
Genomic Analysis of Amphioxus Reveals a Wide Range of Fragments Homologous to Viral Sequences
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Genome Sequencing and De Novo Genome Assembly
2.3. Genome Annotation
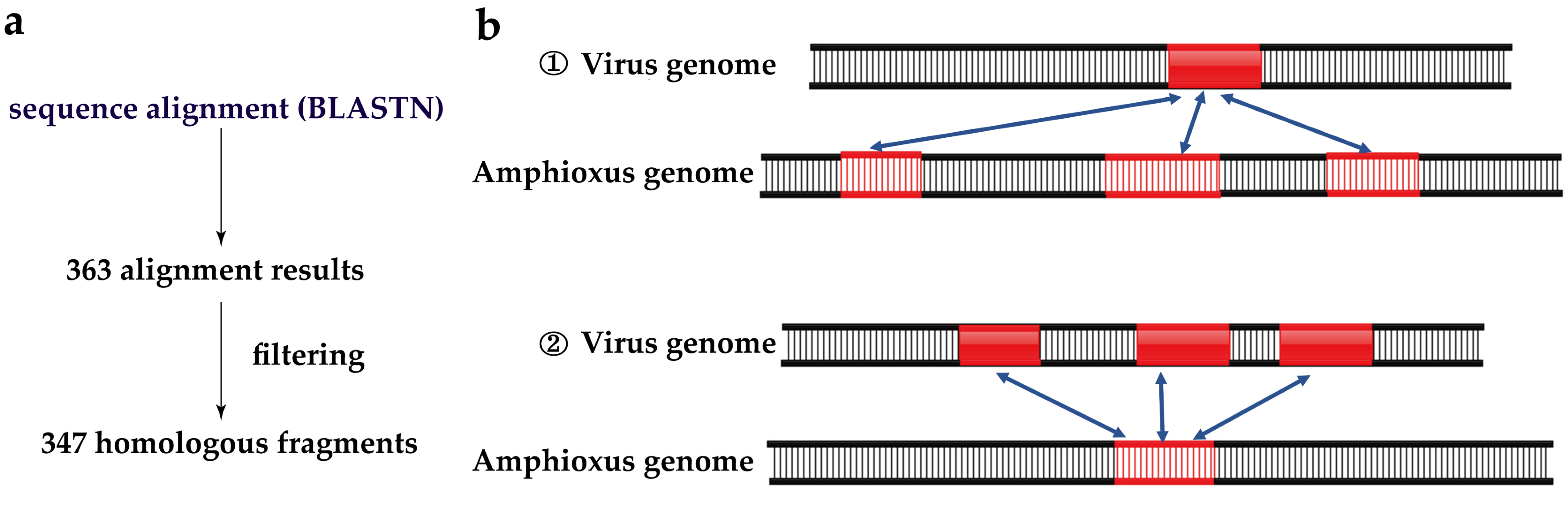
2.4. Identification of HFs in Amphioxus Genome
2.5. Identification of HFs in Viral Genomes
2.6. Data Analysis
3. Results
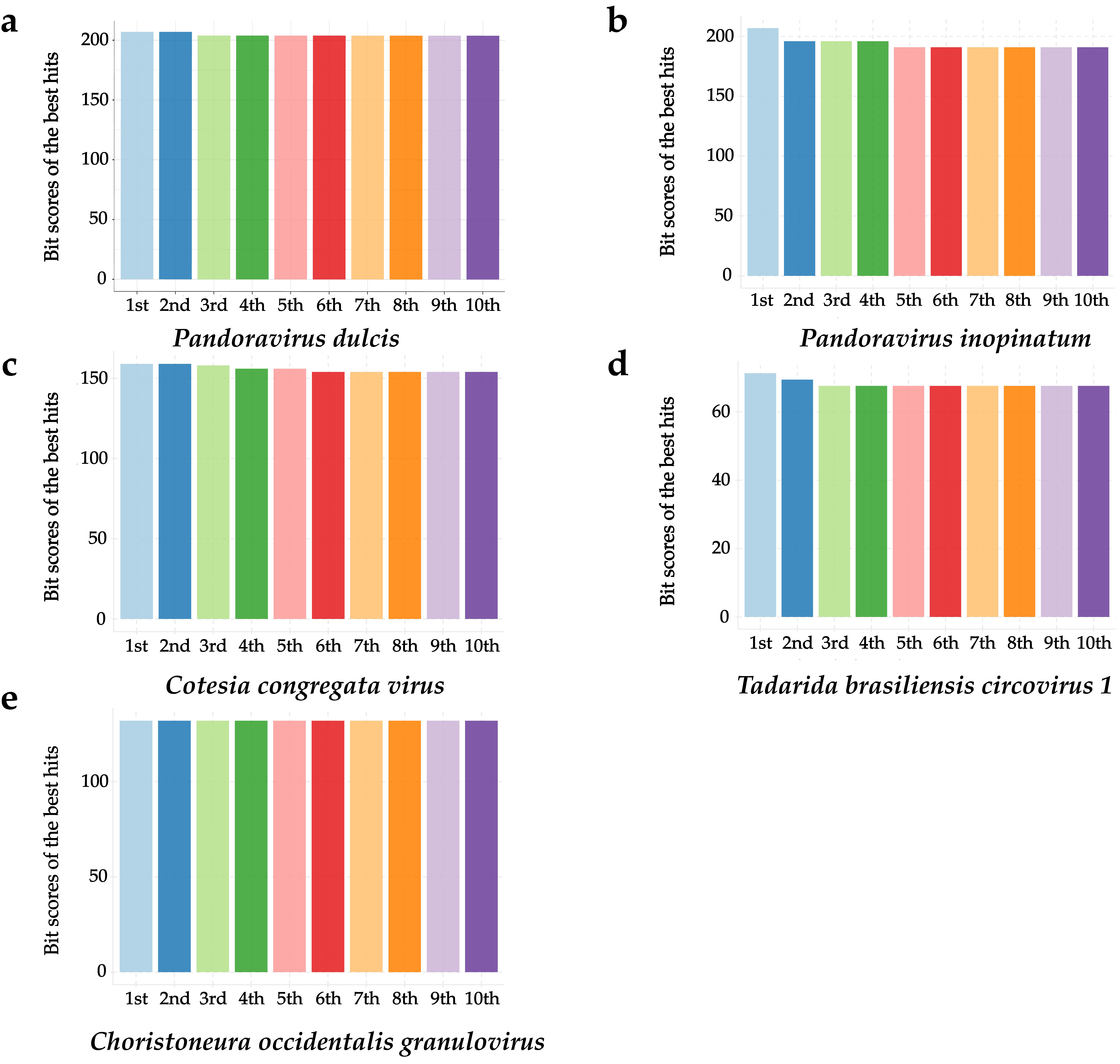
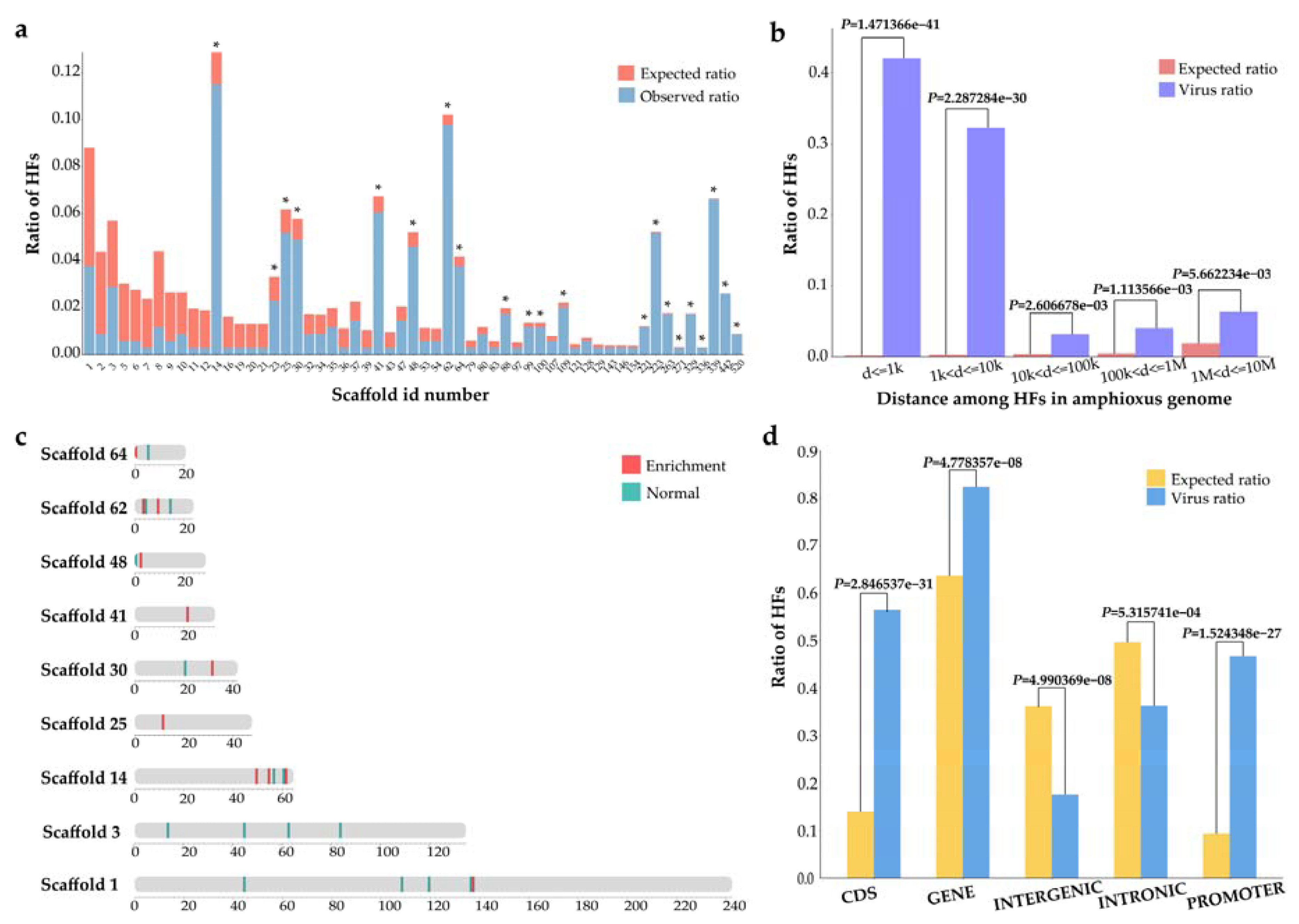
3.1. Identification of Viral HFs in the Amphioxus Genome
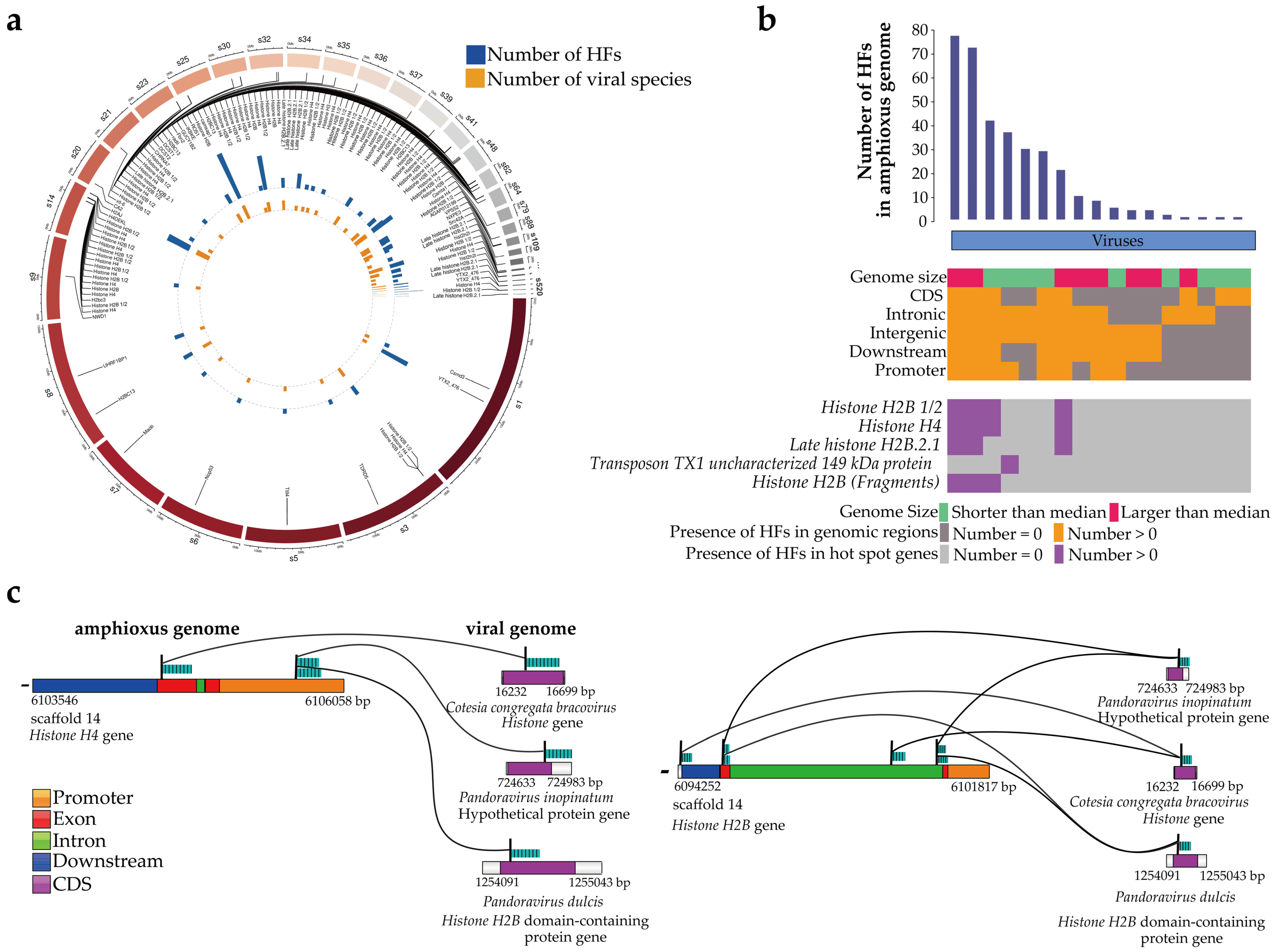
3.2. Gene and Functional Annotation of the Viral HFs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Corrin, B.; Nicholson, A.G. Chapter 12—Tumours. In Pathology of the Lungs, 3rd ed.; Corrin, B., Nicholson, A.G., Eds.; Churchill Livingstone: Edinburgh, UK, 2011; pp. 531–705. [Google Scholar] [CrossRef]
- Shilo, B.Z.; A Weinberg, R. DNA sequences homologous to vertebrate oncogenes are conserved in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 1981, 78, 6789–6792. [Google Scholar] [CrossRef] [PubMed]
- Vennström, B.; Bishop, J.M. Isolation and characterization of chicken DNA homologous to the two putative oncogenes of avian erythroblastosis virus. Cell 1982, 28, 135–143. [Google Scholar] [CrossRef]
- Colby, W.W.; Chen, E.Y.; Smith, D.H.; Levinson, A.D. Identification and nucleotide sequence of a human locus homologous to the v-myc oncogene of avian myelocytomatosis virus MC29. Nature 1983, 301, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Iida, A.; Takemae, H.; Tarigan, R.; Kobayashi, R.; Kato, H.; Shimoda, H.; Omatsu, T.; Supratikno; Basri, C.; Mayasari, N.L.P.I.; et al. Viral-derived DNA invasion and individual variation in an Indonesian population of large flying fox Pteropus vampyrus. J. Veter.-Med. Sci. 2021, 83, 1068–1074. [Google Scholar] [CrossRef]
- Savin, K.W.; Cocks, B.G.; Wong, F.; Sawbridge, T.; Cogan, N.; Savage, D.; Warner, S. A neurotropic herpesvirus infecting the gastropod, abalone, shares ancestry with oyster herpesvirus and a herpesvirus associated with the amphioxus genome. Virol. J. 2010, 7, 308. [Google Scholar] [CrossRef] [PubMed]
