
Molecular Modeling of Viral Type I Fusion Proteins: Inhibitors of Influenza Virus Hemagglutinin and the Spike Protein of Coronavirus
 , , and
, , and 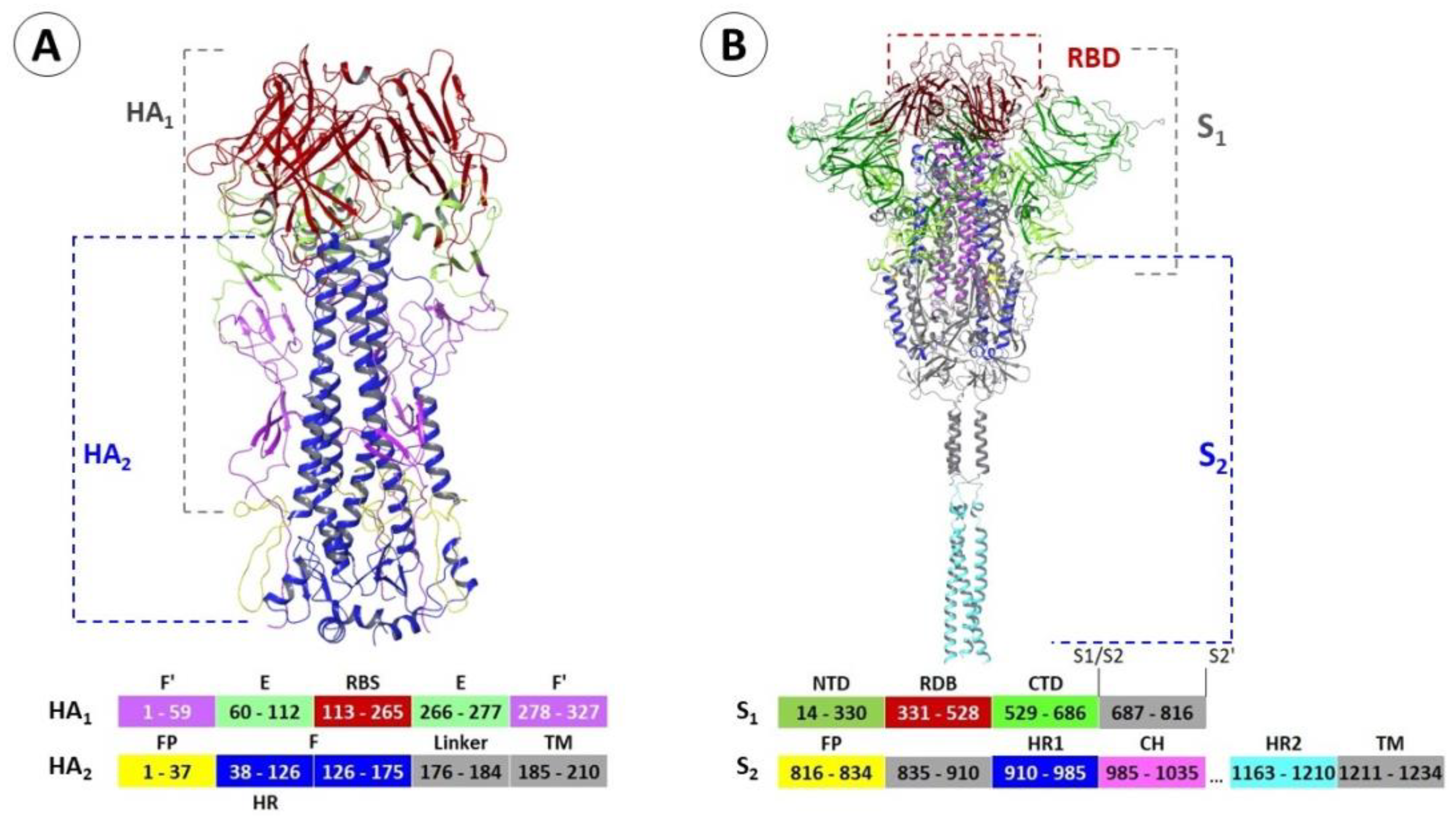{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
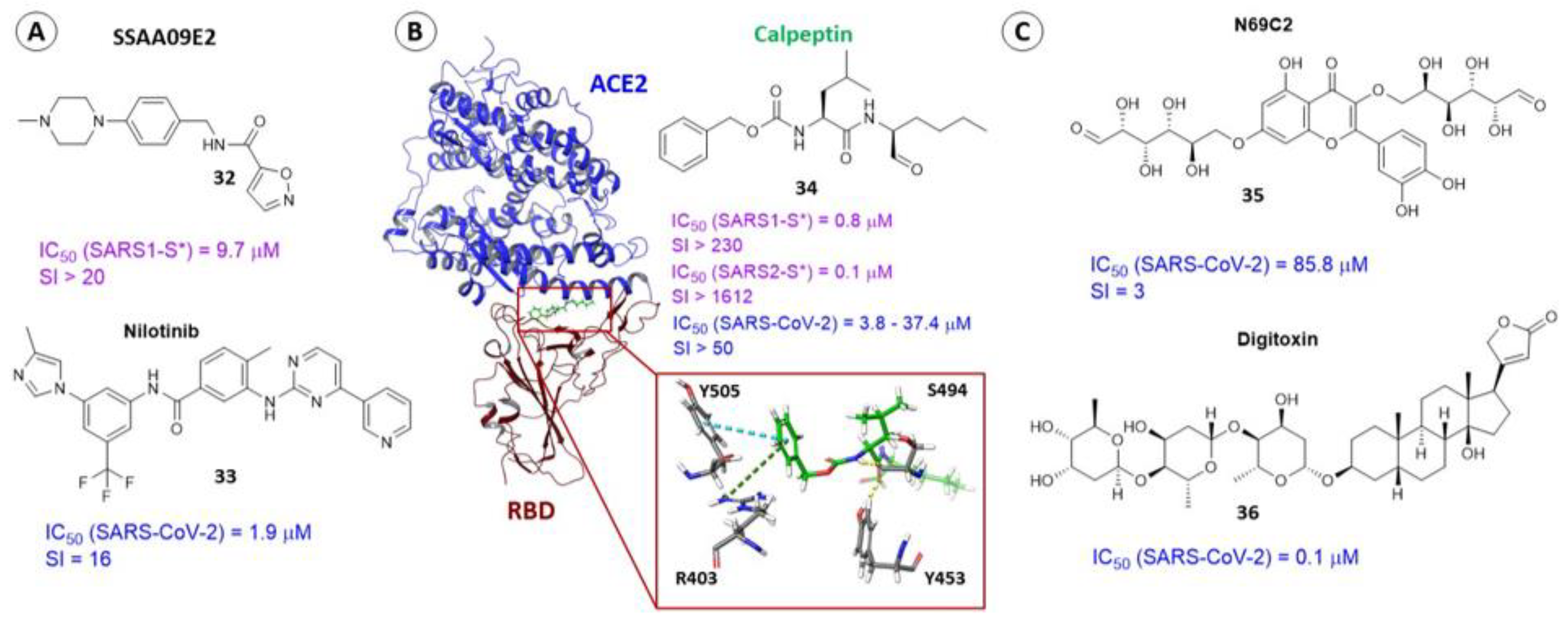{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Surface Viral Proteins
2. Hemagglutinin of Influenza Virus
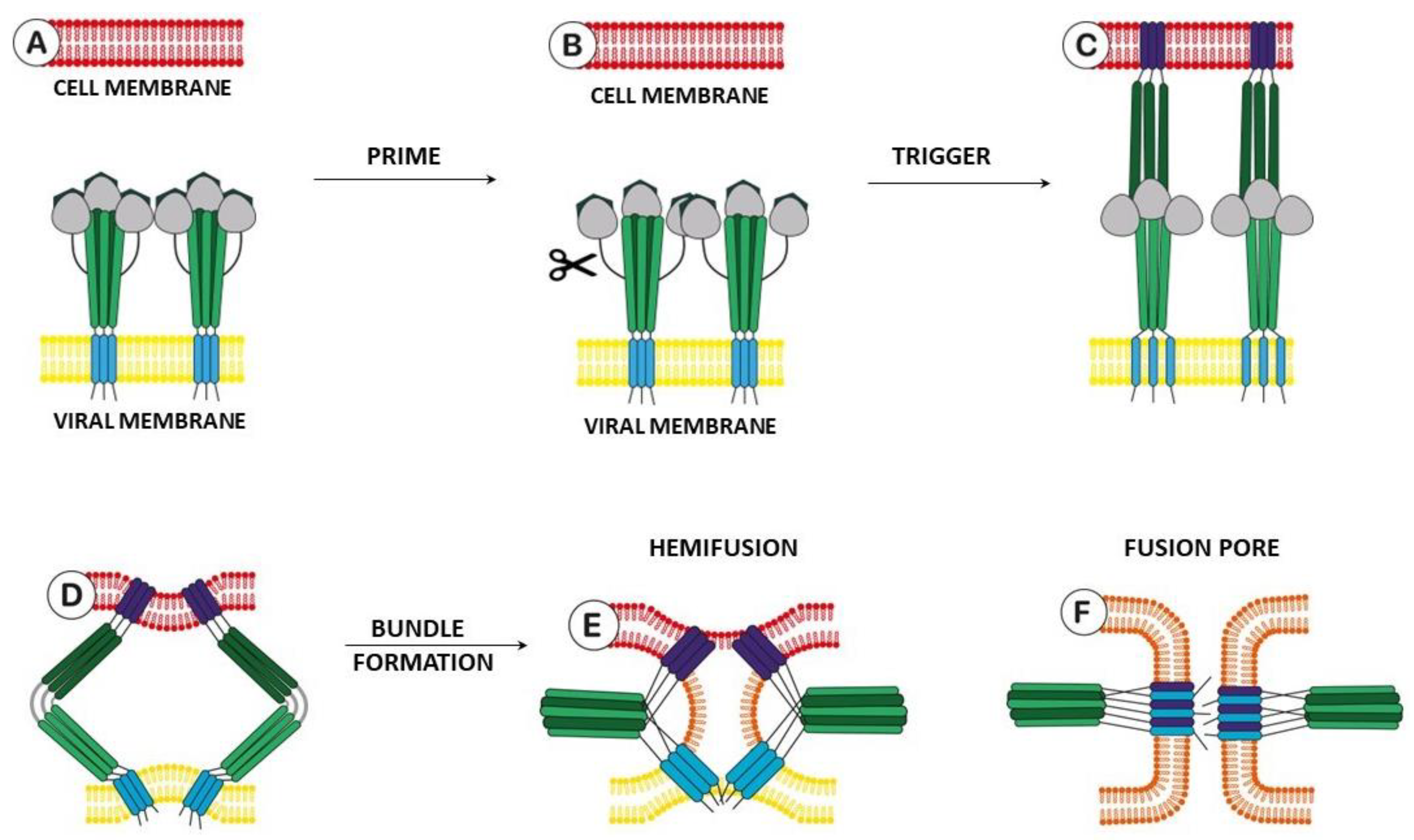
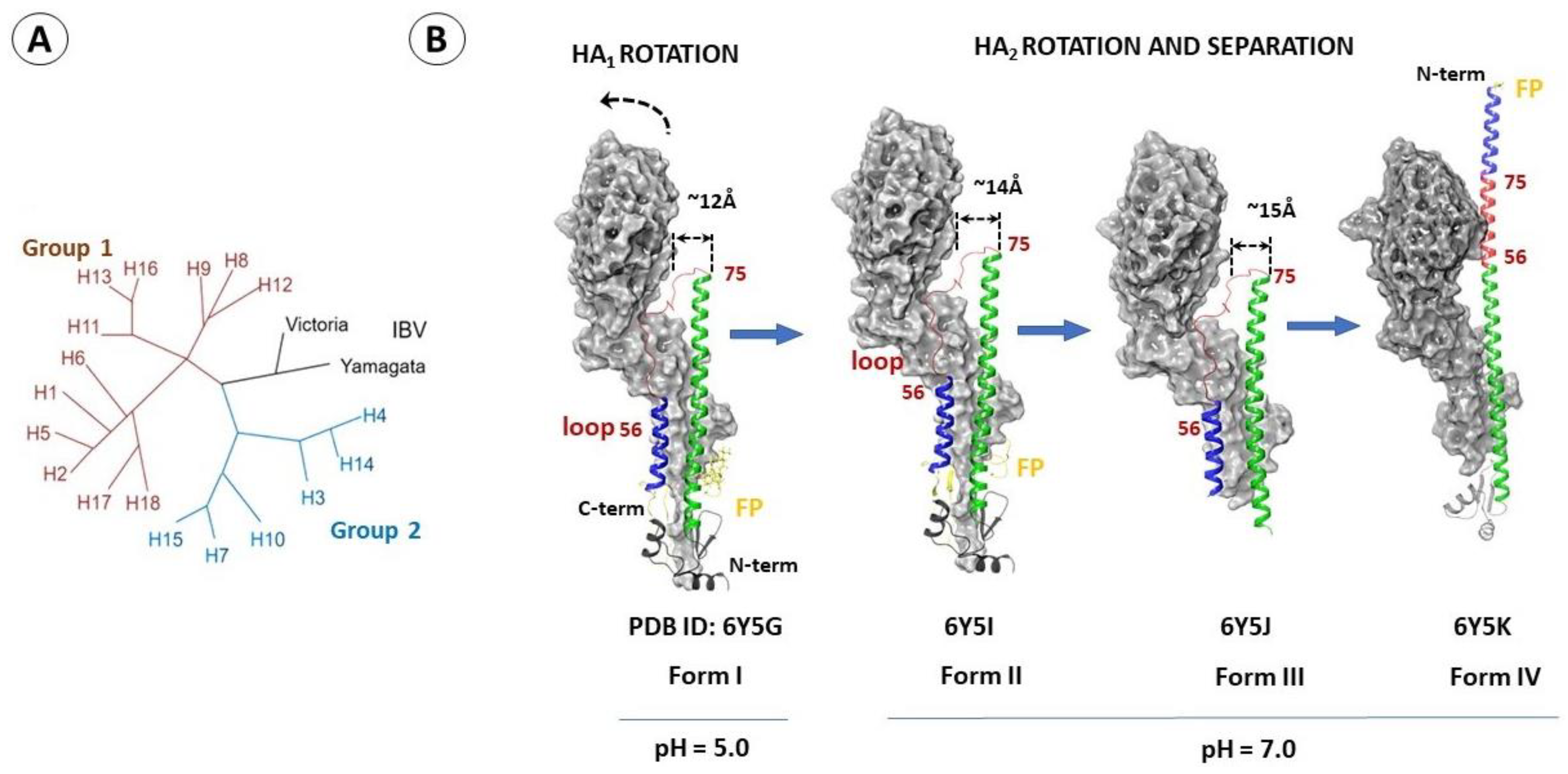
2.1. Structure and Function of Hemagglutinin
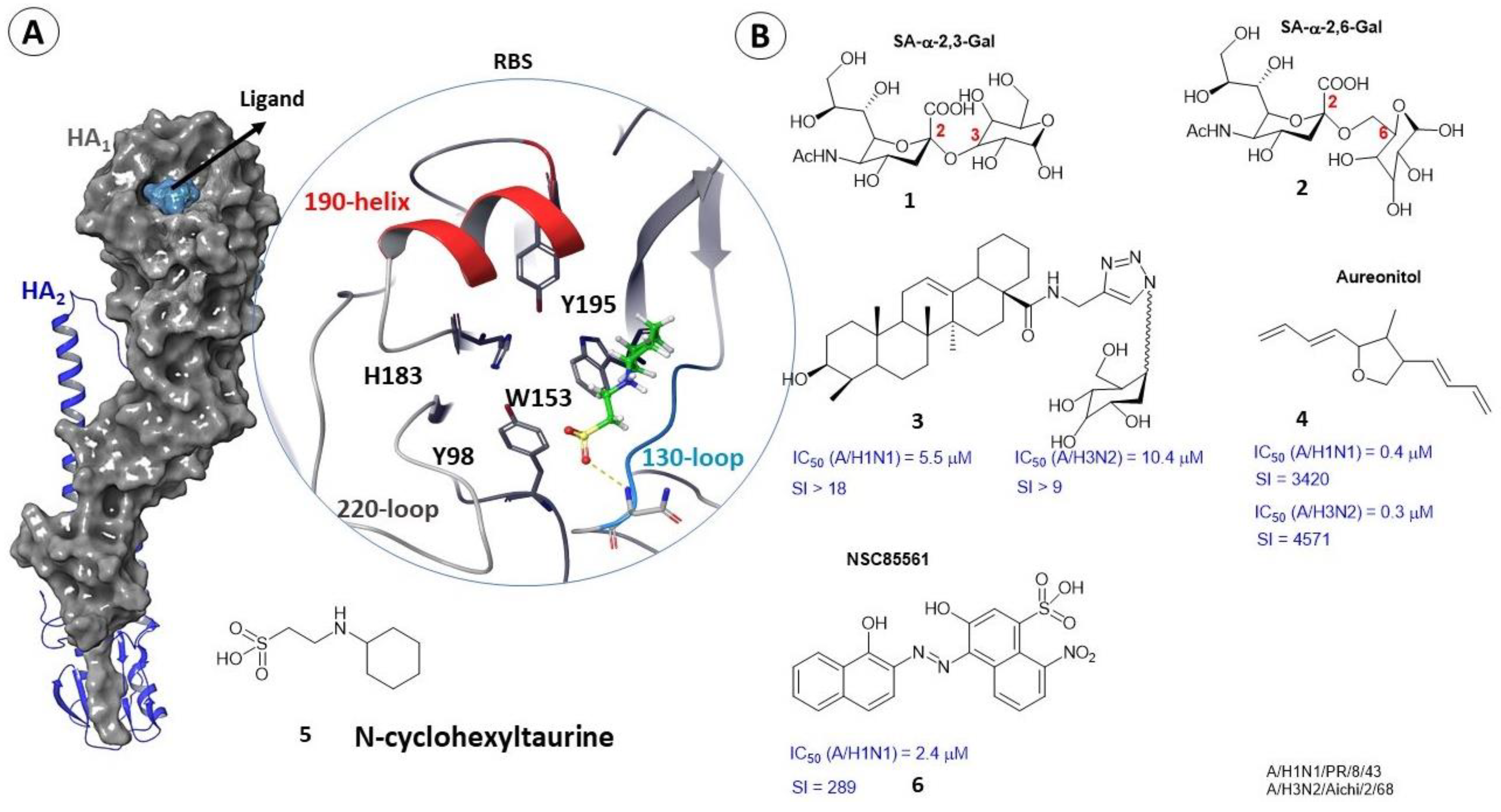
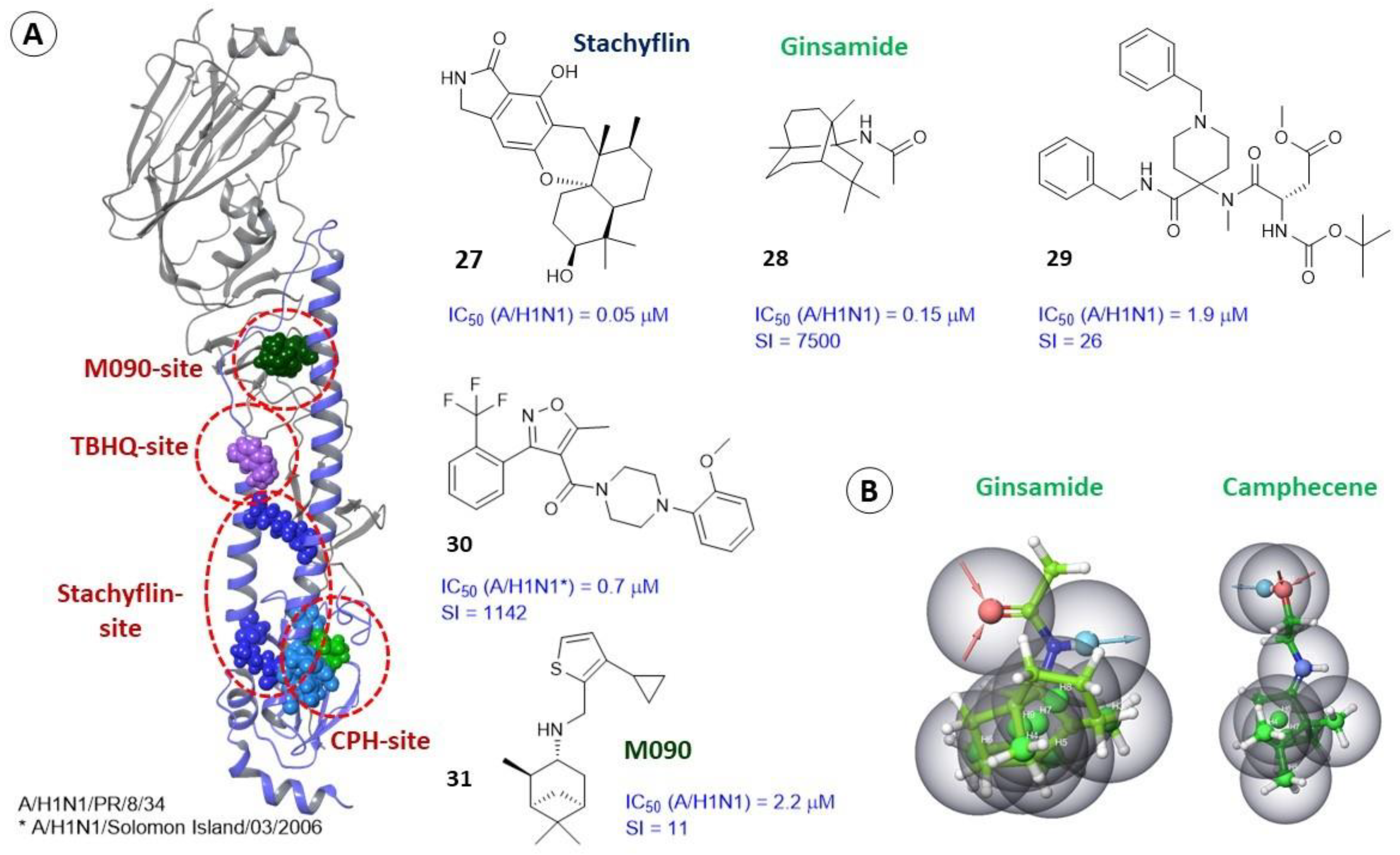
2.2. Binding Sites of Small Molecules in HA1
2.3. Binding Sites of Small Molecules in HA2
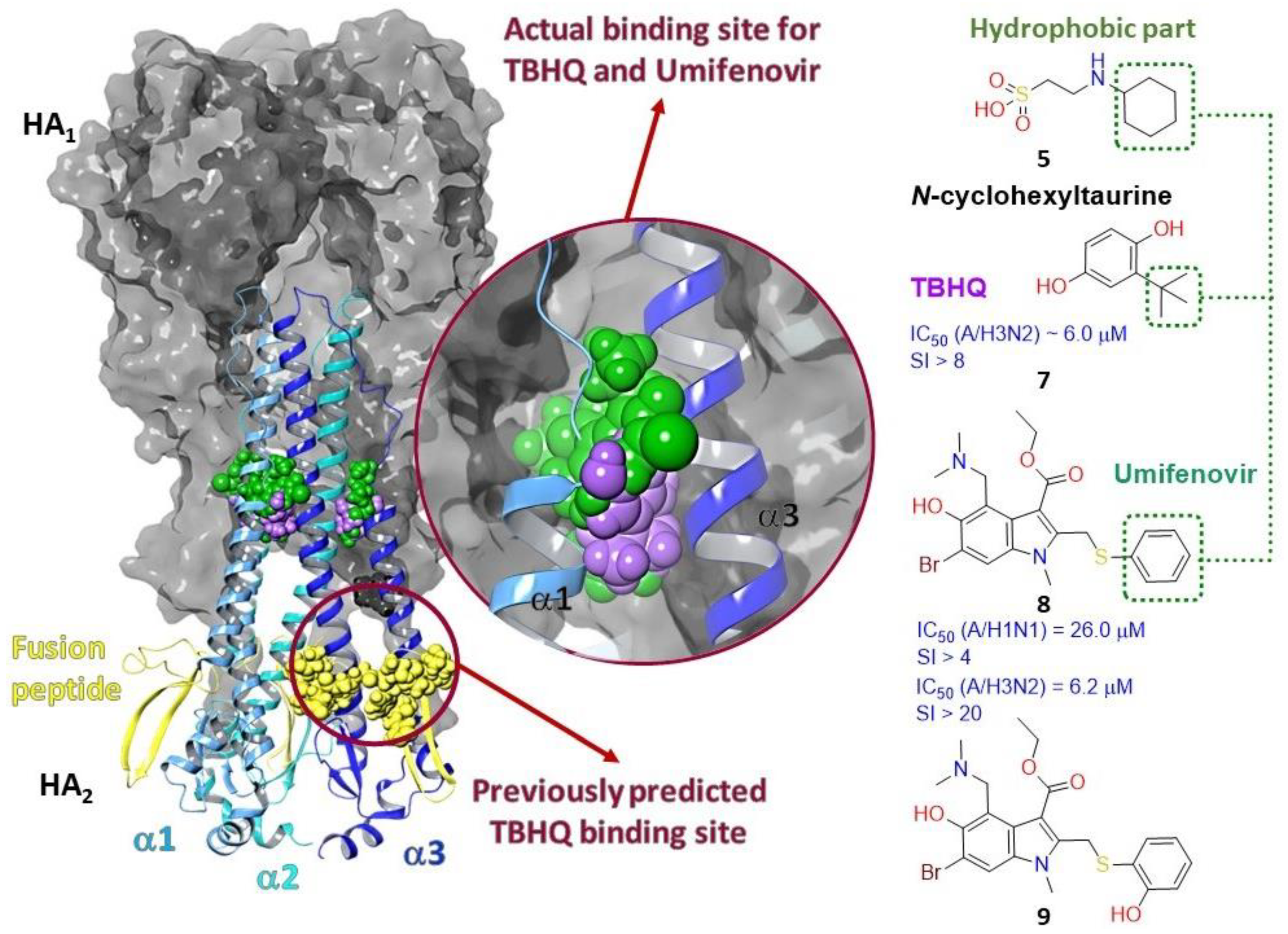
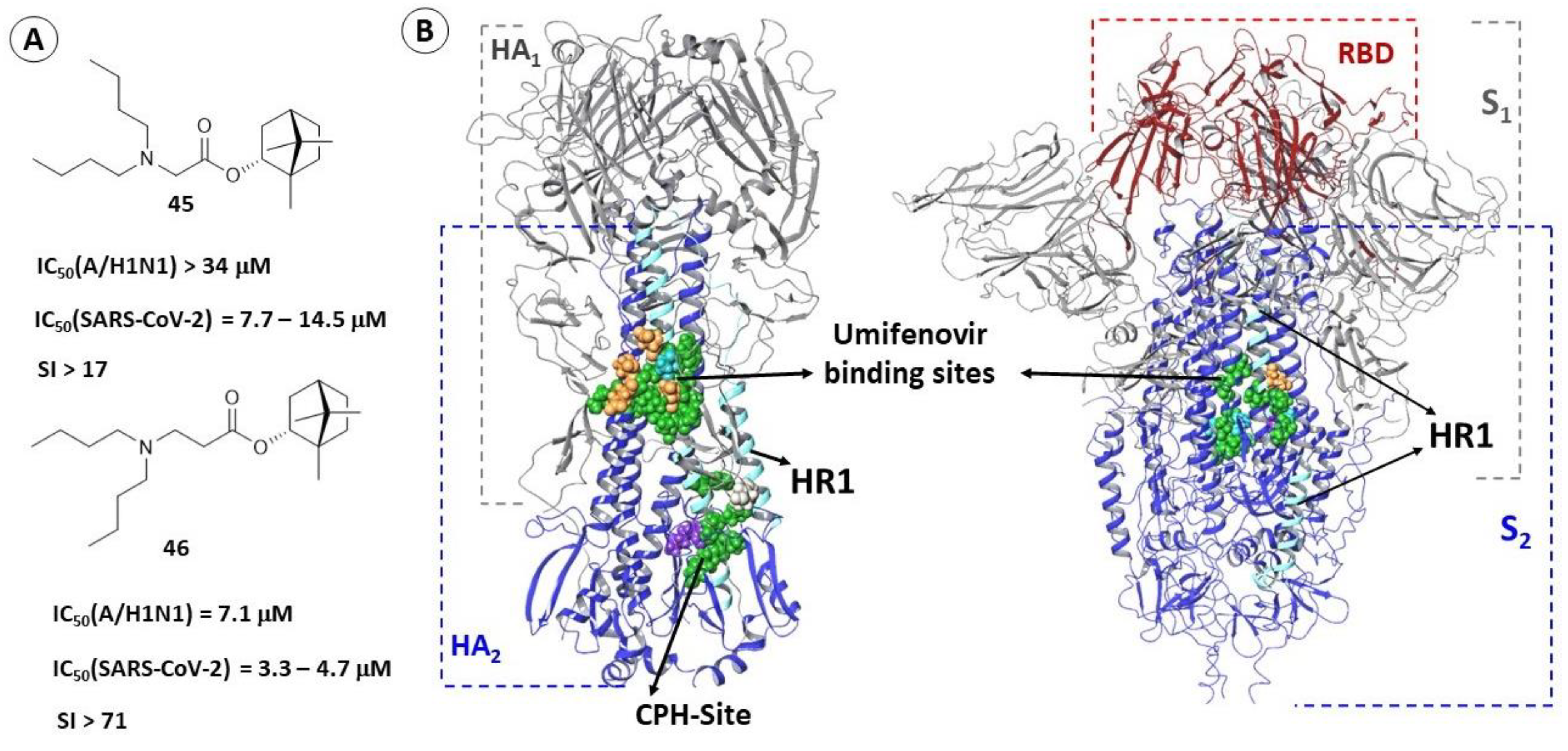
2.3.1. Binding Site of TBQH and Umifenovir
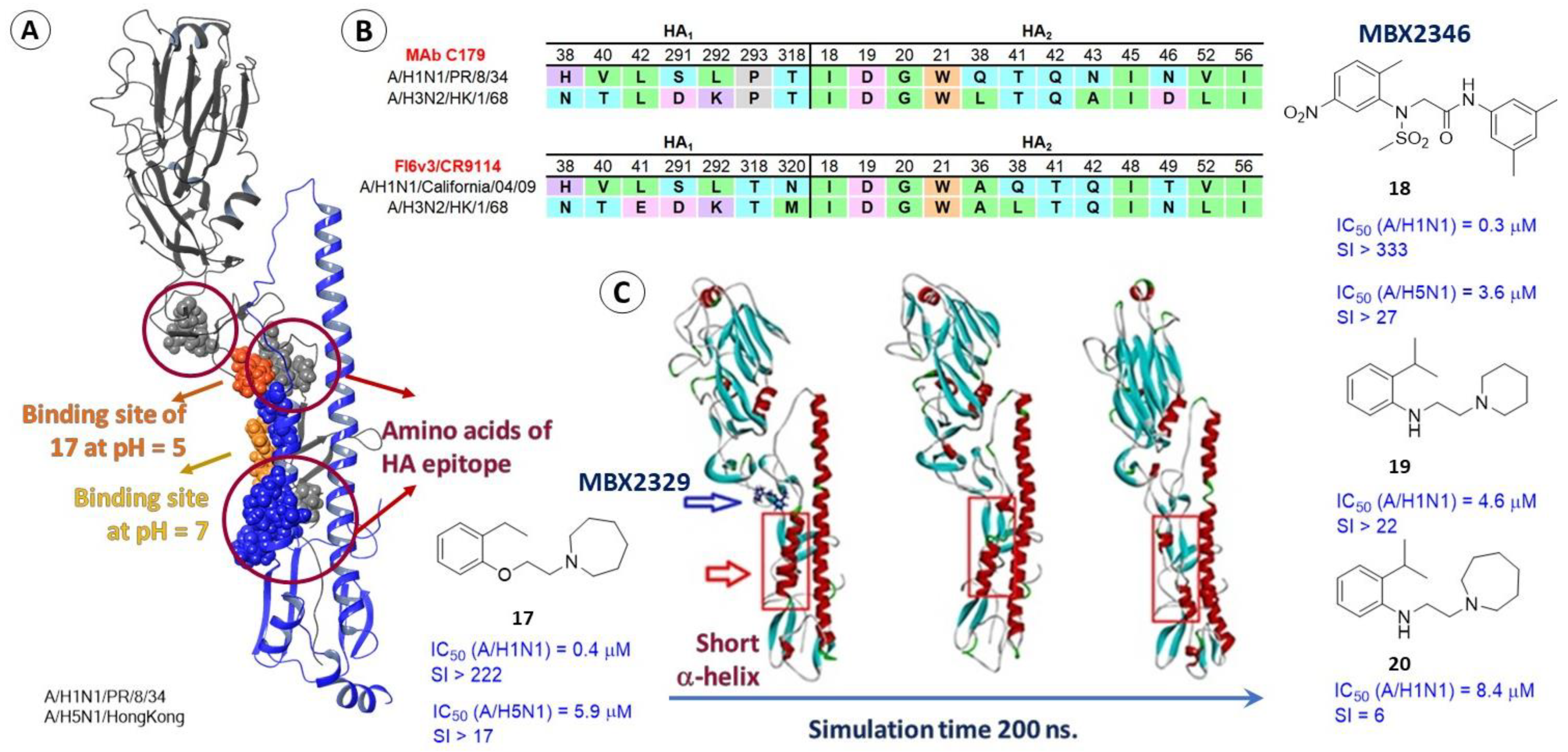
2.3.2. Epitopes of HA as a Possible Binding Site for Inhibitors
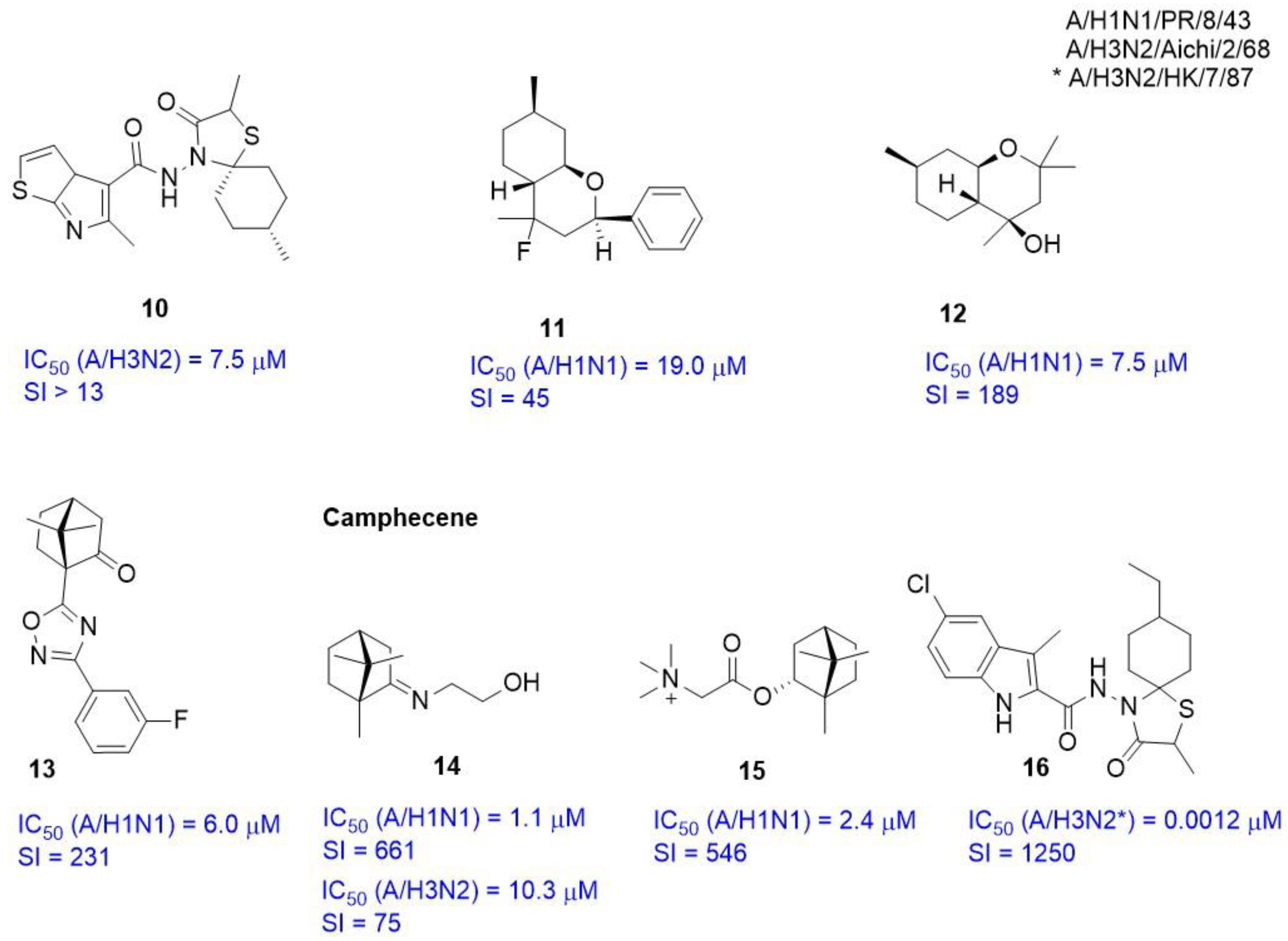
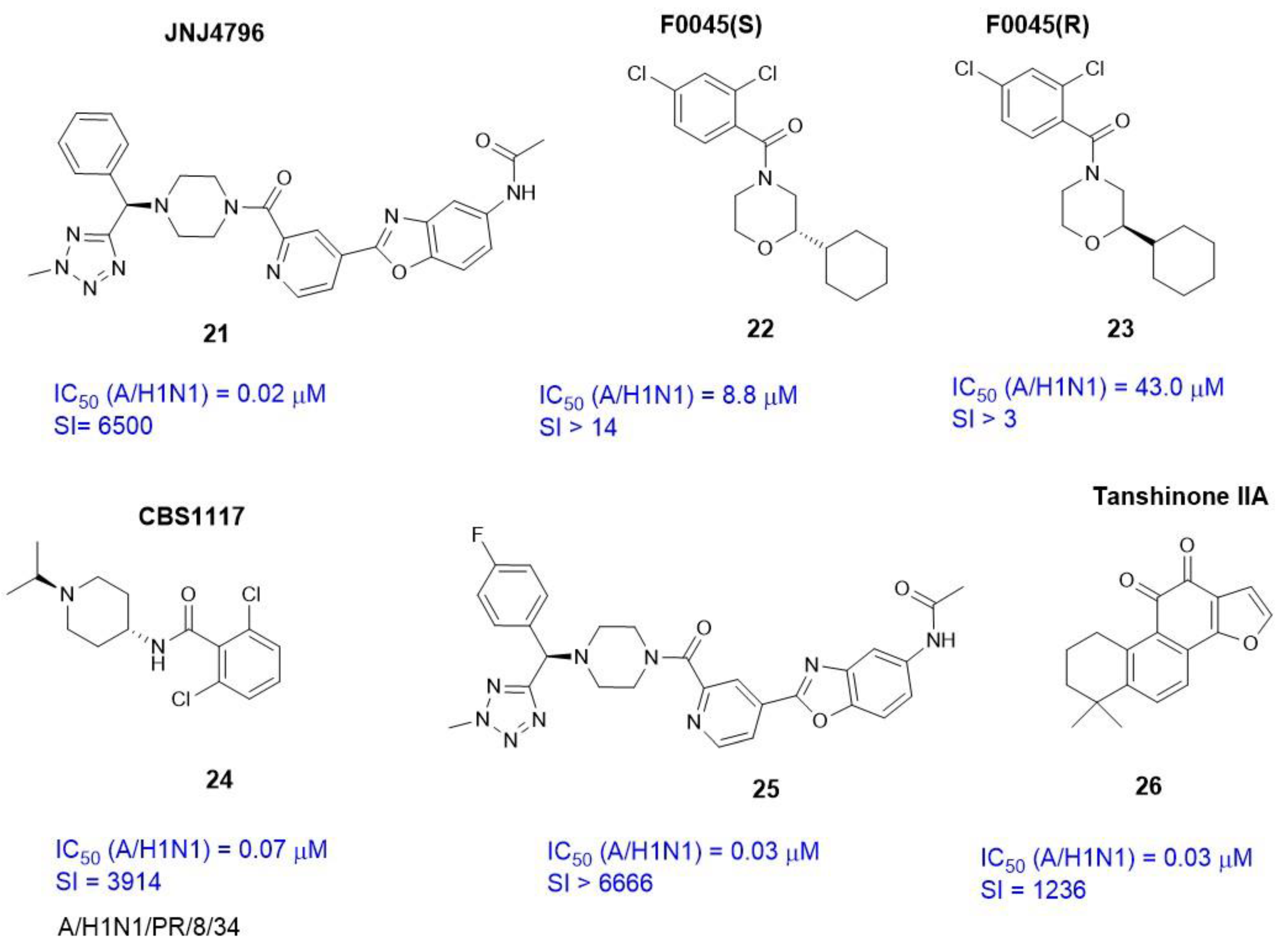
2.3.3. Alternate Binding Sites
2.4. Differences in the Binding Sites of HA2 of Different Phylogenetic Groups
3. Glycoprotein (Spike Protein) of Coronaviruses
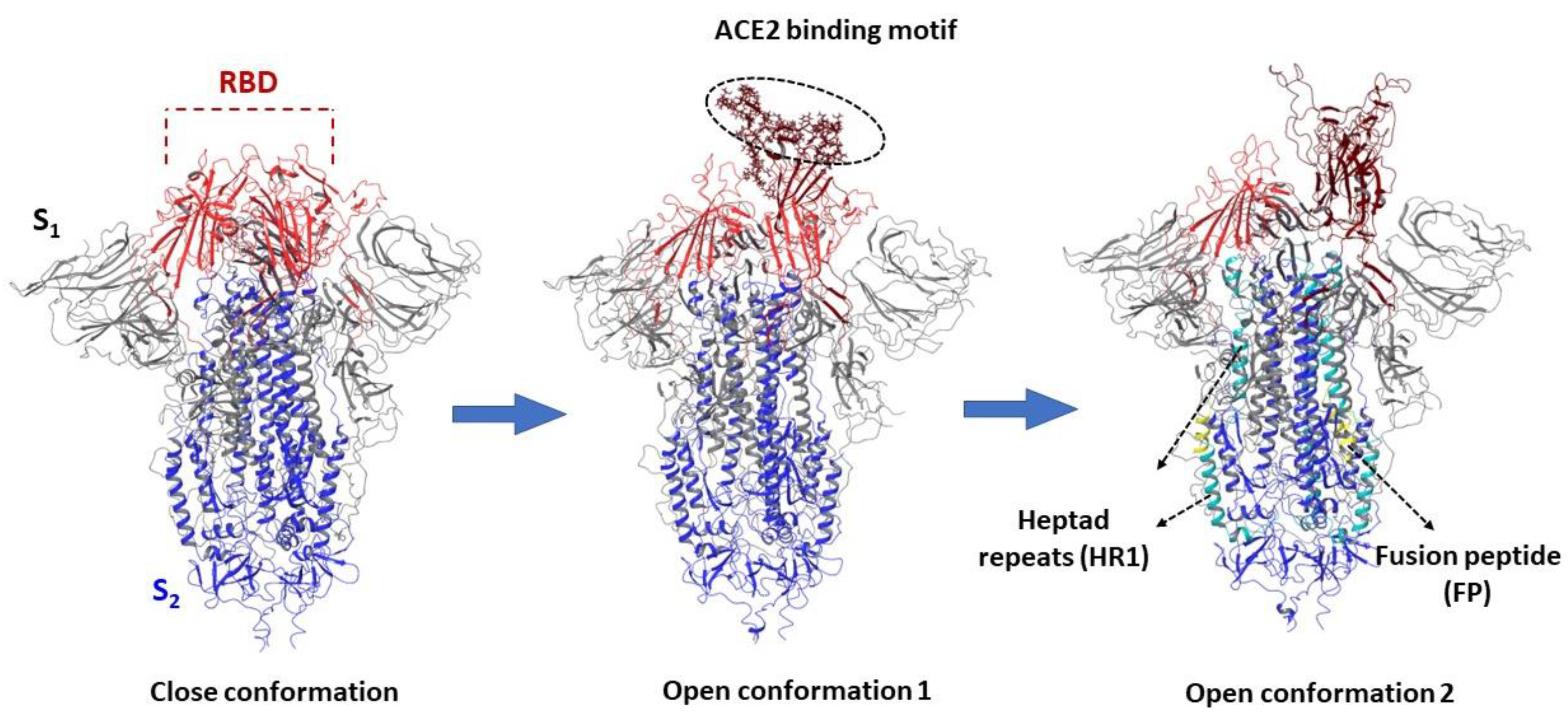
3.1. Structure and Function of the Spike Protein
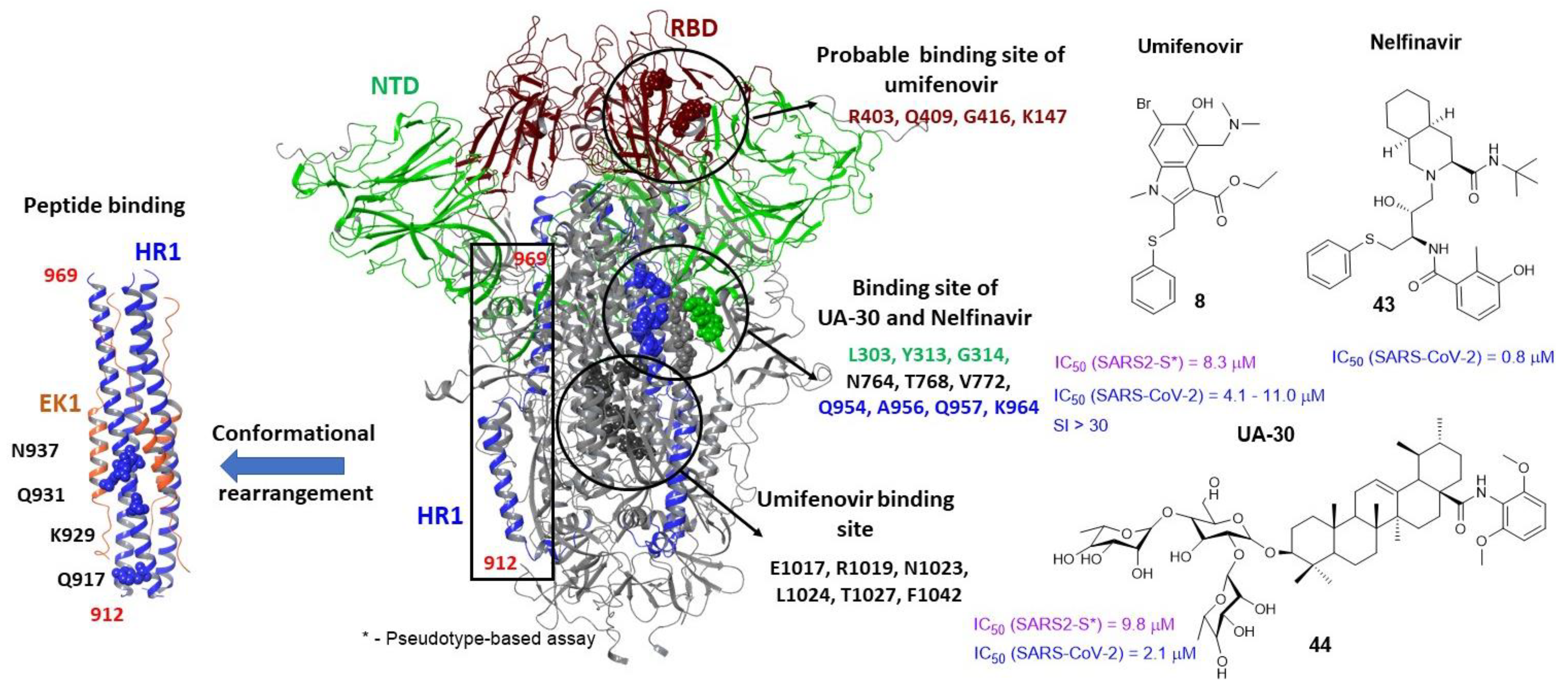
3.2. Small Molecule Binding Sites in S1
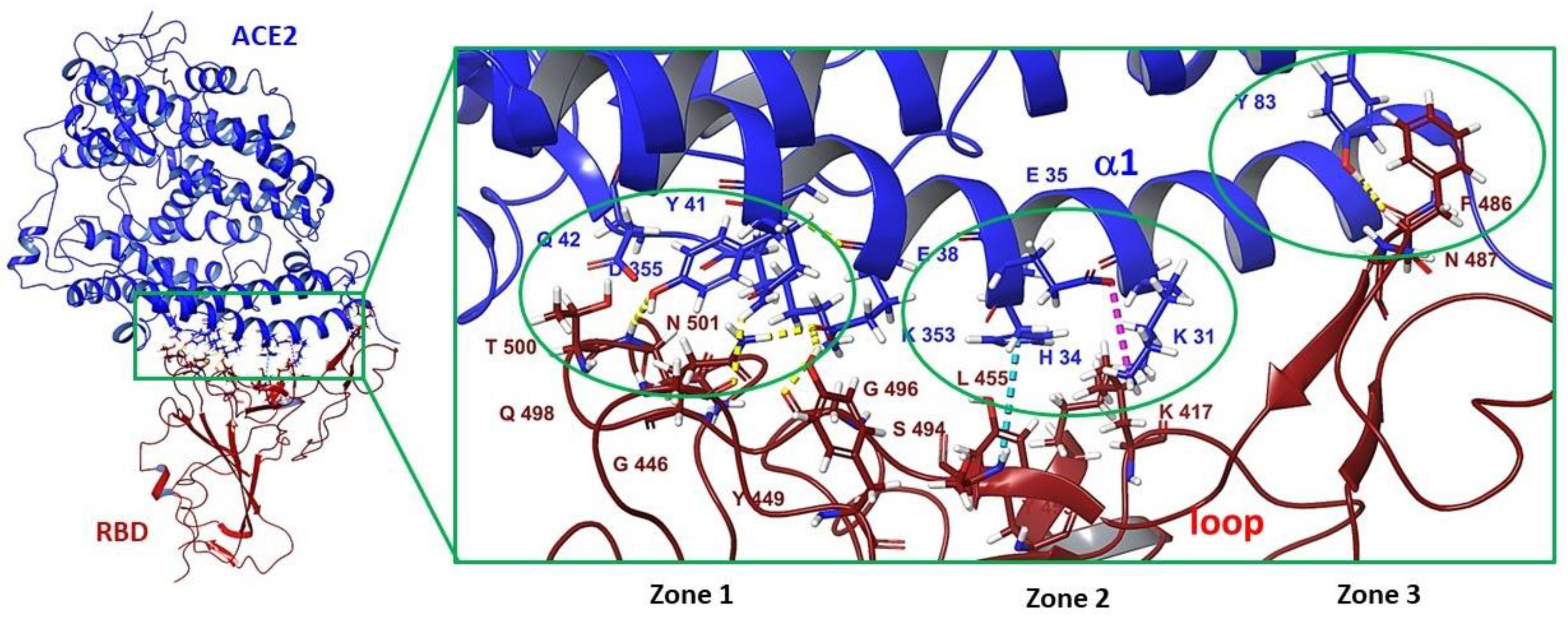
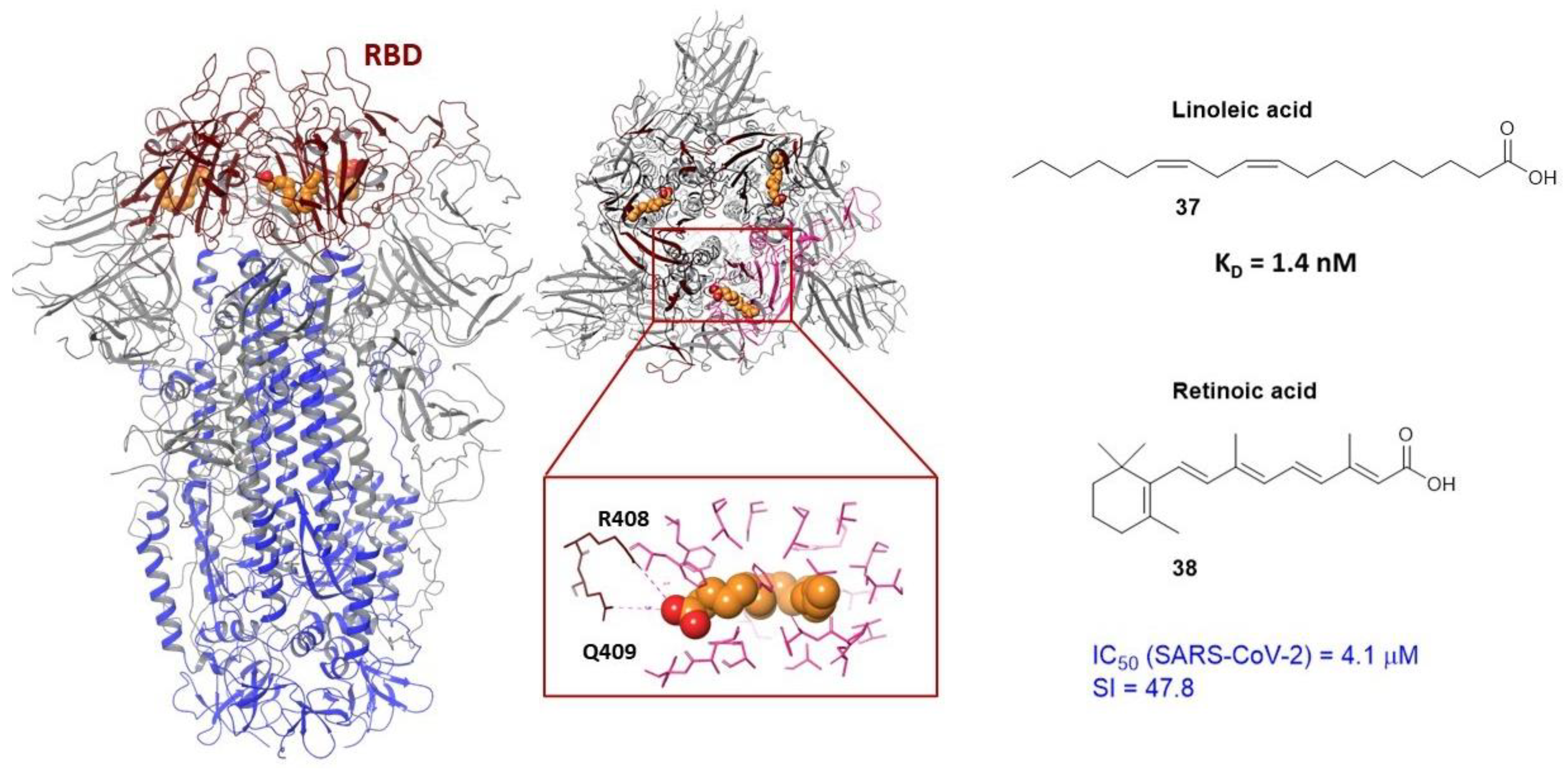
3.3. Binding Site in the Receptor-Binding Domain of S1
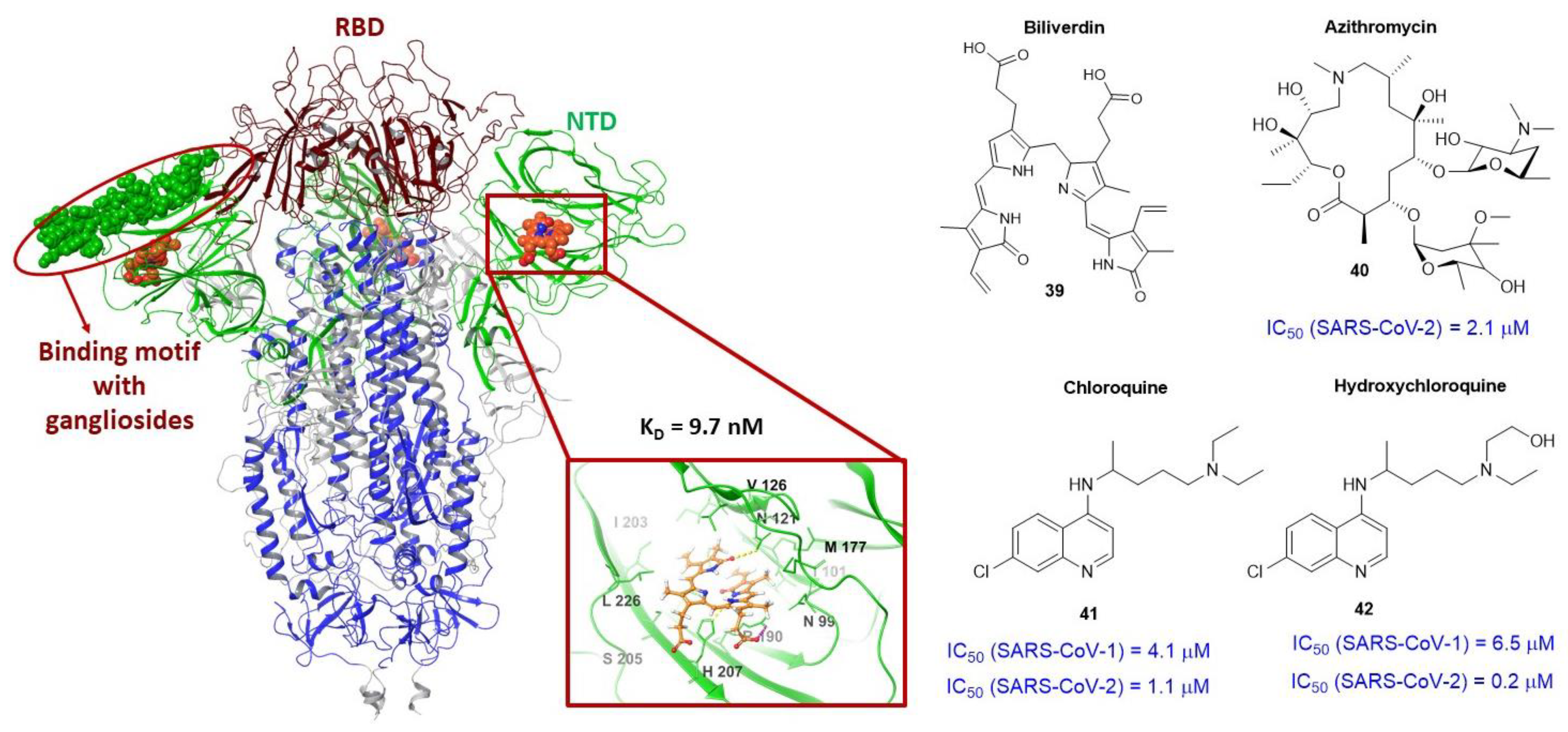
3.4. Binding Site of the N-Terminal Domain of S1
3.5. Binding Sites of the S2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
| a. a. | amino acids |
| ACE2 | angiotensin-converting enzyme 2 |
| CoV | coronavirus |
| CTD | C-terminal domain |
| CPH | camphecene |
| F | fusion protein |
| FP | fusion peptide |
| G | glycoprotein |
| HA | influenza virus hemagglutinin |
| HA1 | first subunits of influenza virus hemagglutinin (global head) |
| HA2 | second subunits of influenza virus hemagglutinin (stem part) |
| HIV-1 | Human immunodeficiency virus |
| HR | heptad repeat |
| HTS | High-throughput screening |
| LA | linoleic acid |
| MERS-CoV | middle east respiratory syndrome |
| RBD | receptor binding domain of coronavirus |
| RBS | hemagglutinin receptor binding site |
| NTD | N-terminal domain |
| PIV | parainfluenza virus |
| QM | quantum mechanic |
| S | S-protein or glycoprotein of coronavirus |
| S1 | first subunit of coronavirus S-protein |
| S2 | second subunit of coronavirus S-protein |
| SI | selectivity index |
| SARS | severe acute respiratory syndrome |
| TBEV | tick-borne encephalitis virus |
| TMD | trans-membrane domain |
| VSV | vesicular stomatitis virus |
References
- White, J.M.; Whittaker, G.R. Fusion of Enveloped Viruses in Endosomes. Traffic 2016, 17, 593–614. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479, 498–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, C.T.; Dutch, R.E. Viral Membrane Fusion and the Transmembrane Domain. Viruses 2020, 12, 693. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and Mechanisms of Viral Membrane Fusion Proteins: Multiple Variations on a Common Theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denolly, S.; Cosset, F.-L. HIV fusion: Catch me if you can. J. Biol. Chem. 2020, 295, 107–113. [Google Scholar] [CrossRef]
- Ward, A.E.; Kiessling, V.; Pornillos, O.; White, J.M.; Ganser-Pornillos, B.K.; Tamm, L.K. HIV-cell membrane fusion intermediates are restricted by Serincs as revealed by cryo-electron and TIRF microscopy. J. Biol. Chem. 2020, 295, 15183–15195. [Google Scholar] [CrossRef]
- Beniac, D.R.; Timothy, B.F. Structure of the Ebola virus glycoprotein spike within the virion envelope at 11 Å resolution. Sci. Rep. 2017, 7, 46374. [Google Scholar] [CrossRef] [Green Version]
- Blijleven, J.S.; Boonstra, S.; Onck, P.R.; van der Giessen, E.; van Oijen, A.M. Mechanisms of influenza viral membrane fusion. Semin. Cell Dev. Biol. 2016, 60, 78–88. [Google Scholar] [CrossRef] [Green Version]
