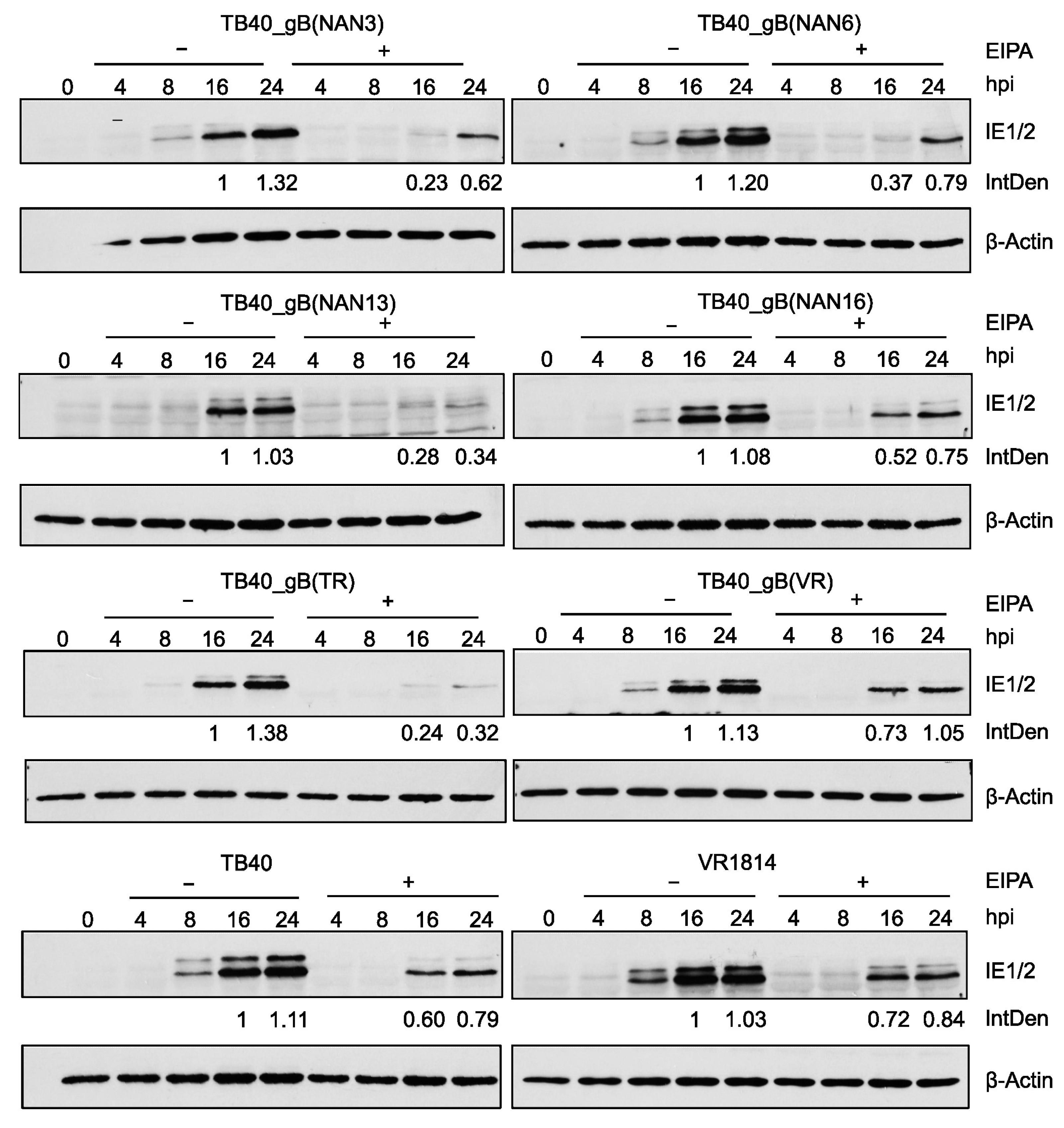
3.5. Influence of gB Variants on Virus Entry
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ambrosini, A.E.; Enquist, L.W. Cell-fusion events induced by α-herpesviruses. Future Virol. 2015, 10, 185–200. [Google Scholar] [CrossRef]
- Chang, A.; Dutch, R.E. Paramyxovirus fusion and entry: Multiple paths to a common end. Viruses 2012, 4, 613–636. [Google Scholar] [CrossRef]
- Cole, N.L.; Grose, C. Membrane fusion mediated by herpesvirus glycoproteins: The paradigm of varicella-zoster virus. Rev. Med. Virol. 2003, 13, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Mohl, B.S.; Chen, J.; Longnecker, R. Gammaherpesvirus entry and fusion: A tale how two human pathogenic viruses enter their host cells. Adv. Virus Res. 2019, 104, 313–343. [Google Scholar] [PubMed]
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Manicklal, S.; Emery, V.C.; Lazzarotto, T.; Boppana, S.B.; Gupta, R.K. The “silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 2013, 26, 86–102. [Google Scholar] [CrossRef]
- Rawlinson, W.D.; Boppana, S.B.; Fowler, K.B.; Kimberlin, D.W.; Lazzarotto, T.; Alain, S.; Daly, K.; Doutre, S.; Gibson, L.; Giles, M.L.; et al. Congenital cytomegalovirus infection in pregnancy and the neonate: Consensus recommendations for prevention, diagnosis, and therapy. Lancet Infect Dis. 2017, 17, e177–e188. [Google Scholar] [CrossRef]
- Foglierini, M.; Marcandalli, J.; Perez, L. HCMV Envelope Glycoprotein Diversity Demystified. Front. Microbiol. 2019, 10, 1005. [Google Scholar] [CrossRef]
- Dobbins, G.C.; Patki, A.; Chen, D.; Tiwari, H.K.; Hendrickson, C.; Britt, W.J.; Fowler, K.; Chen, J.Y.; Boppana, S.B.; Ross, S.A. Association of CMV genomic mutations with symptomatic infection and hearing loss in congenital CMV infection. BMC Infect. Dis. 2019, 19, 1046. [Google Scholar] [CrossRef]
- Paradowska, E.; Jablonska, A.; Studzinska, M.; Kasztelewicz, B.; Wisniewska-Ligier, M.; Dzierzanowska-Fangrat, K.; Wozniakowska-Gesicka, T.; Czech-Kowalska, J. Distribution of the CMV glycoprotein gH/gL/gO and gH/gL/pUL128/pUL130/pUL131A complex variants and associated clinical manifestations in infants infected congenitally or postnatally. Sci. Rep. 2019, 9, 16352. [Google Scholar] [CrossRef]
- Renzette, N.; Bhattacharjee, B.; Jensen, J.D.; Gibson, L.; Kowalik, T.F. Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants. PLoS Pathog. 2011, 7, e1001344. [Google Scholar] [CrossRef]