- Belyi, V.A.; Levine, A.J.; Skalka, A.M. Unexpected Inheritance: Multiple Integrations of Ancient Bornavirus and Ebolavirus/Marburgvirus Sequences in Vertebrate Genomes. PLoS Pathog. 2010, 6, e1001030. [Google Scholar] [CrossRef]
- Rappoport, N.; Linial, M. Viral Proteins Acquired from a Host Converge to Simplified Domain Architectures. PLoS Comput. Biol. 2012, 8, e1002364. [Google Scholar] [CrossRef]
- Kapoor, A.; Simmonds, P.; Lipkin, W.I. Discovery and Characterization of Mammalian Endogenous Parvoviruses. J. Virol. 2010, 84, 12628–12635. [Google Scholar] [CrossRef]
- Horie, M.; Honda, T.; Suzuki, Y.; Kobayashi, Y.; Daito, T.; Oshida, T.; Ikuta, K.; Jern, P.; Gojobori, T.; Coffin, J.M.; et al. Endogenous non-retroviral RNA virus elements in mammalian genomes. Nature 2010, 463, 84–87. [Google Scholar] [CrossRef]
- Zilber-Rosenberg, I.; Rosenberg, E. Role of microorganisms in the evolution of animals and plants: The hologenome theory of evolution. FEMS Microbiol. Rev. 2008, 32, 723–735. [Google Scholar] [CrossRef]
- Wolf, G.; Greenberg, D.; Macfarlan, T.S. Spotting the enemy within: Targeted silencing of foreign DNA in mammalian genomes by the Krüppel-associated box zinc finger protein family. Mob. DNA 2015, 6, 1–20. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef]
- Geisler, C.; Jarvis, D.L. Rhabdovirus-like endogenous viral elements in the genome of Spodoptera frugiperda insect cells are actively transcribed: Implications for adventitious virus detection. Biologicals 2016, 44, 219–225. [Google Scholar] [CrossRef]
- Lequime, S.; Lambrechts, L. Discovery of flavivirus-derived endogenous viral elements in Anopheles mosquito genomes supports the existence of Anopheles-associated insect-specific flaviviruses. Virus Evolut. 2017, 3, vew035. [Google Scholar] [CrossRef]
- Harvey, E.; Holmes, E.C. Diversity and evolution of the animal virome. Nat. Rev. Genet. 2022, 20, 321–334. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Martinez-Gutierrez, C.A.; Weinheimer, A.R.; Aylward, F.O. Dynamic genome evolution and complex virocell metabolism of globally-distributed giant viruses. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Aylward, F.O.; Moniruzzaman, M.; Ha, A.D.; Koonin, E.V. A phylogenomic framework for charting the diversity and evolution of giant viruses. PLoS Biol. 2021, 19, e3001430. [Google Scholar] [CrossRef] [PubMed]