- Benton, D.J.; Gamblin, S.J.; Rosenthal, P.B.; Skehel, J.J. Structural transitions in influenza haemagglutinin at membrane fusion pH. Nature 2020, 583, 150–153. [Google Scholar] [CrossRef]
- Yin, H.-S.; Paterson, R.G.; Wen, X.; Lamb, R.A.; Jardetzky, T.S. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9288–9293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamblin, S.J.; Haire, L.F.; Russell, R.J.; Stevens, D.J.; Xiao, B.; Ha, Y.; Vasisht, N.; Steinhauer, D.A.; Daniels, R.S.; Elliot, A.; et al. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science 2004, 303, 1838–1842. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Chen, Z.; Cui, Q.; Caffrey, M.; Rong, L.; Du, R. Small molecule inhibitors of influenza virus entry. Pharmaceuticals 2021, 14, 587. [Google Scholar] [CrossRef]
- Russell, R.J.; Kerry, P.S.; Stevens, D.J.; Steinhauer, D.A.; Martin, S.R.; Gamblin, S.J.; Skehel, J.J. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. USA 2008, 105, 17736–17741. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Li, M.; Shen, X.; Liu, S. Influenza A Virus Entry Inhibitors Targeting the Hemagglutinin. Viruses 2013, 5, 352–373. [Google Scholar] [CrossRef] [Green Version]
- Petrova, V.N.; Russell, C.A. The evolution of seasonal influenza viruses. Nat. Rev. Microbiol. 2018, 16, 47–60. [Google Scholar] [CrossRef]
- Matrosovich, M.; Herrler, G.; Klenk, H.D. Sialic acid receptors of viruses. Top. Curr. Chem. 2015, 367, 1–28. [Google Scholar] [CrossRef]
- Xu, R.; Wilson, I.A. Structural Characterization of an Early Fusion Intermediate of Influenza Virus Hemagglutinin. J. Virol. 2011, 85, 5172–5182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, B.S.; Whittaker, G.R.; Daniel, S. Influenza Virus-Mediated Membrane Fusion: Determinants of Hemagglutinin Fusogenic Activity and Experimental Approaches for Assessing Virus Fusion. Viruses 2012, 4, 1144–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernomordik, L.V.; Kozlov, M.M. Protein-Lipid Interplay in Fusion and Fission of Biological Membranes. Annu. Rev. Biochem. 2003, 72, 175–207. [Google Scholar] [CrossRef]
- Benton, D.J.; Nans, A.; Calder, L.J.; Turner, J.; Neu, U.; Lin, Y.P.; Ketelaars, E.; Kallewaard, N.L.; Corti, D.; Lanzavecchia, A.; et al. Influenza hemagglutinin membrane anchor. Proc. Natl. Acad. Sci. USA 2018, 115, 10112–10117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazniewski, M.; Dawson, W.K.; Szczepińska, T.; Plewczynski, D. The structural variability of the influenza A hemagglutinin receptor-binding site. Brief. Funct. Genom. 2018, 17, 415–427. [Google Scholar] [CrossRef]
- Luo, M. Influenza Virus Entry. In Viral Molecular Machines; Springer Science+Business Media, LLC: Berlin, Germany, 2012; pp. 201–221. [Google Scholar]
- Priyadarzini, T.R.K.; Selvin, J.F.A.; Gromiha, M.M.; Fukui, K.; Veluraja, K. Theoretical Investigation on the Binding Specificity of Sialyldisaccharides with Hemagglutinins of Influenza A Virus by Molecular Dynamics Simulations. J. Biol. Chem. 2012, 287, 34547–34557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Wang, B. Computational studies of H5N1 hemagglutinin binding with SA-α-2, 3-Gal and SA-α-2, 6-Gal. Biochem. Biophys. Res. Commun. 2006, 347, 662–668. [Google Scholar] [CrossRef]
- Kadam, R.U.; Wilson, I.A. A small-molecule fragment that emulates binding of receptor and broadly neutralizing antibodies to influenza A hemagglutinin. Proc. Natl. Acad. Sci. USA 2018, 115, 4240–4245. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wu, Y.; Yao, S.; Ge, H.; Zhu, Y.; Chen, K.; Chen, W.; Zhang, Y.; Zhu, W.; Wang, H.Y.; et al. Discovery of potential small molecular SARS-CoV-2 entry blockers targeting the spike protein. Acta Pharmacol. Sin. 2021, 43, 788–796. [Google Scholar] [CrossRef]
