
Receptor Binding-Induced Conformational Changes in Herpes Simplex Virus Glycoprotein D Permit Interaction with the gH/gL Complex to Activate Fusion
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
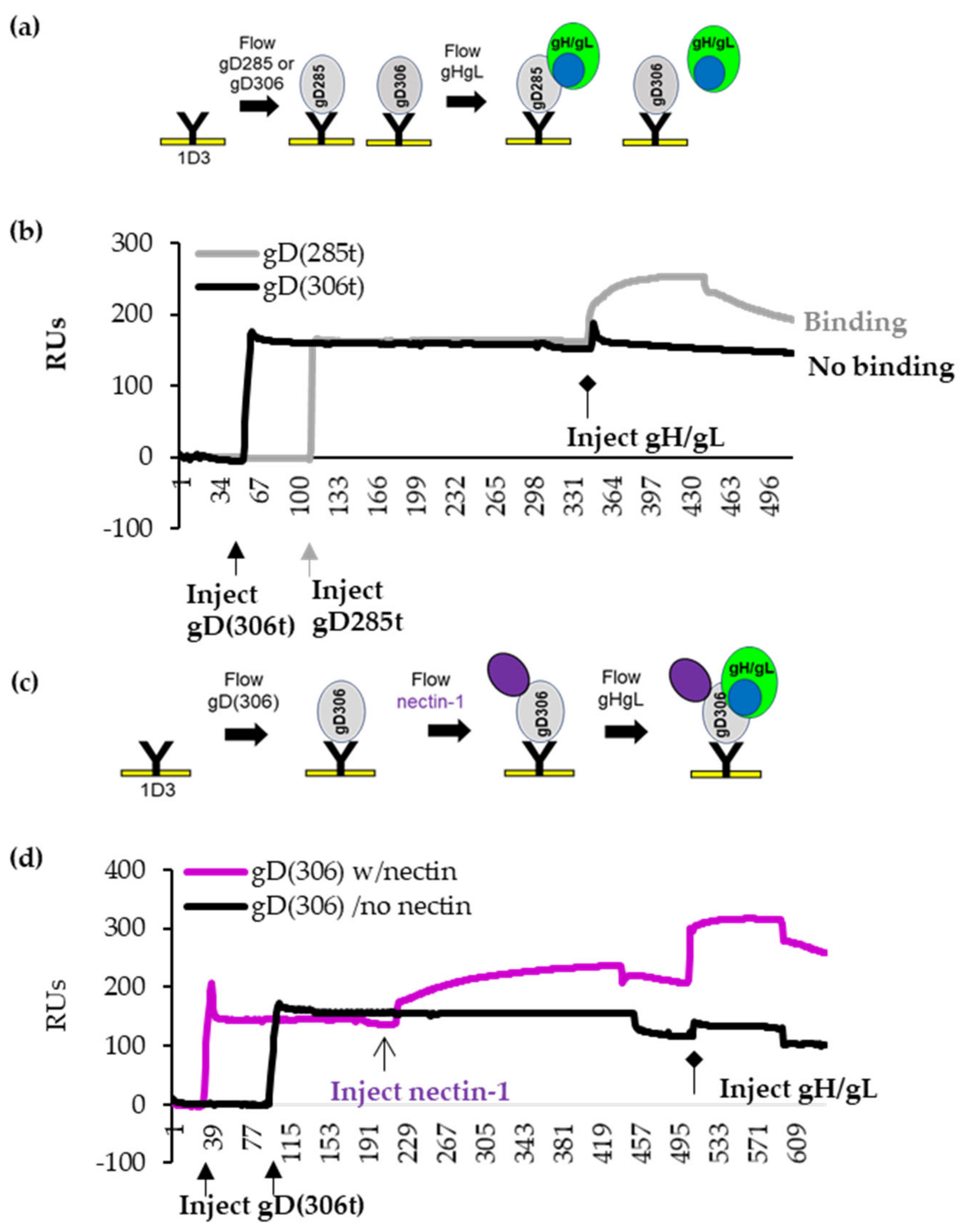
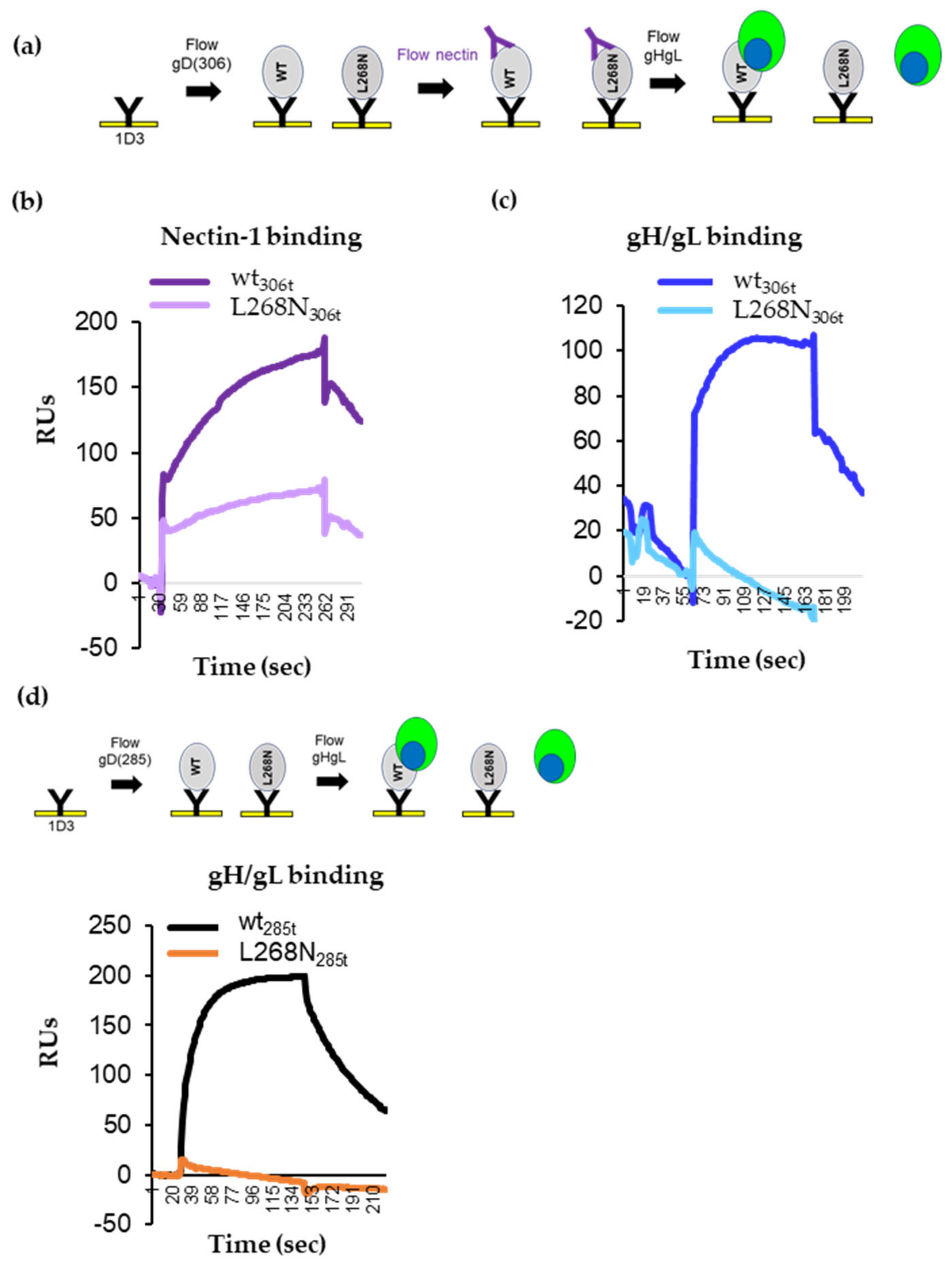
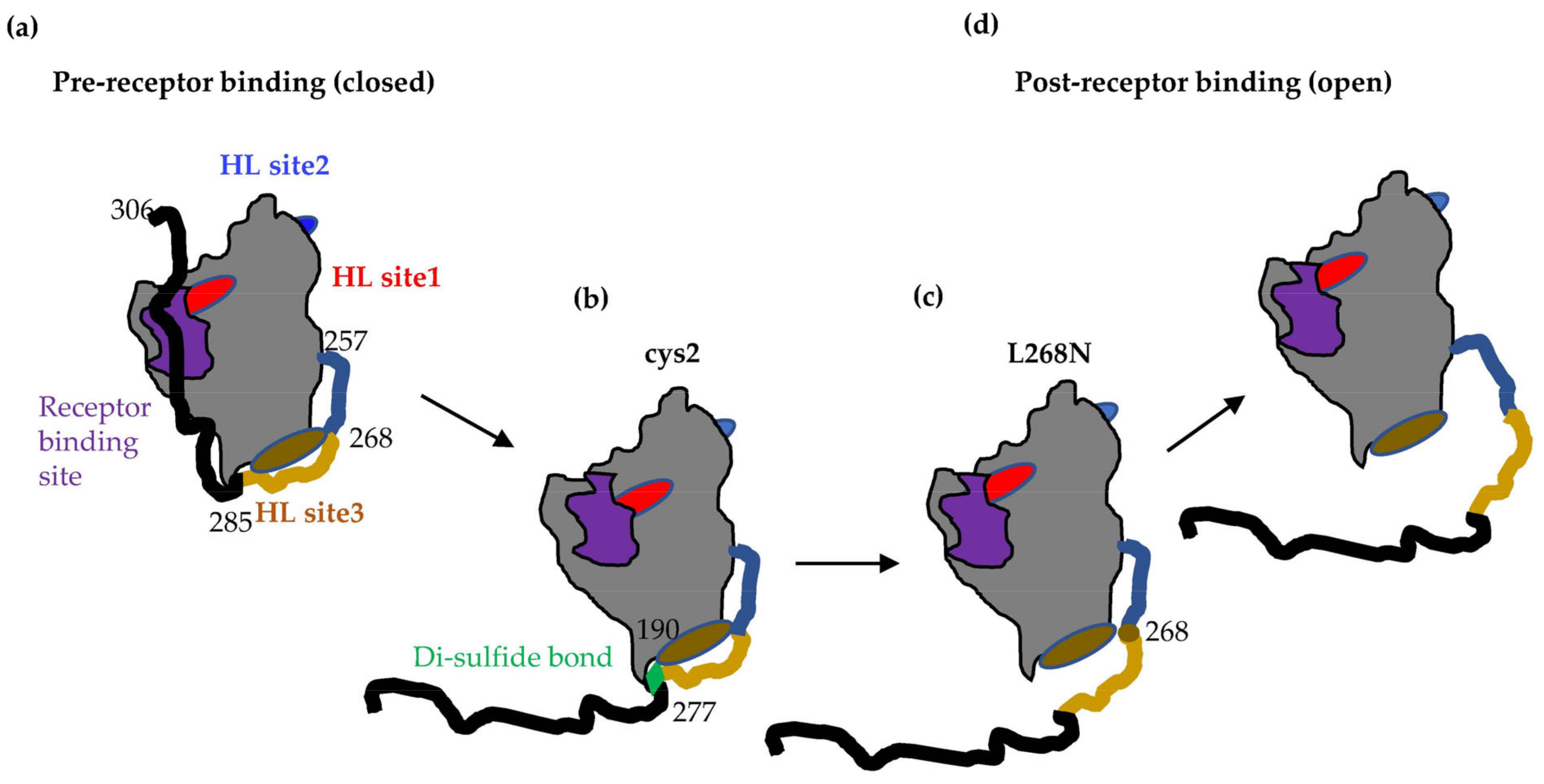
3.1. Binding of Nectin-1 Induces Conformational Changes in gD306t (Closed Conformation) That Allow for gH/gL Binding
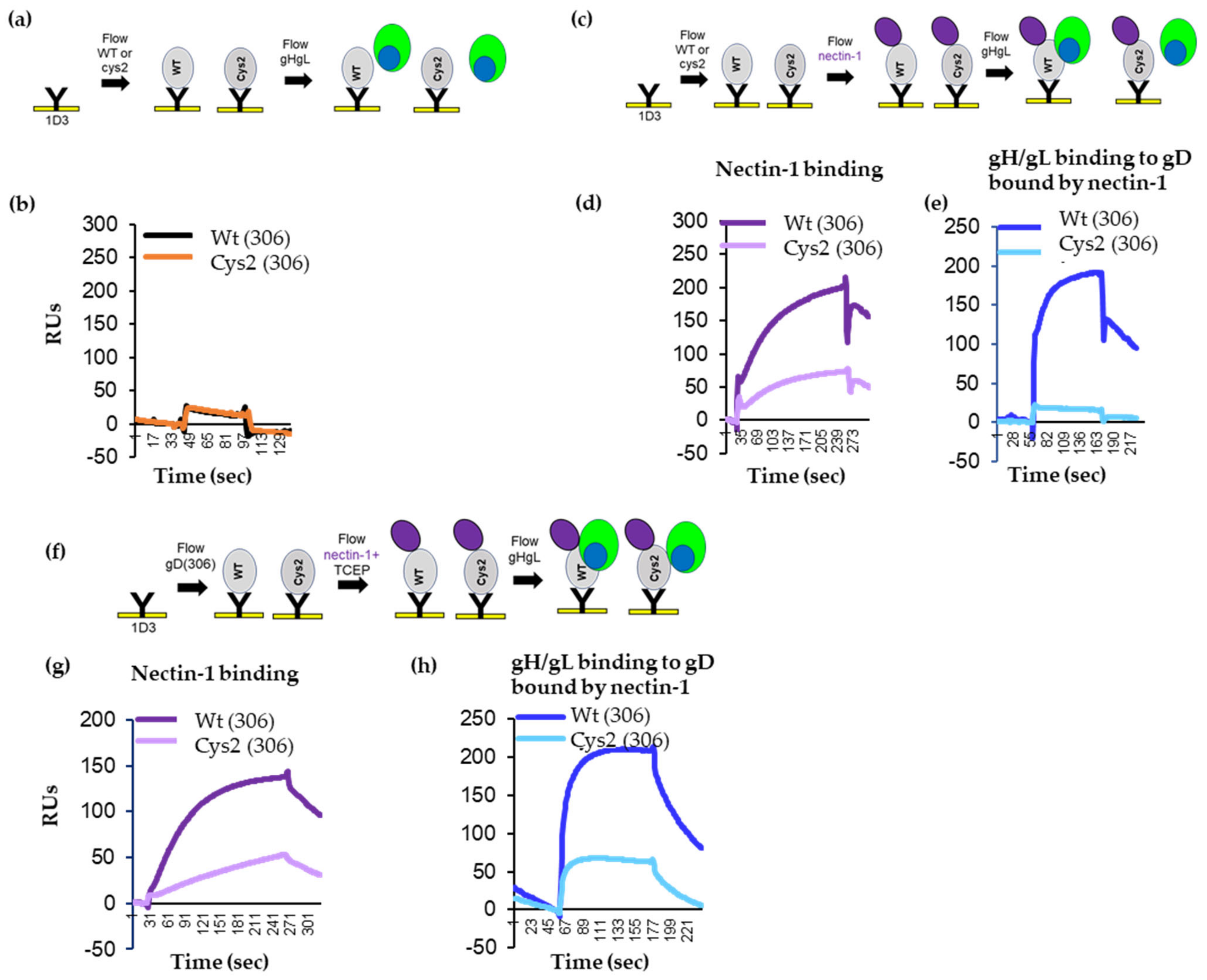
3.2. Tethering of the C-Term by a Disulfide Bond in gD Prevents Movement Required for Fusion Function
3.3. Reduction of the Disulfide Bond with TCEP Restores Fusion Function of Cys2
3.4. Locking of C-Term to the Core of gD Allows for Nectin-1 Binding but Prevents the gD–gH/gL Interaction
3.5. TCEP Treatment of Soluble Cys2 Truncated Protein Allows for the Interaction with gH/gL
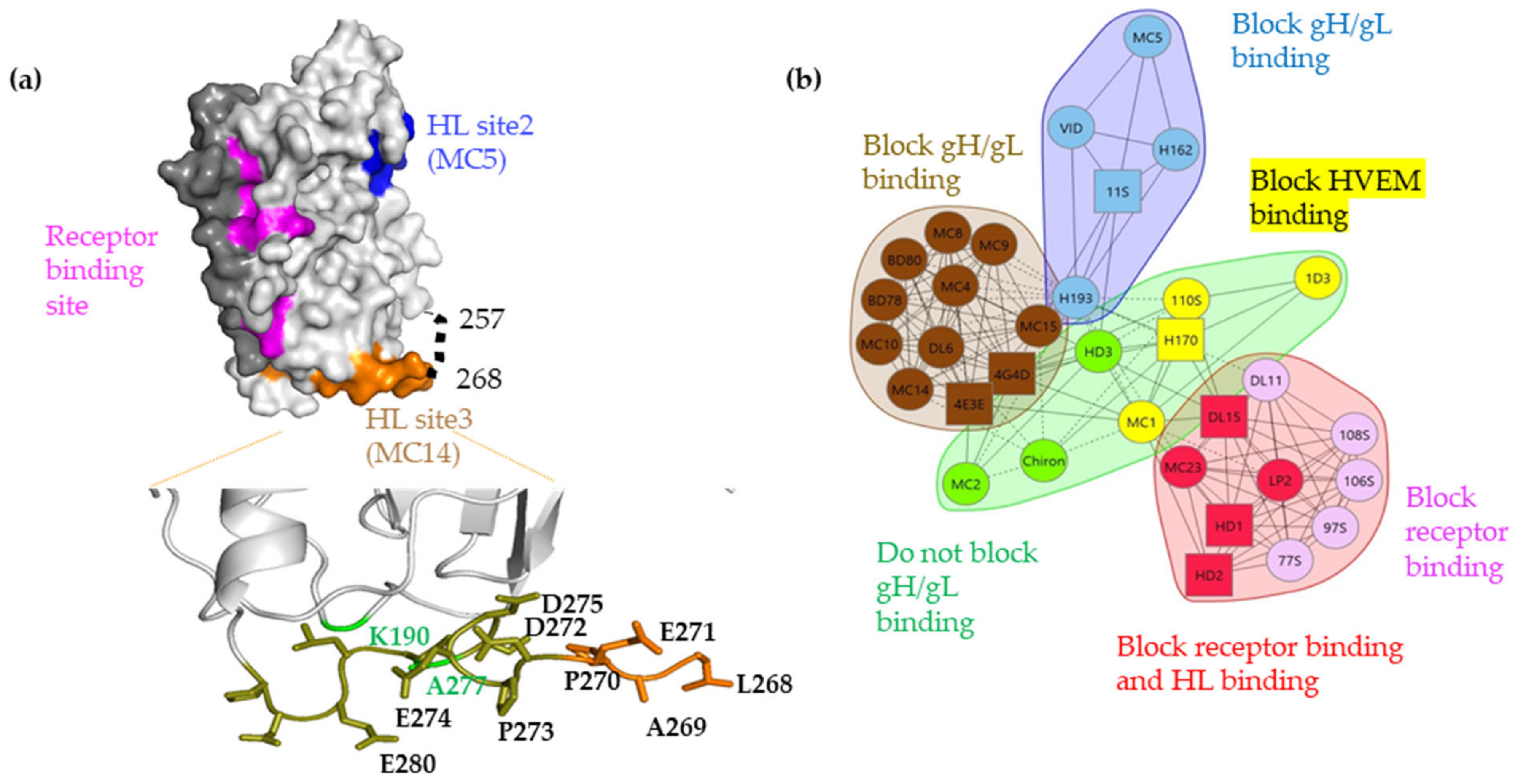
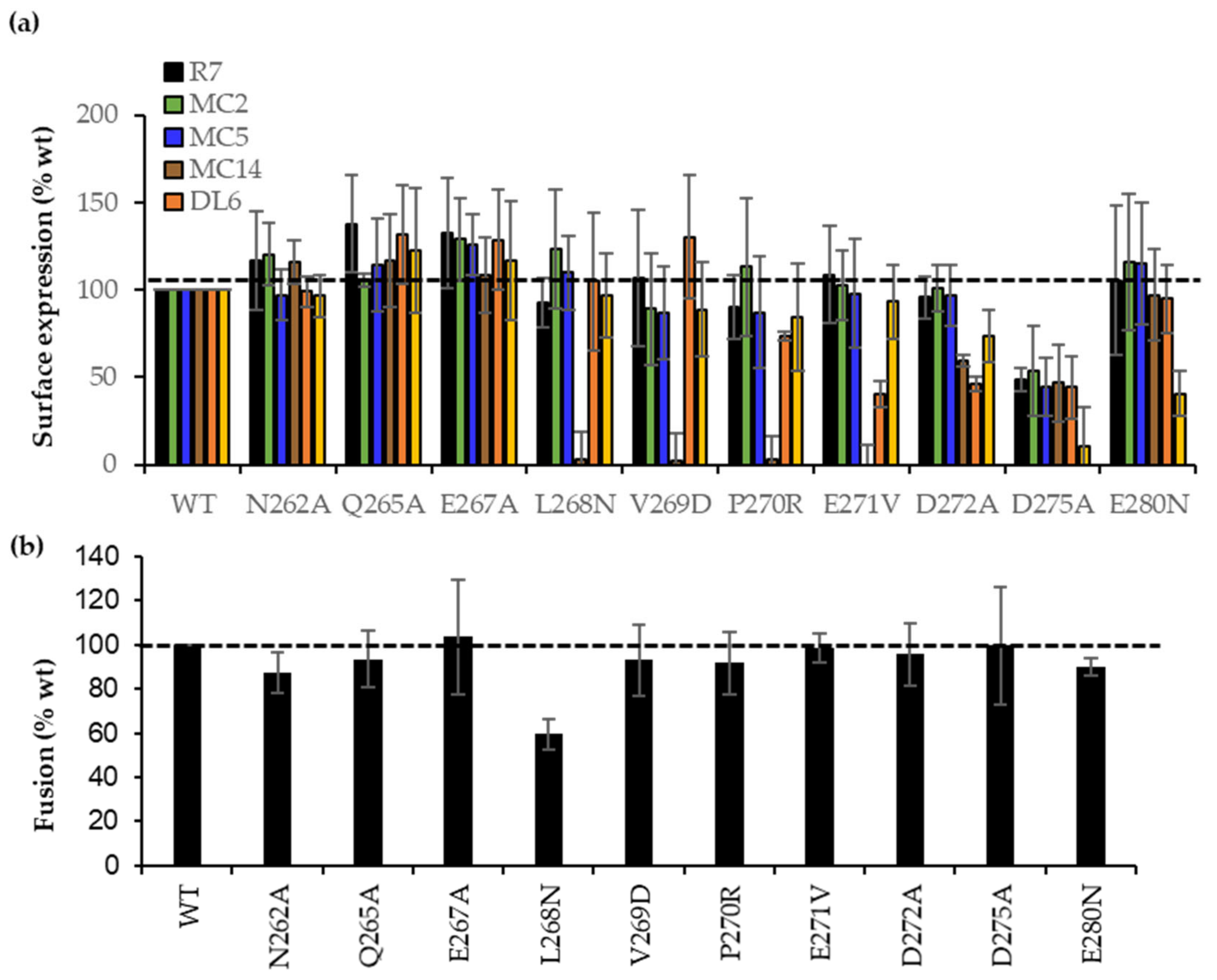
3.6. Site-Directed Mutagenesis in the 262–282 Region of gD Identifies Residues Important for gH/gL Binding
3.7. Characterization of Soluble gD L268N
3.8. gD L268N Mutant Does Not Bind gH/gL
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spear, P.G.; Eisenberg, R.J.; Cohen, G.H. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 2000, 275, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carfi, A.; Willis, S.H.; Whitbeck, J.C.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Wiley, D.C. Herpes simplex virus glyco-protein D bound to the human receptor HveA. Mol. Cell 2001, 8, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.A.; Landsburg, D.J.; Carfi, A.; Whitbeck, J.C.; Zuo, Y.; Wiley, D.C.; Cohen, G.H.; Eisenberg, R.J. Potential nectin-1 binding site on herpes simplex virus glycoprotein d. J. Virol. 2005, 79, 1282–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, M.; Zago, A.; Shukla, D.; Spear, P.G. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J. Virol. 