- Thomas, V.; Bertelli, C.; Collyn, F.; Casson, N.; Telenti, A.; Goesmann, A.; Croxatto, A.; Greub, G. Lausannevirus, a giant amoebal virus encoding histone doublets. Environ. Microbiol. 2011, 13, 1454–1466. [Google Scholar] [CrossRef]
- Yoshikawa, G.; Blanc-Mathieu, R.; Song, C.; Kayama, Y.; Mochizuki, T.; Murata, K.; Ogata, H.; Takemura, M. Medusavirus, a Novel Large DNA Virus Discovered from Hot Spring Water. J. Virol. 2019, 93, e02130-18. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.R.; Palfy, M.; Hilbert, L.; Kumar, M.; Karschau, J.; Zaburdaev, V.; Shevchenko, A.; Vastenhouw, N.L. Competition between histone and transcription factor binding regulates the onset of transcription in zebrafish embryos. eLife 2017, 6, e23326. [Google Scholar] [CrossRef] [PubMed]
- Gad, W.; Kim, Y. N-terminal tail of a viral histone H4 encoded in Cotesia plutellae bracovirus is essential to suppress gene expression of host histone H4. Insect Mol. Biol. 2009, 18, 111–118. [Google Scholar] [CrossRef]
- Hepat, R.; Song, J.-J.; Lee, D.; Kim, Y. A Viral Histone H4 Joins to Eukaryotic Nucleosomes and Alters Host Gene Expression. J. Virol. 2013, 87, 11223–11230. [Google Scholar] [CrossRef]
- Hepat, R.; Kim, Y. Transient expression of a viral histone H4 inhibits expression of cellular and humoral immune-associated genes in Tribolium castaneum. Biochem. Biophys. Res. Commun. 2011, 415, 279–283. [Google Scholar] [CrossRef]
- Gad, W.; Kim, Y. A viral histone H4 encoded by Cotesia plutellae bracovirus inhibits haemocyte-spreading behaviour of the diamondback moth, Plutella xylostella. J. Gen. Virol. 2008, 89, 931–938. [Google Scholar] [CrossRef]
- Kumar, S.; Gu, X.; Kim, Y. A viral histone H4 suppresses insect insulin signal and delays host development. Dev. Comp. Immunol. 2016, 63, 66–77. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y. A viral histone H4 suppresses expression of a transferrin that plays a role in the immune response of the diamondback moth, Plutella xylostella. Insect Mol. Biol. 2010, 19, 567–574. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Dzik, J.; Edgecombe, G.D.; Ramsköld, L.; Zhou, G.-Q. A possible Early Cambrian chordate. Nature 1995, 377, 720–722. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Huang, D.-Y.; Li, C.-W. An early Cambrian craniate-like chordate. Nature 1999, 402, 518–522. [Google Scholar] [CrossRef]