- Lu, W.; Du, W.; Somovilla, V.J.; Yu, G.; Haksar, D.; De Vries, E.; Boons, G.J.; De Vries, R.P.; De Haan, C.A.M.; Pieters, R.J. Enhanced Inhibition of Influenza A Virus Adhesion by Di- and Trivalent Hemagglutinin Inhibitors. J. Med. Chem. 2019, 62, 6398–6404. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Su, Y.; Yang, F.; Xiao, S.; Yin, Z.; Liu, J.; Zhong, J.; Zhou, D.; Yu, F. Design, synthesis and biological evaluation of amino acids-oleanolic acid conjugates as influenza virus inhibitors. Bioorganic Med. Chem. 2019, 27, 115147. [Google Scholar] [CrossRef]
- Sacramento, C.Q.; Marttorelli, A.; Fintelman-Rodrigues, N.; De Freitas, C.S.; De Melo, G.R.; Rocha, M.E.N.; Kaiser, C.R.; Rodrigues, K.F.; Da Costa, G.L.; Alves, C.M.; et al. Aureonitol, a fungi-derived tetrahydrofuran, inhibits influenza replication by targeting its surface glycoprotein hemagglutinin. PLoS ONE 2015, 10, e0139236. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-J.; Yeh, C.-Y.; Cheng, J.-C.; Huang, Y.-Q.; Hsu, K.-C.; Lin, Y.-F.; Lu, C.-H. Potent sialic acid inhibitors that target influenza A virus hemagglutinin. Sci. Rep. 2021, 11, 8637. [Google Scholar] [CrossRef]
- Le, K.P.; Do, P.-C.; Amaro, R.E.; Le, L. Molecular Docking of Broad-Spectrum Antibodies on Hemagglutinins of Influenza A Virus. Evol. Bioinforma. 2019, 15, 1176934319876938. [Google Scholar] [CrossRef]
- Strauch, E.M.; Bernard, S.M.; La, D.; Bohn, A.J.; Lee, P.S.; Anderson, C.E.; Nieusma, T.; Holstein, C.A.; Garcia, N.K.; Hooper, K.A.; et al. Computational design of trimeric influenza-neutralizing proteins targeting the hemagglutinin receptor binding site. Nat. Biotechnol. 2017, 35, 667–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, S.; Wang, T.; Kuai, Z.; Qian, M.; Tian, X.; Zhang, X.; Yu, Y.; Wang, S.; Zhang, H.; Li, H.; et al. Exploration of binding and inhibition mechanism of a small molecule inhibitor of influenza virus H1N1 hemagglutinin by molecular dynamics simulation. Sci. Rep. 2017, 7, 3786. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Zhu, Z.; Ding, Y.; Wu, W.; Yang, J.; Liu, S. An oligothiophene compound neutralized influenza A viruses by interfering with hemagglutinin. Biochim. Biophys. Acta-Biomembr. 2018, 1860, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Cheng, H.; Cui, Q.; Peet, N.P.; Gaisina, I.N.; Rong, L. Identification of a novel inhibitor targeting influenza A virus group 2 hemagglutinins. Antivir. Res. 2021, 186, 105013. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Abouzid, M. Arbidol targeting influenza virus A Hemagglutinin; A comparative study. Biophys. Chem. 2021, 277, 106663. [Google Scholar] [CrossRef]
- Bodian, D.L.; Yamasaki, R.B.; Buswell, R.L.; Stearns, J.F.; White, J.M.; Kuntz, I.D. Inhibition of the Fusion-Inducing Conformational Change of Influenza Hemagglutinin by Benzoquinones and Hydroquinones. Biochemistry 1993, 32, 2967–2978. [Google Scholar] [CrossRef]
- Hoffman, L.R.; Kuntz, I.D.; White, J.M. Structure-based identification of an inducer of the low-pH conformational change in the influenza virus hemagglutinin: Irreversible inhibition of infectivity. J. Virol. 1997, 71, 8808–8820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boriskin, Y.; Leneva, I.; Pecheur, E.-I.; Polyak, S. Arbidol: A Broad-Spectrum Antiviral Compound that Blocks Viral Fusion. Curr. Med. Chem. 2008, 15, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Kadam, R.U.; Wilson, I.A. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc. Natl. Acad. Sci. USA 2017, 114, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Wright, Z.V.F.; Wu, N.C.; Kadam, R.U.; Wilson, I.A.; Wolan, D.W. Structure-based optimization and synthesis of antiviral drug Arbidol analogues with significantly improved affinity to influenza hemagglutinin. Bioorganic Med. Chem. Lett. 2017, 27, 3744–3748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderlinden, E.; Göktaş, F.; Cesur, Z.; Froeyen, M.; Reed, M.L.; Russell, C.J.; Cesur, N.; Naesens, L. Novel Inhibitors of Influenza Virus Fusion: Structure-Activity Relationship and Interaction with the Viral Hemagglutinin. J. Virol. 2010, 84, 4277–4288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilyina, I.V.; Zarubaev, V.V.; Lavrentieva, I.N.; Shtro, A.A.; Esaulkova, I.L.; Korchagina, D.V.; Borisevich, S.S.; Volcho, K.P.; Salakhutdinov, N.F. Highly potent activity of isopulegol-derived substituted octahydro-2H-chromen-4-ols against influenza A and B viruses. Bioorganic Med. Chem. Lett. 2018, 28, 2061–2067. [Google Scholar] [CrossRef] [PubMed]