2003, 77, 9221–9231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, T.M.; Atanasiu, D.; Saw, W.T.; Lou, H.; Whitbeck, J.C.; Ditto, N.T.; Bruun, B.; Browne, H.; Bennett, L.; Wu, C.; et al. Localization of the Interaction Site of Herpes Simplex Virus Glycoprotein D (gD) on the Membrane Fusion Regulator, gH/gL. J. Virol. 2020, 94, e00983-20. [Google Scholar] [CrossRef]
- Cairns, T.M.; Ditto, N.T.; Atanasiu, D.; Lou, H.; Brooks, B.D.; Saw, W.T.; Eisenberg, R.J.; Cohen, G.H. Surface Plasmon Resonance Reveals Direct Binding of Herpes Simplex Virus Glycoproteins gH/gL to gD and Locates a gH/gL Binding Site on gD. J. Virol. 2019, 93, e00289-19. [Google Scholar] [CrossRef] [Green Version]
- Atanasiu, D.; Saw, W.T.; Cairns, T.M.; Eisenberg, R.J.; Cohen, G.H. Using Split Luciferase Assay and anti-HSV Glycoprotein Monoclonal Antibodies to Predict a Functional Binding Site between gD and gH/gL. J. Virol. 2021, 95, e00053-21. [Google Scholar] [CrossRef]
- Krummenacher, C.; Supekar, V.M.; Whitbeck, J.C.; Lazear, E.; Connolly, S.A.; Eisenberg, R.J.; Cohen, G.H.; Wiley, D.C.; Carfí, A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 2005, 24, 4144–4153. [Google Scholar] [CrossRef]
- Cocchi, F.; Fusco, D.; Menotti, L.; Gianni, T.; Eisenberg, R.J.; Cohen, G.H.; Campadelli-Fiume, G. The soluble ectodomain of herpes simplex virus gD contains a membrane-proximal pro-fusion domain and suffices to mediate virus entry. Proc. Natl. Acad. Sci. USA 2004, 101, 7445–7450. [Google Scholar] [CrossRef] [Green Version]
- Zago, A.; Jogger, C.R.; Spear, P.G. Use of herpes simplex virus and pseudorabies virus chimeric glycoprotein D molecules to identify regions critical for membrane fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 17498–17503. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.; Longnecker, R.; Connolly, S.A. Substitution of herpes simplex virus 1 entry glycoproteins with those of saimiriine herpesvirus 1 reveals a gD-gH/gL functional interaction and a region within the gD profusion domain that is critical for fusion. J. Virol. 2014, 88, 6470–6482. [Google Scholar] [CrossRef] [Green Version]