- Buscher, N.; Paulus, C.; Nevels, M.; Tenzer, S.; Plachter, B. The proteome of human cytomegalovirus virions and dense bodies is conserved across different strains. Med. Microbiol. Immunol. 2015, 204, 285–293. [Google Scholar] [CrossRef]
- Nguyen, C.C.; Kamil, J.P. Pathogen at the Gates: Human Cytomegalovirus Entry and Cell Tropism. Viruses 2018, 10, 704. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Frascaroli, G.; Zhou, X.; Knickmann, J.; Brune, W. Cell Fusion and Syncytium Formation in Betaherpesvirus Infection. Viruses 2021, 13, 1973. [Google Scholar] [CrossRef] [PubMed]
- Burke, H.G.; Heldwein, E.E. Crystal Structure of the Human Cytomegalovirus Glycoprotein B. PLoS Pathog. 2015, 11, e1005227. [Google Scholar] [CrossRef] [PubMed]
- Ruzsics, Z.; Borst, E.M.; Bosse, J.B.; Brune, W.; Messerle, M. Manipulating CMV Genomes by BAC Mutagenesis: Strategies and Applications. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norwich, UK, 2013; Volume 1, pp. 38–58. [Google Scholar]
- Tang, J.; Frascaroli, G.; Lebbink, R.J.; Ostermann, E.; Brune, W. Human cytomegalovirus glycoprotein B variants affect viral entry, cell fusion, and genome stability. Proc. Natl. Acad. Sci. USA 2019, 116, 18021–18030. [Google Scholar] [CrossRef]
- Dargan, D.J.; Douglas, E.; Cunningham, C.; Jamieson, F.; Stanton, R.J.; Baluchova, K.; McSharry, B.P.; Tomasec, P.; Emery, V.C.; Percivalle, E.; et al. Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J. Gen. Virol. 2010, 91, 1535–1546. [Google Scholar] [CrossRef]
- Galitska, G.; Biolatti, M.; De Andrea, M.; Leone, A.; Coscia, A.; Bertolotti, L.; Ala, U.; Bertino, E.; Dell’Oste, V.; Landolfo, S. Biological relevance of Cytomegalovirus genetic variability in congenitally and postnatally infected children. J. Clin. Virol. 2018, 108, 132–140. [Google Scholar] [CrossRef]
- Galitska, G.; Coscia, A.; Forni, D.; Steinbrueck, L.; De Meo, S.; Biolatti, M.; De Andrea, M.; Cagliani, R.; Leone, A.; Bertino, E.; et al. Genetic Variability of Human Cytomegalovirus Clinical Isolates Correlates With Altered Expression of Natural Killer Cell-Activating Ligands and IFN-gamma. Front. Immunol. 2021, 12, 532484. [Google Scholar] [CrossRef]
- Revello, M.G.; Baldanti, F.; Percivalle, E.; Sarasini, A.; De-Giuli, L.; Genini, E.; Lilleri, D.; Labo, N.; Gerna, G. In vitro selection of human cytomegalovirus variants unable to transfer virus and virus products from infected cells to polymorphonuclear leukocytes and to grow in endothelial cells. J. Gen. Virol. 2001, 82, 1429–1438. [Google Scholar] [CrossRef]