- Mallatt, J.; Chen, J.-Y. Fossil sister group of craniates: Predicted and found. J. Morphol. 2003, 258, 1–31. [Google Scholar] [CrossRef]
- Blair, J.E.; Hedges, S.B. Molecular Phylogeny and Divergence Times of Deuterostome Animals. Mol. Biol. Evol. 2005, 22, 2275–2284. [Google Scholar] [CrossRef]
- Delsuc, F.; Brinkmann, H.; Chourrout, D.; Philippe, H. Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature 2006, 439, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Vienne, A.; Pontarotti, P. Metaphylogeny of 82 gene families sheds a new light on chordate evolution. Int. J. Biol. Sci. 2006, 2, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Putnam, N.H.; Butts, T.; Ferrier, D.E.K.; Furlong, R.F.; Hellsten, U.; Kawashima, T.; Robinson-Rechavi, M.; Shoguchi, E.; Terry, A.; Yu, J.-K.; et al. The amphioxus genome and the evolution of the chordate karyotype. Nature 2008, 453, 1064–1071. [Google Scholar] [CrossRef]
- Schubert, M.; Escriva, H.; Xavier-Neto, J.; Laudet, V. Amphioxus and tunicates as evolutionary model systems. Trends Ecol. Evol. 2006, 21, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Holland, L.Z.; Laudet, V.; Schubert, M. The chordate amphioxus: An emerging model organism for developmental biology. Cell. Mol. Life Sci. 2004, 61, 2290–2308. [Google Scholar] [CrossRef]
- Xiong, Q.; Yang, K.Y.; Zeng, X.; Wang, M.; Ng, P.K.-S.; Zhou, J.-W.; Ng, J.K.-W.; Law, C.T.-Y.; Du, Q.; Xu, K.; et al. Massive Horizontal Gene Transfer in Amphioxus Illuminates the Early Evolution of Deuterostomes. bioRxiv 2022. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2009, 25, 4.10.1–4.10.14. [Google Scholar] [CrossRef]
- Bao, Z.; Eddy, S.R. Automated De Novo Identification of Repeat Sequence Families in Sequenced Genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, i351–i358. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Korf, I.; Robb, S.M.C.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Alvarado, A.S.; Yandell, M. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008, 18, 188–196. [Google Scholar] [CrossRef]