- Cairns, T.M.; Connolly, S.A. Entry of Alphaherpesviruses. Curr. Issues Mol. Biol. 2021, 41, 63–124. [Google Scholar] [CrossRef]
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The structural basis of herpesvirus entry. Nat. Reviews. Microbiol. 2021, 19, 110–121. [Google Scholar] [CrossRef]
- Eisenberg, R.J.; Atanasiu, D.; Cairns, T.M.; Gallagher, J.R.; Krummenacher, C.; Cohen, G.H. Herpes virus fusion and entry: A story with many characters. Viruses 2012, 4, 800–832. [Google Scholar] [CrossRef]
- Lazear, E.; Carfi, A.; Whitbeck, J.C.; Cairns, T.M.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J. Engineered disulfide bonds in herpes simplex virus type 1 gD separate receptor binding from fusion initiation and viral entry. J. Virol. 2008, 82, 700–709. [Google Scholar] [CrossRef] [Green Version]
- Hook, L.M.; Cairns, T.M.; Awasthi, S.; Brooks, B.D.; Ditto, N.T.; Eisenberg, R.J.; Cohen, G.H.; Friedman, H.M. Vaccine-induced antibodies to herpes simplex virus glycoprotein D epitopes involved in virus entry and cell-to-cell spread correlate with pro-tection against genital disease in guinea pigs. PLoS Pathog. 2018, 14, e1007095. [Google Scholar] [CrossRef] [Green Version]
- Dix, R.D.; Pereira, L.; Baringer, J.R. Use of monoclonal antibody directed against herpes simplex virus glycoproteins to protect mice against acute virus-induced neurological disease. Infect. Immun. 1981, 34, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.G.; Krummenacher, C.; Eisenberg, R.J.; Cohen, G.H.; Fraser, N.W. Development of a syngenic murine B16 cell line-derived melanoma susceptible to destruction by neuroattenuated HSV-1. Mol. Ther. 2001, 3, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Pertel, P.E. Human herpesvirus 8 glycoprotein B (gB), gH, and gL can mediate cell fusion. J. Virol. 