- Ilyina, I.V.; Patrusheva, O.S.; Zarubaev, V.V.; Misiurina, M.A.; Slita, A.V.; Esaulkova, I.L.; Korchagina, D.V.; Gatilov, Y.V.; Borisevich, S.S.; Volcho, K.P.; et al. Influenza antiviral activity of F- and OH-containing isopulegol-derived octahydro-2H-chromenes. Bioorganic Med. Chem. Lett. 2021, 31, 127677. [Google Scholar] [CrossRef]
- Chernyshov, V.V.; Yarovaya, O.I.; Esaulkova, I.L.; Sinegubova, E.; Borisevich, S.S.; Popadyuk, I.I.; Zarubaev, V.V.; Salakhutdinov, N.F. Novel O–acylated amidoximes and substituted 1,2,4–oxadiazoles synthesised from (+)–ketopinic acid possessing potent virus-inhibiting activity against phylogenetically distinct influenza A viruses. Bioorganic Med. Chem. Lett. 2021, 55, 128465. [Google Scholar] [CrossRef] [PubMed]
- Zarubaev, V.V.; Garshinina, A.V.; Tretiak, T.S.; Fedorova, V.A.; Shtro, A.A.; Sokolova, A.S.; Yarovaya, O.I.; Salakhutdinov, N.F. Broad range of inhibiting action of novel camphor-based compound with anti-hemagglutinin activity against influenza viruses in vitro and in vivo. Antivir. Res. 2015, 120, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Borisevich, S.S.; Gureev, M.A.; Yarovaya, O.I.; Zarubaev, V.V.; Kostin, G.A.; Porozov, Y.B.; Salakhutdinov, N.F. Can molecular dynamics explain decreased pathogenicity in mutant camphecene-resistant influenza virus? J. Biomol. Struct. Dyn. 2021, 40, 5481–5492. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.S.; Yarovaya, O.I.; Baranova, D.V.; Galochkina, A.V.; Shtro, A.A.; Kireeva, M.V.; Borisevich, S.S.; Gatilov, Y.V.; Zarubaev, V.V.; Salakhutdinov, N.F. Quaternary ammonium salts based on (-)-borneol as effective inhibitors of influenza virus. Arch. Virol. 2021, 166, 1965–1976. [Google Scholar] [CrossRef] [PubMed]
- Cihan-Üstündağ, G.; Zopun, M.; Vanderlinden, E.; Ozkirimli, E.; Persoons, L.; Çapan, G.; Naesens, L. Superior inhibition of influenza virus hemagglutinin-mediated fusion by indole-substituted spirothiazolidinones. Bioorg. Med. Chem. 2020, 28, 115130. [Google Scholar] [CrossRef] [PubMed]
- Yarovaya, O.I.; Salakhutdinov, N.F. Mono- and sesquiterpenes as a starting platform for the development of antiviral drugs. Russ. Chem. Rev. 2021, 90, 488–510. [Google Scholar] [CrossRef]
- Laursen, N.S.; Wilson, I.A. Broadly neutralizing antibodies against influenza viruses. Antivir. Res. 2013, 98, 476–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreyfus, C.; Ekiert, D.C.; Wilson, I.A. Structure of a Classical Broadly Neutralizing Stem Antibody in Complex with a Pandemic H2 Influenza Virus Hemagglutinin. J. Virol. 2013, 87, 7149–7154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, A.; Antanasijevic, A.; Wang, M.; Li, B.; Mills, D.M.; Ames, J.A.; Nash, P.J.; Williams, J.D.; Peet, N.P.; Moir, D.T.; et al. New Small Molecule Entry Inhibitors Targeting Hemagglutinin-Mediated Influenza A Virus Fusion. J. Virol. 2014, 88, 1447–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Korte, T.; Rachakonda, P.S.; Knapp, E.-W.; Herrmann, A. Energetics of the loop-to-helix transition leading to the coiled-coil structure of influenza virus hemagglutinin HA2 subunits. Proteins Struct. Funct. Bioinforma. 2009, 74, 291–303. [Google Scholar] [CrossRef]
- Leiva, R.; Barniol-Xicota, M.; Codony, S.; Ginex, T.; Vanderlinden, E.; Montes, M.; Caffrey, M.; Luque, F.J.; Naesens, L.; Vázquez, S. Aniline-Based Inhibitors of Influenza H1N1 Virus Acting on Hemagglutinin-Mediated Fusion. J. Med. Chem. 2018, 61, 98–118. [Google Scholar] [CrossRef]