- Sinzger, C.; Hahn, G.; Digel, M.; Katona, R.; Sampaio, K.L.; Messerle, M.; Hengel, H.; Koszinowski, U.; Brune, W.; Adler, B. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J. Gen. Virol. 2008, 89, 359–368. [Google Scholar] [CrossRef]
- Murphy, E.; Yu, D.; Grimwood, J.; Schmutz, J.; Dickson, M.; Jarvis, M.A.; Hahn, G.; Nelson, J.A.; Myers, R.M.; Shenk, T.E. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2003, 100, 14976–14981. [Google Scholar] [CrossRef]
- Andreoni, M.; Faircloth, M.; Vugler, L.; Britt, W.J. A rapid microneutralization assay for the measurement of neutralizing antibody reactive with human cytomegalovirus. J. Virol. Methods 1989, 23, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Wang, J.; Wang, Z.; Ru, T.; Dai, Y.; Li, J.; Zhu, X.; Liu, J.; Ye, X.; Zhu, B.; et al. Rare detection of cytomegalovirus in severe fetal malformations in China. J. Clin. Virol. 2016, 79, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Thakur, N.; Conceicao, C.; Isaacs, A.; Human, S.; Modhiran, N.; McLean, R.K.; Pedrera, M.; Tan, T.K.; Rijal, P.; Townsend, A.; et al. Micro-fusion inhibition tests: Quantifying antibody neutralization of virus-mediated cell-cell fusion. J. Gen. Virol. 2020, 102, jgv001506. [Google Scholar] [CrossRef]
- Tischer, B.K.; Smith, G.A.; Osterrieder, N. En passant mutagenesis: A two step markerless red recombination system. Methods Mol. Biol. 2010, 634, 421–430. [Google Scholar]
- Ishikawa, H.; Meng, F.; Kondo, N.; Iwamoto, A.; Matsuda, Z. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng. Des. Sel. 2012, 25, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Brixel, R.; Brune, W. Copy-Paste Mutagenesis: A Method for Large-Scale Alteration of Viral Genomes. Int. J. Mol. Sci. 2019, 20, 913. [Google Scholar] [CrossRef]
- Wang, D.; Shenk, T. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J. Virol. 2005, 79, 10330–10338. [Google Scholar] [CrossRef]
- Gerna, G.; Percivalle, E.; Perez, L.; Lanzavecchia, A.; Lilleri, D. Monoclonal Antibodies to Different Components of the Human Cytomegalovirus (HCMV) Pentamer gH/gL/pUL128L and Trimer gH/gL/gO as well as Antibodies Elicited during Primary HCMV Infection Prevent Epithelial Cell Syncytium Formation. J. Virol. 2016, 90, 6216–6223. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.D.; de Harven, E. Herpes simplex virus and human cytomegalovirus replication in WI-38 cells. II. An ultrastructural study of viral penetration. J. Virol. 1974, 14, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Compton, T.; Nepomuceno, R.R.; Nowlin, D.M. Human cytomegalovirus penetrates host cells by pH-independent fusion at the cell surface. Virology 1992, 191, 387–395. [Google Scholar] [CrossRef]