2002, 76, 4390–4400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, T.M.; Milne, R.S.; Ponce-de-Leon, M.; Tobin, D.K.; Cohen, G.H.; Eisenberg, R.J. Structure-function analysis of herpes simplex virus type 1 gD and gH-gL: Clues from gDgH chimeras. J. Virol. 2003, 77, 6731–6742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, T.M.; Friedman, L.S.; Lou, H.; Whitbeck, J.C.; Shaner, M.S.; Cohen, G.H.; Eisenberg, R.J. N-terminal mutants of herpes simplex virus type 2 gH are transported without gL but require gL for function. J. Virol. 2007, 81, 5102–5111. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Meng, F.; Kondo, N.; Iwamoto, A.; Matsuda, Z. Generation of a dual-functional split-reporter protein for moni-toring membrane fusion using self-associating split GFP. Protein Eng. Des. Sel. 2012, 25, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Krummenacher, C.; Nicola, A.V.; Whitbeck, J.C.; Lou, H.; Hou, W.; Lambris, J.D.; Geraghty, R.J.; Spear, P.G.; Cohen, G.H.; Ei-senberg, R.J. Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry medi-ator, two structurally unrelated mediators of virus entry. J. Virol. 1998, 72, 7064–7074. [Google Scholar] [CrossRef] [Green Version]
- Hannah, B.P.; Cairns, T.M.; Bender, F.C.; Whitbeck, J.C.; Lou, H.; Eisenberg, R.J.; Cohen, G.H. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J. Virol. 2009, 83, 6825–6836. [Google Scholar] [CrossRef] [Green Version]
- Sisk, W.P.; Bradley, J.D.; Leipold, R.J.; Stoltzfus, A.M.; Ponce de Leon, M.; Hilf, M.; Peng, C.; Cohen, G.H.; Eisenberg, R.J. High-level expression and purification of secreted forms of herpes simplex virus type 1 glycoprotein gD synthesized by baculovirus-infected insect cells. J. Virol. 1994, 68, 766–775. [Google Scholar] [CrossRef] [Green Version]