- Slater, G.S.C.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef]
- Borodovsky, M.; Lomsadze, A. Eukaryotic Gene Prediction Using GeneMark.hmm-E and GeneMark-ES. Curr. Protoc. Bioinform. 2011, 35, Unit 4.6.1–Unit 4.6.10. [Google Scholar] [CrossRef]
- Hoff, K.J.; Stanke, M. Predicting Genes in Single Genomes with AUGUSTUS. Curr. Protoc. Bioinform. 2018, 65, e57. [Google Scholar] [CrossRef]
- Scott, M.G.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004, 32 (Suppl. S2), W20–W25. [Google Scholar]
- R Team. R: A language and environment for statistical computing. Computing 2011, 1, 12–21. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Zhao, L.-H.; Liu, X.; Yan, H.-X.; Li, W.-Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.P.; Zhuang, X.-H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992, Erratum in Nat. Commun. 2016, 7, 13591. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, Y.; Tian, T.; Rao, X.; Dong, W.; Zhang, J.; Yang, Y.; Tao, Q.; Peng, F.; Shen, C.; et al. Analysis of viral integration reveals new insights of oncogenic mechanism in HBV-infected intrahepatic cholangiocarcinoma and combined hepatocellular-cholangiocarcinoma. Hepatol. Int. 2022, 16, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Tsui, J.C.-C.; Shi, M.; Peng, J.; Cao, C.Y.; Kan, L.L.-Y.; Lau, C.P.-Y.; Liang, Y.; Wang, L.; Liu, L.; et al. Genome-Wide Characterization of Host Transcriptional and Epigenetic Alterations During HIV Infection of T Lymphocytes. Front. Immunol. 2020, 11, 2131. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef]
- Liu, W.; Xie, Y.; Ma, J.; Luo, X.; Nie, P.; Zuo, Z.; Lahrmann, U.; Zhao, Q.; Zheng, Y.; Zhao, Y.; et al. IBS: An illustrator for the presentation and visualization of biological sequences. Bioinformatics 2015, 31, 3359–3361. [Google Scholar] [CrossRef]
- Ren, J.; Wen, L.; Gao, X.; Jin, C.; Xue, Y.; Yao, X. DOG 1.0: Illustrator of protein domain structures. Cell Res. 2009, 19, 271–273. [Google Scholar] [CrossRef]
- Akashi, M.; Takemura, M. Co-Isolation and Characterization of Two Pandoraviruses and a Mimivirus from a Riverbank in Japan. Viruses 2019, 11, 1123. [Google Scholar] [CrossRef]