- Hetzenecker, S.; Helenius, A.; Krzyzaniak, M.A. HCMV Induces Macropinocytosis for Host Cell Entry in Fibroblasts. Traffic 2016, 17, 351–368. [Google Scholar] [CrossRef] [PubMed]
- Koivusalo, M.; Welch, C.; Hayashi, H.; Scott, C.C.; Kim, M.; Alexander, T.; Touret, N.; Hahn, K.M.; Grinstein, S. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell Biol. 2010, 188, 547–563. [Google Scholar] [CrossRef]
- Day, L.Z.; Stegmann, C.; Schultz, E.P.; Lanchy, J.M.; Yu, Q.; Ryckman, B.J. Polymorphisms in Human Cytomegalovirus Glycoprotein O (gO) Exert Epistatic Influences on Cell-Free and Cell-to-Cell Spread and Antibody Neutralization on gH Epitopes. J. Virol. 2020, 94, e02051-19. [Google Scholar] [CrossRef]
- Zhou, M.; Lanchy, J.M.; Ryckman, B.J. Human Cytomegalovirus gH/gL/gO Promotes the Fusion Step of Entry into All Cell Types, whereas gH/gL/UL128-131 Broadens Virus Tropism through a Distinct Mechanism. J. Virol. 2015, 89, 8999–9009. [Google Scholar] [CrossRef]
- Vo, M.; Aguiar, A.; McVoy, M.A.; Hertel, L. Cytomegalovirus Strain TB40/E Restrictions and Adaptations to Growth in ARPE-19 Epithelial Cells. Microorganisms 2020, 8, 615. [Google Scholar] [CrossRef] [PubMed]
- Al Qaffas, A.; Camiolo, S.; Vo, M.; Aguiar, A.; Ourahmane, A.; Sorono, M.; Davison, A.J.; McVoy, M.A.; Hertel, L. Genome sequences of human cytomegalovirus strain TB40/E variants propagated in fibroblasts and epithelial cells. Virol J. 2021, 18, 112. [Google Scholar] [CrossRef] [PubMed]
- Vanarsdall, A.L.; Johnson, D.C. Human cytomegalovirus entry into cells. Curr. Opin. Virol. 2012, 2, 37–42. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Cimato, G.; Zhou, Y.; Frascaroli, G.; Brune, W. A Virus Genetic System to Analyze the Fusogenicity of Human Cytomegalovirus Glycoprotein B Variants. Viruses 2023, 15, 979. https://doi.org/10.3390/v15040979
Zhou X, Cimato G, Zhou Y, Frascaroli G, Brune W. A Virus Genetic System to Analyze the Fusogenicity of Human Cytomegalovirus Glycoprotein B Variants. Viruses. 2023; 15(4):979. https://doi.org/10.3390/v15040979
Chicago/Turabian StyleZhou, Xuan, Giorgia Cimato, Yihua Zhou, Giada Frascaroli, and Wolfram Brune. 2023. "A Virus Genetic System to Analyze the Fusogenicity of Human Cytomegalovirus Glycoprotein B Variants" Viruses 15, no. 4: 979. https://doi.org/10.3390/v15040979