- Kadam, R.U.; Juraszek, J.; Brandenburg, B.; Buyck, C.; Schepens, W.B.G.; Kesteleyn, B.; Stoops, B.; Vreeken, R.J.; Vermond, J.; Goutier, W.; et al. Potent peptidic fusion inhibitors of influenza virus. Science 2017, 358, 496–502. [Google Scholar] [CrossRef] [Green Version]
- Ekiert, D.C.; Bhabha, G.; Elsliger, M.-A.; Friesen, R.H.E.; Jongeneelen, M.; Throsby, M.; Goudsmit, J.; Wilson, I.A. Antibody Recognition of a Highly Conserved Influenza Virus Epitope. Science 2009, 324, 246–251. [Google Scholar] [CrossRef] [Green Version]
- van Dongen, M.J.P.; Kadam, R.U.; Juraszek, J.; Lawson, E.; Brandenburg, B.; Schmitz, F.; Schepens, W.B.G.; Stoops, B.; van Diepen, H.A.; Jongeneelen, M.; et al. A small-molecule fusion inhibitor of influenza virus is orally active in mice. Science 2019, 363, eaar6221. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.; Casciaro, B.; Genovese, A.; Brancaccio, D.; Marcocci, M.E.; Novellino, E.; Carotenuto, A.; Palamara, A.T.; Mangoni, M.L.; Nencioni, L. Temporin g, an amphibian antimicrobial peptide against influenza and parainfluenza respiratory viruses: Insights into biological activity and mechanism of action. FASEB J. 2021, 35, e21358. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Kadam, R.U.; Lee, C.C.D.; Woehl, J.L.; Wu, N.C.; Zhu, X.; Kitamura, S.; Wilson, I.A.; Wolan, D.W. An influenza A hemagglutinin small-molecule fusion inhibitor identified by a new high-throughput fluorescence polarization screen. Proc. Natl. Acad. Sci. USA 2020, 117, 18431–18438. [Google Scholar] [CrossRef] [PubMed]
- Antanasijevic, A.; Durst, M.A.; Cheng, H.; Gaisina, I.N.; Perez, J.T.; Manicassamy, B.; Rong, L.; Lavie, A.; Caffrey, M. Structure of avian influenza hemagglutinin in complex with a small molecule entry inhibitor. Life Sci. Alliance 2020, 3, e202000724. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Li, Y.; Lv, K.; Gao, R.; Wang, A.; Yan, H.; Qin, X.; Xu, S.; Ma, C.; Jiang, J.; et al. Optimization and SAR research at the piperazine and phenyl rings of JNJ4796 as new anti-influenza A virus agents, part 1. Eur. J. Med. Chem. 2021, 222, 113591. [Google Scholar] [CrossRef] [PubMed]
- Elebeedy, D.; Badawy, I.; Elmaaty, A.A.; Saleh, M.M.; Kandeil, A.; Ghanem, A.; Kutkat, O.; Alnajjar, R.; Abd El Maksoud, A.I.; Al-karmalawy, A.A. In vitro and computational insights revealing the potential inhibitory effect of Tanshinone IIA against influenza A virus. Comput. Biol. Med. 2022, 141, 105149. [Google Scholar] [CrossRef]
- Motohashi, Y.; Igarashi, M.; Okamatsu, M.; Noshi, T.; Sakoda, Y.; Yamamoto, N.; Ito, K.; Yoshida, R.; Kida, H. Antiviral activity of stachyflin on influenza A viruses of different hemagglutinin subtypes. Virol. J. 2013, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Sokolova, A.S.; Yarovaya, O.I.; Shernyukov, A.V.; Gatilov, Y.V.; Razumova, Y.V.; Zarubaev, V.V.; Tretiak, T.S.; Pokrovsky, A.G.; Kiselev, O.I.; Salakhutdinov, N.F. Discovery of a new class of antiviral compounds: Camphor imine derivatives. Eur. J. Med. Chem. 2015, 105, 263–273. [Google Scholar] [CrossRef]
- Zarubaev, V.V.; Pushkina, E.A.; Borisevich, S.S.; Galochkina, A.V.; Garshinina, A.V.; Shtro, A.A.; Egorova, A.A.; Sokolova, A.S.; Khursan, S.L.; Yarovaya, O.I.; et al. Selection of influenza virus resistant to the novel camphor-based antiviral camphecene results in loss of pathogenicity. Virology 2018, 524, 69–77. [Google Scholar] [CrossRef]
- Yang, H.; Chang, J.C.; Guo, Z.; Carney, P.J.; Shore, D.A.; Donis, R.O.; Cox, N.J.; Villanueva, J.M.; Klimov, A.I.; Stevens, J. Structural Stability of Influenza A(H1N1)pdm09 Virus Hemagglutinins. J. Virol. 2014, 88, 4828–4838. [Google Scholar] [CrossRef] [Green Version]
- Volobueva, A.S.; Yarovaya, O.I.; Kireeva, M.V.; Borisevich, S.S.; Kovaleva, K.S.; Mainagashev, I.Y.; Gatilov, Y.V.; Ilyina, M.G.; Zarubaev, V.V.; Salakhutdinov, N.F. Discovery of New Ginsenol-Like Compounds with High Antiviral Activity. Molecules 2021, 26, 6794. [Google Scholar] [CrossRef] [PubMed]