- Lima, F.E.S.; Cibulski, S.P.; Bello, A.G.D.; Mayer, F.Q.; Witt, A.A.; Roehe, P.M.; D’Azevedo, P.A. A Novel Chiropteran Circovirus Genome Recovered from a Brazilian Insectivorous Bat Species. Genome Announc. 2015, 3, e01393-15. [Google Scholar] [CrossRef]
- Wiederkehr, M.A.; Qi, W.; Schoenbaechler, K.; Fraefel, C.; Kubacki, J. Virus Diversity, Abundance, and Evolution in Three Different Bat Colonies in Switzerland. Viruses 2022, 14, 1911. [Google Scholar] [CrossRef]
- Rhee, S.S.; Hunter, E. Myristylation is required for intracellular transport but not for assembly of D-type retrovirus capsids. J. Virol. 1987, 61, 1045–1053. [Google Scholar] [CrossRef]
- Sonigo, P.; Barker, C.; Hunter, E.; Wain-Hobson, S. Nucleotide sequence of Mason-Pfizer monkey virus: An immunosuppressive D-type retrovirus. Cell 1986, 45, 375–385. [Google Scholar] [CrossRef]
- Hanson, L.; Dishon, A.; Kotler, M. Herpesviruses that Infect Fish. Viruses 2011, 3, 2160–2191. [Google Scholar] [CrossRef]
- Zhang, Q.-Y.; Gui, J.-F. Diversity, evolutionary contribution and ecological roles of aquatic viruses. Sci. China Life Sci. 2018, 61, 1486–1502. [Google Scholar] [CrossRef]
- Crane, M.; Hyatt, A. Viruses of Fish: An Overview of Significant Pathogens. Viruses 2011, 3, 2025–2046. [Google Scholar] [CrossRef]
- Carvalho, J.E.; Lahaye, F.; Schubert, M. Keeping amphioxus in the laboratory: An update on available husbandry methods. Int. J. Dev. Biol. 2017, 61, 773–783. [Google Scholar] [CrossRef]
- Etchegaray, E.; Naville, M.; Volff, J.-N.; Haftek-Terreau, Z. Transposable element-derived sequences in vertebrate development. Mob. DNA 2021, 12, 1–24. [Google Scholar] [CrossRef]
- Nikiforov, M.A.; Gudkov, A.V. ART-CH: A VL30 in chickens? J. Virol. 1994, 68, 846–853. [Google Scholar] [CrossRef]
- Mount, S.M.; Rubin, G.M. Complete nucleotide sequence of the Drosophila transposable element copia: Homology between copia and retroviral proteins. Mol. Cell. Biol. 1985, 5, 1630–1638. [Google Scholar] [CrossRef]
- Nair, S.; Bist, P.; Dikshit, N.; Krishnan, M.N. Global functional profiling of human ubiquitome identifies E3 ubiquitin ligase DCST1 as a novel negative regulator of Type-I interferon signaling. Sci. Rep. 2016, 6, 36179. [Google Scholar] [CrossRef]