- Rux, A.H.; Willis, S.H.; Nicola, A.V.; Hou, W.; Peng, C.; Lou, H.; Cohen, G.H.; Eisenberg, R.J. Functional region IV of glycopro-tein D from herpes simplex virus modulates glycoprotein binding to the herpesvirus entry mediator. J. Virol. 1998, 72, 7091–7098. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, R.J.; Ponce de Leon, M.; Pereira, L.; Long, D.; Cohen, G.H. Purification of glycoprotein gD of herpes simplex virus types 1 and 2 by use of monoclonal antibody. J. Virol. 1982, 41, 1099–1104. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, R.J.; Long, D.; Ponce de Leon, M.; Matthews, J.T.; Spear, P.G.; Gibson, M.G.; Lasky, L.A.; Berman, P.; Golub, E.; Cohen, G.H. Localization of epitopes of herpes simplex virus type 1 glycoprotein D. J. Virol. 1985, 53, 634–644. [Google Scholar] [CrossRef] [Green Version]
- Lazear, E.; Whitbeck, J.C.; Ponce-de-Leon, M.; Cairns, T.M.; Willis, S.H.; Zuo, Y.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J. Antibody-induced conformational changes in herpes simplex virus glycoprotein gD reveal new targets for virus neutrali-zation. J. Virol. 2012, 86, 1563–1576. [Google Scholar] [CrossRef] [Green Version]
- Atanasiu, D.; Saw, W.T.; Gallagher, J.R.; Hannah, B.P.; Matsuda, Z.; Whitbeck, J.C.; Cohen, G.H.; Eisenberg, R.J. Dual Split Pro-tein-Based Fusion Assay Reveals that Mutations to Herpes Simplex Virus (HSV) Glycoprotein gB Alter the Kinetics of Cell-Cell Fusion Induced by HSV Entry Glycoproteins. J. Virol. 2013, 87, 11332–11345. [Google Scholar] [CrossRef] [Green Version]
- Saw, W.T.; Matsuda, Z.; Eisenberg, R.J.; Cohen, G.H.; Atanasiu, D. Using a split luciferase assay (SLA) to measure the kinetics of cell-cell fusion mediated by herpes simplex virus glycoproteins. Methods 2015, 90, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, G.H.; Isola, V.J.; Kuhns, J.; Berman, P.W.; Eisenberg, R.J. Localization of discontinuous epitopes of herpes simplex virus glycoprotein D: Use of a nondenaturing (“native” gel) system of polyacrylamide gel electrophoresis coupled with Western blot-ting. J. Virol. 1986, 60, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krummenacher, C.; Baribaud, I.; Ponce de Leon, M.; Whitbeck, J.C.; Lou, H.; Cohen, G.H.; Eisenberg, R.J. Localization of a binding site for herpes simplex virus glycoprotein D on herpesvirus entry mediator C by using antireceptor monoclonal anti-bodies. J. Virol. 