- de Castro, S.; Ginex, T.; Vanderlinden, E.; Laporte, M.; Stevaert, A.; Cumella, J.; Gago, F.; Camarasa, M.J.; Luque, F.J.; Naesens, L.; et al. N-benzyl 4,4-disubstituted piperidines as a potent class of influenza H1N1 virus inhibitors showing a novel mechanism of hemagglutinin fusion peptide interaction. Eur. J. Med. Chem. 2020, 194, 112223. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, S.; Lee, G.Y.; Park, S.; Bae, J.-Y.; Heo, J.; Kim, H.-Y.; Woo, S.-H.; Lee, H.U.; Ahn, C.A.; et al. Novel Small Molecule Targeting the Hemagglutinin Stalk of Influenza Viruses. J. Virol. 2019, 93, e00878-19. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Li, R.; Zhou, Y.; Xiao, M.; Ma, C.; Yang, Z.; Zeng, S.; Du, Q.; Yang, C.; Jiang, H.; et al. Discovery of Highly Potent Pinanamine-Based Inhibitors against Amantadine- and Oseltamivir-Resistant Influenza A Viruses. J. Med. Chem. 2018, 61, 5187–5198. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef]
- Carino, A.; Moraca, F.; Fiorillo, B.; Marchianò, S.; Sepe, V.; Biagioli, M.; Finamore, C.; Bozza, S.; Francisci, D.; Distrutti, E.; et al. Hijacking SARS-CoV-2/ACE2 receptor interaction by natural and semi-synthetic steroidal agents acting on functional pockets on the receptor binding domain. Front. Chem. 2020, 8, 572885. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Roustan, C.; Borg, A.; Xu, P.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. The effect of the D614G substitution on the structure of the spike glycoprotein of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2022586118. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020, 5, 283. [Google Scholar] [CrossRef] [PubMed]
- Xiu, S.; Dick, A.; Ju, H.; Mirzaie, S.; Abdi, F.; Cocklin, S.; Zhan, P.; Liu, X. Inhibitors of SARS-CoV-2 Entry: Current and Future Opportunities. J. Med. Chem. 2020, 63, 12256–12274. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adedeji, A.O.; Severson, W.; Jonsson, C.; Singh, K.; Weiss, S.R.; Sarafianos, S.G. Novel Inhibitors of Severe Acute Respiratory Syndrome Coronavirus Entry That Act by Three Distinct Mechanisms. J. Virol. 2013, 87, 8017–8028. [Google Scholar] [CrossRef] [Green Version]
- Razizadeh, M.; Nikfar, M.; Liu, Y. Small molecule therapeutics to destabilize the ACE2-RBD complex: A molecular dynamics study. Biophys. J. 2021, 120, 2793–2804. [Google Scholar] [CrossRef]
- Cagno, V.; Magliocco, G.; Tapparel, C.; Daali, Y. The tyrosine kinase inhibitor nilotinib inhibits SARS-CoV-2 in vitro. Basic Clin. Pharmacol. Toxicol. 2021, 128, 621–624. [Google Scholar] [CrossRef]
- Banerjee, S.; Yadav, S.; Banerjee, S.; Fakayode, S.O.; Parvathareddy, J.; Reichard, W.; Surendranathan, S.; Mahmud, F.; Whatcott, R.; Thammathong, J.; et al. Drug Repurposing to Identify Nilotinib as a Potential SARS-CoV-2 Main Protease Inhibitor: Insights from a Computational and In Vitro Study. J. Chem. Inf. Model. 2021, 61, 5469–5483. [Google Scholar] [CrossRef]
- Roth, S.; Danielli, A. Rapid and sensitive inhibitor screening using magnetically modulated biosensors. Sensors 2021, 21, 4814. [Google Scholar] [CrossRef]
- Deganutti, G.; Prischi, F.; Reynolds, C.A. Supervised molecular dynamics for exploring the druggability of the SARS-CoV-2 spike protein. J. Comput. Aided Mol. Des. 2021, 35, 195–207. [Google Scholar] [CrossRef]
- Ruan, Z.; Liu, C.; Guo, Y.; He, Z.; Huang, X.; Jia, X.; Yang, T. SARS-CoV-2 and SARS-CoV: Virtual screening of potential inhibitors targeting RNA-dependent RNA polymerase activity (NSP12). J. Med. Virol. 2021, 93, 389–400. [Google Scholar] [CrossRef]
- Galimberti, S.; Petrini, M.; Baratè, C.; Ricci, F.; Balducci, S.; Grassi, S.; Guerrini, F.; Ciabatti, E.; Mechelli, S.; Di Paolo, A.; et al. Tyrosine Kinase Inhibitors Play an Antiviral Action in Patients Affected by Chronic Myeloid Leukemia: A Possible Model Supporting Their Use in the Fight Against SARS-CoV-2. Front. Oncol. 2020, 10, 1428. [Google Scholar] [CrossRef]