- Bartl, S.; Baish, M.; Weissman, I.L.; Diaz, M. Did the molecules of adaptive immunity evolve from the innate immune system? Integr. Comp. Biol. 2003, 43, 338–346. [Google Scholar] [CrossRef]
- Cao, Y.; Fang, T.; Fan, M.; Wang, L.; Lv, C.; Song, X.; Jin, P.; Ma, F. Functional characterization of STATa/b genes encoding transcription factors from Branchiostoma belcheri. Dev. Comp. Immunol. 2020, 114, 103838. [Google Scholar] [CrossRef] [PubMed]
- Horie, M. The biological significance of bornavirus-derived genes in mammals. Curr. Opin. Virol. 2017, 25, 1–6. [Google Scholar] [CrossRef]
- Feschotte, C.; Gilbert, C. Endogenous viruses: Insights into viral evolution and impact on host biology. Nat. Rev. Genet. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.J.; Lock, W.M.; Mager, D.L. Endogenous retroviral LTRs as promoters for human genes: A critical assessment. Gene 2009, 448, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Jern, P.; Coffin, J.M. Effects of Retroviruses on Host Genome Function. Annu. Rev. Genet. 2008, 42, 709–732. [Google Scholar] [CrossRef]
- Bézier, A.; Annaheim, M.; Herbinière, J.; Wetterwald, C.; Gyapay, G.; Bernard-Samain, S.; Wincker, P.; Roditi, I.; Heller, M.; Belghazi, M. Polydnaviruses of braconid wasps derive from an ancestral nudivirus. Science 2009, 323, 926–930. [Google Scholar] [CrossRef]
- Debyser, Z.; Christ, F.; De Rijck, J.; Gijsbers, R. Host factors for retroviral integration site selection. Trends Biochem. Sci. 2015, 40, 108–116. [Google Scholar] [CrossRef]
- Talbert, P.B.; Armache, K.-J.; Henikoff, S. Viral histones: Pickpocket’s prize or primordial progenitor? Epigenet. Chromatin 2022, 15, 21. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Accession | Number of HFs in Amphioxus | Virus Name |
|---|---|---|
| NC_021858.1 | 78 | Pandoravirus dulcis |
| NC_026440.1 | 73 | Pandoravirus inopinatum |
| NC_006639.1 | 42 | Cotesia congregata bracovirus |
| NC_028045.1 | 37 | Tadarida brasiliensis circovirus 1 |
| NC_008168.1 | 30 | Choristoneura occidentalis granulovirus |
| NC_001550.1 | 29 | Mason-Pfizer monkey virus |
| NC_022098.1 | 21 | Pandoravirus salinus |
| NC_026421.1 | 10 | Equid gammaherpesvirus 5 |
| NC_028094.1 | 8 | Chrysochromulina ericina virus |
| NC_026141.2 | 5 | Adelie penguin polyomavirus |