2000, 74, 10863–10872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, T.M.; Ditto, N.T.; Lou, H.; Brooks, B.D.; Atanasiu, D.; Eisenberg, R.J.; Cohen, G.H. Global sensing of the antigenic struc-ture of herpes simplex virus gD using high-throughput array-based SPR imaging. PLoS Pathog. 2017, 13, e1006430. [Google Scholar] [CrossRef] [Green Version]
- Atanasiu, D.; Saw, W.T.; Eisenberg, R.J.; Cohen, G.H. Regulation of HSV glycoprotein induced cascade of events governing cell-cell fusion. J. Virol. 2016, 90, 10535–10544. [Google Scholar] [CrossRef] [Green Version]
- Navaratnarajah, C.K.; Kumar, S.; Generous, A.; Apte-Sengupta, S.; Mateo, M.; Cattaneo, R. The measles virus hemagglutinin stalk: Structures and functions of the central fusion activation and membrane-proximal segments. J. Virol. 2014, 88, 6158–6167. [Google Scholar] [CrossRef] [Green Version]
- Navaratnarajah, C.K.; Negi, S.; Braun, W.; Cattaneo, R. Membrane fusion triggering: Three modules with different structure and function in the upper half of the measles virus attachment protein stalk. J. Biol. Chem. 2012, 287, 38543–38551. [Google Scholar] [CrossRef] [Green Version]
- Cline, D.J.; Redding, S.E.; Brohawn, S.G.; Psathas, J.N.; Schneider, J.P.; Thorpe, C. New water-soluble phosphines as reductants of peptide and protein disulfide bonds: Reactivity and membrane permeability. Biochemistry 2004, 43, 15195–15203. [Google Scholar] [CrossRef]
- Long, D.; Wilcox, W.C.; Abrams, W.R.; Cohen, G.H.; Eisenberg, R.J. Disulfide bond structure of glycoprotein D of herpes simplex virus types 1 and 2. J. Virol. 1992, 66, 6668–6685. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, W.C.; Long, D.; Sodora, D.L.; Eisenberg, R.J.; Cohen, G.H. The contribution of cysteine residues to antigenicity and extent of processing of herpes simplex virus type 1 glycoprotein D. J. Virol. 1988, 62, 1941–1947. [Google Scholar] [CrossRef] [Green Version]