- Bouchlarhem, A.; Haddar, L.; Lamzouri, O.; Onci-Es-Saad; Nasri, S.; Aichouni, N.; Bkiyar, H.; Mebrouk, Y.; Skiker, I.; Housni, B. Multiple cranial nerve palsies revealing blast crisis in patient with chronic myeloid leukemia in the accelerated phase under nilotinib during severe infection with SARS-COV-19 virus: Case report and review of literature. Radiol. Case Rep. 2021, 16, 3602–3609. [Google Scholar] [CrossRef] [PubMed]
- Mediouni, S.; Mou, H.; Otsuka, Y.; Jablonski, J.A.; Adcock, R.S.; Batra, L.; Chung, D.-H.; Rood, C.; de Vera, I.M.S.; Rahaim, R., Jr.; et al. Identification of potent small molecule inhibitors of SARS-CoV-2 entry. SLAS Discov. 2022, 27, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalhor, H.; Sadeghi, S.; Abolhasani, H.; Kalhor, R.; Rahimi, H. Repurposing of the approved small molecule drugs in order to inhibit SARS-CoV-2 S protein and human ACE2 interaction through virtual screening approaches. J. Biomol. Struct. Dyn. 2022, 40, 1299–1315. [Google Scholar] [CrossRef]
- Wei, T.; Wang, H.; Wu, X.Q.; Lu, Y.; Guan, S.H.; Dong, F.Q.; Dong, C.L.; Zhu, G.L.; Bao, Y.Z.; Zhang, J.; et al. In Silico Screening of Potential Spike Glycoprotein Inhibitors of SARS-CoV-2 with Drug Repurposing Strategy. Chin. J. Integr. Med. 2020, 26, 663–669. [Google Scholar] [CrossRef]
- Aherfi, S.; Pradines, B.; Devaux, C.; Honore, S.; Colson, P.; La Scola, B.; Raoult, D. Drug repurposing against SARS-CoV-1, SARS-CoV-2 and MERS-CoV. Future Microbiol. 2021, 16, 1341–1370. [Google Scholar] [CrossRef]
- Toelzer, C.; Gupta, K.; Yadav, S.K.N.; Borucu, U.; Davidson, A.D.; Williamson, M.K.; Shoemark, D.K.; Garzoni, F.; Staufer, O.; Milligan, R.; et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science 2020, 370, 725–730. [Google Scholar] [CrossRef]
- Rosa, A.; Pye, V.E.; Graham, C.; Muir, L.; Seow, J.; Ng, K.W.; Cook, N.J.; Rees-Spear, C.; Parker, E.; dos Santos, M.S.; et al. SARS-CoV-2 can recruit a heme metabolite to evade antibody immunity. Sci. Adv. 2021, 7, eabg7607. [Google Scholar] [CrossRef]
- Fantini, J.; Di Scala, C.; Chahinian, H.; Yahi, N. Structural and molecular modelling studies reveal a new mechanism of action of chloroquine and hydroxychloroquine against SARS-CoV-2 infection. Int. J. Antimicrob. Agents 2020, 55, 105960. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Chahinian, H.; Yahi, N. Synergistic antiviral effect of hydroxychloroquine and azithromycin in combination against SARS-CoV-2: What molecular dynamics studies of virus-host interactions reveal. Int. J. Antimicrob. Agents 2020, 56, 106020. [Google Scholar] [CrossRef] [PubMed]
- Braz, H.L.B.; de Moraes Silveira, J.A.; Marinho, A.D.; de Moraes, M.E.A.; de Moraes Filho, M.O.; Monteiro, H.S.A.; Jorge, R.J.B. In silico study of azithromycin, chloroquine and hydroxychloroquine and their potential mechanisms of action against SARS-CoV-2 infection. Int. J. Antimicrob. Agents 2020, 56, 106119. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; Rolain, J.-M.; Lagier, J.-C.; Brouqui, P.; Raoult, D. Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. Int. J. Antimicrob. Agents 2020, 55, 105932. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Yan, L.; Xu, W.; Agrawal, A.S.; Algaissi, A.; Tseng, C.-T.K.; Wang, Q.; Du, L.; Tan, W.; Wilson, I.A.; et al. A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci. Adv. 2019, 5, eaav4580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Cao, R.; Zhang, H.; Liu, J.; Xu, M.; Hu, H.; Li, Y.; Zhao, L.; Li, W.; Sun, X.; et al. The anti-influenza virus drug, arbidol is an efficient inhibitor of SARS-CoV-2 in vitro. Cell Discov. 2020, 6, 28. [Google Scholar] [CrossRef]
- Cai, L.; Guo, X.; Cao, Y.; Ying, P.; Hong, L.; Zhang, Y.; Yi, G.; Fu, M. Determining available strategies for prevention and therapy: Exploring COVID-19 from the perspective of ACE2 (Review). Int. J. Mol. Med. 2021, 47, 43. [Google Scholar] [CrossRef]
- Vankadari, N. Arbidol: A potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int. J. Antimicrob. Agents 2020, 56, 105998. [Google Scholar] [CrossRef] [PubMed]