| NC_008603.1 | 4 | Paramecium bursaria Chlorella virus FR483 |
| NC_008724.1 | 4 | Acanthocystis turfacea Chlorella virus 1 |
| NC_019491.1 | 2 | Cyprinid herpesvirus 1 |
| NC_000852.5 | 1 | Paramecium bursaria Chlorella virus 1 |
| NC_001716.2 | 1 | Human betaherpesvirus 7 |
| NC_008094.1 | 1 | Y73 sarcoma virus |
| NC_023006.1 | 1 | Pseudomonas phage PPpW-3 |
| Virus Name | Number of HFs in Viruses | Number of HFs in Amphioxus | Virus Gene | Ratio of HFs in Viruses | Ratio of HFs in Amphioxus |
|---|---|---|---|---|---|
| Cotesia congregate bracovirus | 9 | 39 | Histone | 55.4% (32.5%–59.1%) | 50.4% (0.500%–88.4%) |
| Pandoravirus dulcis | 5 | 70 | Histone H2B domain-containing protein | 25.7% (17.0%–26.4%) | 49.5% (1.60%–77.7%) |
| Pandoravirus salinus | 1 | 16 | Histone H2B domain | 23.4% | 12.4% (7.80%–76.4%) |
| Pandoravirus inopinatum | 3 | 68 | hypothetical protein | -- | -- |
| Scaffold Id | The Length of HFs (bp) | Amphioxus Genes | Virus Name | Virus Genes |
|---|---|---|---|---|
| Scaffold 14 | 276 | Histone H4 | Cotesia congregata bracovirus | Histone |
| scaffold 14 | 275 | Histone H4 | Cotesia congregata bracovirus | Histone |
| scaffold 14 | 275 | Histone H2B | Cotesia congregata bracovirus | Histone |
| scaffold 14 | 272 | Histone H4 | Cotesia congregata bracovirus | Histone |
| scaffold 14 | 271 | Histone H2B | Cotesia congregata bracovirus | Histone |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, Q.; Peng, F.; Xiong, Q.; Xu, K.; Yang, K.Y.; Wang, M.; Wu, Z.; Li, S.; Cheng, X.; Rao, X.; et al. Genomic Analysis of Amphioxus Reveals a Wide Range of Fragments Homologous to Viral Sequences. Viruses 2023, 15, 909. https://doi.org/10.3390/v15040909
Du Q, Peng F, Xiong Q, Xu K, Yang KY, Wang M, Wu Z, Li S, Cheng X, Rao X, et al. Genomic Analysis of Amphioxus Reveals a Wide Range of Fragments Homologous to Viral Sequences. Viruses. 2023; 15(4):909. https://doi.org/10.3390/v15040909
Chicago/Turabian StyleDu, Qiao, Fang Peng, Qing Xiong, Kejin Xu, Kevin Yi Yang, Mingqiang Wang, Zhitian Wu, Shanying Li, Xiaorui Cheng, Xinjie Rao, and et al. 2023. "Genomic Analysis of Amphioxus Reveals a Wide Range of Fragments Homologous to Viral Sequences" Viruses 15, no. 4: 909. https://doi.org/10.3390/v15040909