- Di Giovine, P.; Settembre, E.C.; Bhargava, A.K.; Luftig, M.A.; Lou, H.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C.; Carfi, A. Structure of herpes simplex virus glycoprotein D bound to the human receptor nectin-1. PLoS Pathog. 2011, 7, e1002277. [Google Scholar] [CrossRef] [Green Version]
- Isola, V.J.; Eisenberg, R.J.; Siebert, G.R.; Heilman, C.J.; Wilcox, W.C.; Cohen, G.H. Fine mapping of antigenic site II of herpes simplex virus glycoprotein D. J. Virol. 1989, 63, 2325–2334. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, R.J.; Cerini, C.P.; Heilman, C.J.; Joseph, A.D.; Dietzschold, B.; Golub, E.; Long, D.; Ponce de Leon, M.; Cohen, G.H. Synthetic glycoprotein D-related peptides protect mice against herpes simplex virus challenge. J. Virol. 1985, 56, 1014–1017. [Google Scholar] [CrossRef] [Green Version]
- Heber-Katz, E.; Dietzschold, B. Immune response to synthetic herpes simplex virus peptides: The feasibility of a synthetic vac-cine. Curr. Top. Microbiol. Immunol. 1986, 130, 51–64. [Google Scholar] [CrossRef]
- Atanasiu, D.; Saw, W.T.; Lazear, E.; Whitbeck, J.C.; Cairns, T.M.; Lou, H.; Eisenberg, R.J.; Cohen, G.H. Using Antibodies and Mutants To Localize the Presumptive gH/gL Binding Site on Herpes Simplex Virus gD. J. Virol. 2018, 92, e01694-18. [Google Scholar] [CrossRef] [Green Version]
- Chiang, H.Y.; Cohen, G.H.; Eisenberg, R.J. Identification of functional regions of herpes simplex virus glycoprotein gD by using linker-insertion mutagenesis. J. Virol. 1994, 68, 2529–2543. [Google Scholar] [CrossRef] [Green Version]
- Milne, R.S.; Hanna, S.L.; Rux, A.H.; Willis, S.H.; Cohen, G.H.; Eisenberg, R.J. Function of herpes simplex virus type 1 gD mutants with different receptor-binding affinities in virus entry and fusion. J. Virol. 2003, 77, 8962–8972. [Google Scholar] [CrossRef] [Green Version]