- Padhi, A.K.; Seal, A.; Khan, J.M.; Ahamed, M.; Tripathi, T. Unraveling the mechanism of arbidol binding and inhibition of SARS-CoV-2: Insights from atomistic simulations. Eur. J. Pharmacol. 2021, 894, 173836. [Google Scholar] [CrossRef]
- Borisevich, S.S.; Khamitov, E.M.; Gureev, M.A.; Yarovaya, O.I.; Rudometova, N.B.; Zybkina, A.V.; Mordvinova, E.D.; Shcherbakov, D.N.; Maksyutov, R.A.; Salakhutdinov, N.F. Simulation of Molecular Dynamics of SARS-CoV-2 S-Protein in the Presence of Multiple Arbidol Molecules: Interactions and Binding Mode Insights. Viruses 2022, 14, 119. [Google Scholar] [CrossRef]
- Huynh, T.; Wang, H.; Luan, B. In Silico Exploration of the Molecular Mechanism of Clinically Oriented Drugs for Possibly Inhibiting SARS-CoV-2’s Main Protease. J. Phys. Chem. Lett. 2020, 11, 4413–4420. [Google Scholar] [CrossRef] [PubMed]
- Ghasemlou, A.; Uskoković, V.; Sefidbakht, Y. Exploration of potential inhibitors for SARS-CoV-2 Mpro considering its mutants via structure-based drug design, molecular docking, MD simulations, MM/PBSA, and DFT calculations. Biotechnol. Appl. Biochem. 2022, 70, 439–457. [Google Scholar] [CrossRef] [PubMed]
- Musarrat, F.; Chouljenko, V.; Dahal, A.; Nabi, R.; Chouljenko, T.; Jois, S.D.; Kousoulas, K.G. The anti-HIV drug nelfinavir mesylate (Viracept) is a potent inhibitor of cell fusion caused by the SARSCoV-2 spike (S) glycoprotein warranting further evaluation as an antiviral against COVID-19 infections. J. Med. Virol. 2020, 92, 2087–2095. [Google Scholar] [CrossRef]
- Li, H.; Cheng, C.; Shi, S.; Wu, Y.; Gao, Y.; Liu, Z.; Liu, M.; Li, Z.; Huo, L.; Pan, X.; et al. Identification, optimization, and biological evaluation of 3-O-β-chacotriosyl ursolic acid derivatives as novel SARS-CoV-2 entry inhibitors by targeting the prefusion state of spike protein. Eur. J. Med. Chem. 2022, 238, 114426. [Google Scholar] [CrossRef] [PubMed]
- Yarovaya, O.I.; Shcherbakov, D.N.; Borisevich, S.S.; Sokolova, A.S.; Gureev, M.A.; Khamitov, E.M.; Rudometova, N.B.; Zybkina, A.V.; Mordvinova, E.D.; Zaykovskaya, A.V.; et al. Borneol Ester Derivatives as Entry Inhibitors of a Wide Spectrum of SARS-CoV-2 Viruses. Viruses 2022, 14, 1295. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Yarovaya, O.I.; Semenova, M.D.; Shtro, A.A.; Orshanskaya, I.R.; Zarubaev, V.V.; Salakhutdinov, N.F. Synthesis and in vitro study of novel borneol derivatives as potent inhibitors of the influenza A virus. Medchemcomm 2017, 8, 960–963. [Google Scholar] [CrossRef] [Green Version]
- Tong, L.; Wang, L.; Liao, S.; Xiao, X.; Qu, J.; Wu, C.; Zhu, Y.; Tai, W.; Huang, Y.; Wang, P.; et al. A Retinol Derivative Inhibits SARS-CoV-2 Infection by Interrupting Spike-Mediated Cellular Entry. mBio 2022, 13, e01485-22. [Google Scholar] [CrossRef]
- Xia, S.; Lan, Q.; Zhu, Y.; Wang, C.; Xu, W.; Li, Y.; Wang, L.; Jiao, F.; Zhou, J.; Hua, C.; et al. Structural and functional basis for pan-CoV fusion inhibitors against SARS-CoV-2 and its variants with preclinical evaluation. Signal Transduct. Target. Ther. 2021, 6, 288. [Google Scholar] [CrossRef]
















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borisevich, S.S.; Zarubaev, V.V.; Shcherbakov, D.N.; Yarovaya, O.I.; Salakhutdinov, N.F. Molecular Modeling of Viral Type I Fusion Proteins: Inhibitors of Influenza Virus Hemagglutinin and the Spike Protein of Coronavirus. Viruses 2023, 15, 902. https://doi.org/10.3390/v15040902
Borisevich SS, Zarubaev VV, Shcherbakov DN, Yarovaya OI, Salakhutdinov NF. Molecular Modeling of Viral Type I Fusion Proteins: Inhibitors of Influenza Virus Hemagglutinin and the Spike Protein of Coronavirus. Viruses. 2023; 15(4):902. https://doi.org/10.3390/v15040902
Chicago/Turabian StyleBorisevich, Sophia S., Vladimir V. Zarubaev, Dmitriy N. Shcherbakov, Olga I. Yarovaya, and Nariman F. Salakhutdinov. 2023. "Molecular Modeling of Viral Type I Fusion Proteins: Inhibitors of Influenza Virus Hemagglutinin and the Spike Protein of Coronavirus" Viruses 15, no. 4: 902. https://doi.org/10.3390/v15040902