- Ishizaka, S.T.; Piacente, P.; Silva, J.; Mishkin, E.M. IgG subtype is correlated with efficiency of passive protection and effector function of anti-herpes simplex virus glycoprotein D monoclonal antibodies. J. Infect. Dis. 1995, 172, 1108–1111. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Ab Recognition (%wt) | Fusion Activity (%wt) | ||||
|---|---|---|---|---|---|---|
| MC2 | MC5 | MC14 | DL6 | 4E3E | ||
| N262A | 120 ± 17 | 97 ± 14 | 115 ± 14 | 100 ± 8 | 96 ± 12 | 80 ± 22 |
| Q265A | 105 ± 4 | 114 ± 26 | 116 ± 26 | 130 ± 28 | 122 ± 35 | 105 ± 12 |
| E267A | 128 ± 23 | 125 ± 17 | 108 ± 21 | 128 ± 28 | 116 ± 33 | 88 ± 8 |
| L268N | 123 ± 34 | 109 ± 21 | 3 ± 15 | 104 ± 39 | 96 ± 23 | 59 ± 9 |
| V269D | 88 ± 31 | 87 ± 26 | 2 ± 15 | 130 ± 35 | 88 ± 27 | 120 ± 29 |
| P270R | 87 ± 35 | 72 ± 30 | 2 ± 13 | 84 ± 2 | 88 ± 30 | 100 ± 18 |
| E271V | 102 ± 20 | 97 ± 31 | 1 ± 12 | 40 ± 7 | 93 ± 21 | 98 ± 8 |
| D272A | 100 ± 13 | 96 ± 17 | 60 ± 3 | 46 ± 4 | 73 ± 15 | 87 ± 19 |
| D275A | 53 ± 25 | 44 ± 16 | 46 ± 21 | 44 ± 17 | 10 ± 22 | 81 ± 18 |
| E280N | 115 ± 40 | 115 ± 45 | 96 ± 20 | 94 ± 3 | 40 ± 10 | 85 ± 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atanasiu, D.; Saw, W.T.; Cairns, T.M.; Friedman, H.M.; Eisenberg, R.J.; Cohen, G.H. Receptor Binding-Induced Conformational Changes in Herpes Simplex Virus Glycoprotein D Permit Interaction with the gH/gL Complex to Activate Fusion. Viruses 2023, 15, 895. https://doi.org/10.3390/v15040895
Atanasiu D, Saw WT, Cairns TM, Friedman HM, Eisenberg RJ, Cohen GH. Receptor Binding-Induced Conformational Changes in Herpes Simplex Virus Glycoprotein D Permit Interaction with the gH/gL Complex to Activate Fusion. Viruses. 2023; 15(4):895. https://doi.org/10.3390/v15040895
Chicago/Turabian StyleAtanasiu, Doina, Wan Ting Saw, Tina M. Cairns, Harvey M. Friedman, Roselyn J. Eisenberg, and Gary H. Cohen. 2023. "Receptor Binding-Induced Conformational Changes in Herpes Simplex Virus Glycoprotein D Permit Interaction with the gH/gL Complex to Activate Fusion" Viruses 15, no. 4: 895. https://doi.org/10.3390/v15040895