
Discovery of Potential Inhibitors of SARS-CoV-2 Main Protease by a Transfer Learning Method

Abstract
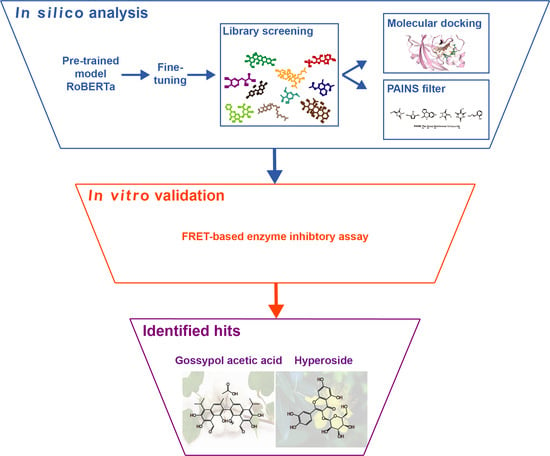
:
1. Introduction
2. Materials and Methods
2.1. Dataset Preparation
2.2. Chemical Space Analysis
2.3. Model Performance Evaluation
2.4. Compound Libraries and Compounds
2.5. PAINS Filtering
2.6. Molecular Docking Protocol
2.7. Protein Expression and Purification of SARS-CoV-2 Mpro
2.8. FRET-Based Mpro Enzyme Activity Inhibition Assay
3. Results
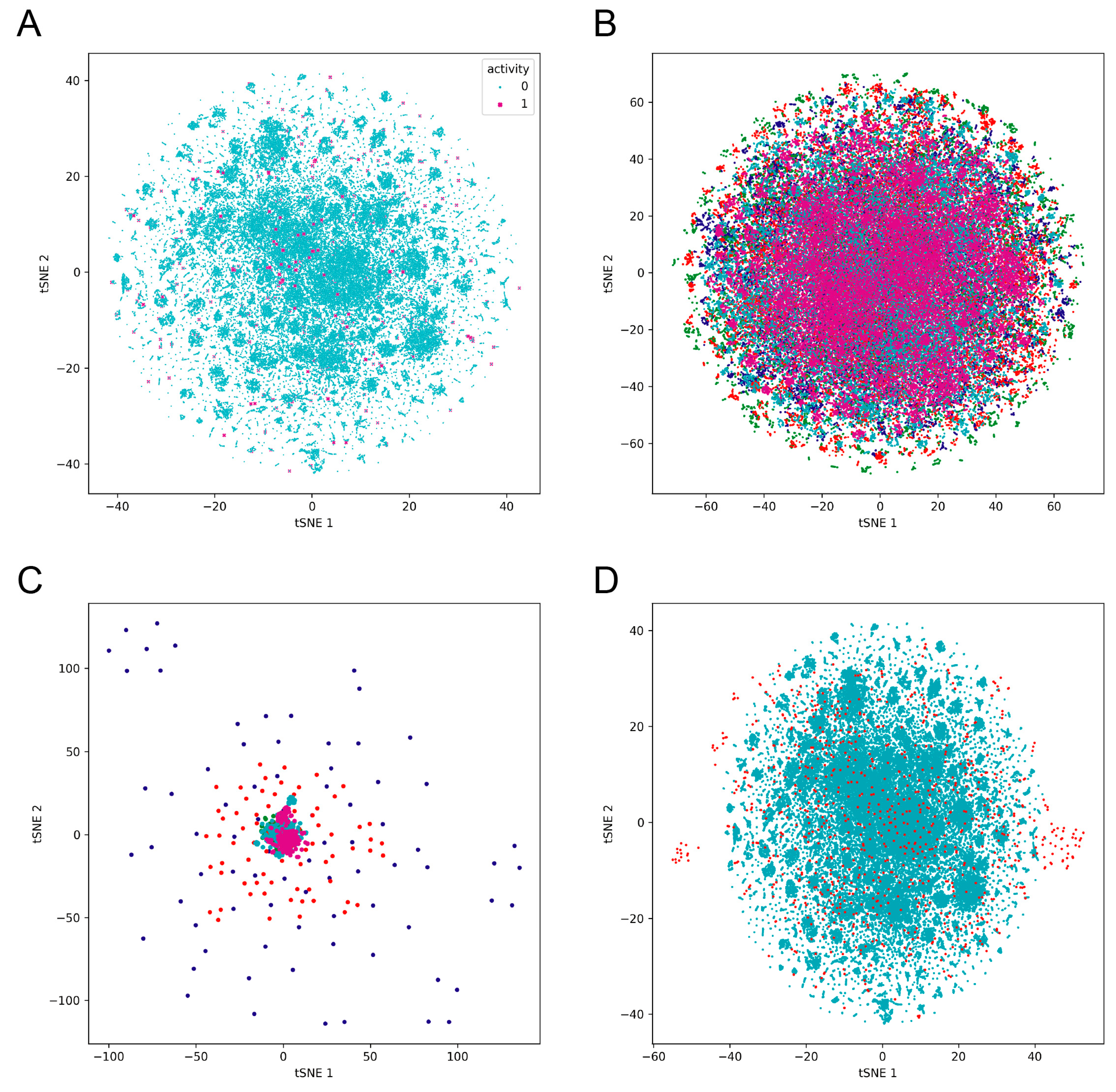
3.1. Dataset Preprocessing and Chemical Space Analysis
3.2. Performance of the Fine-Tuned Model
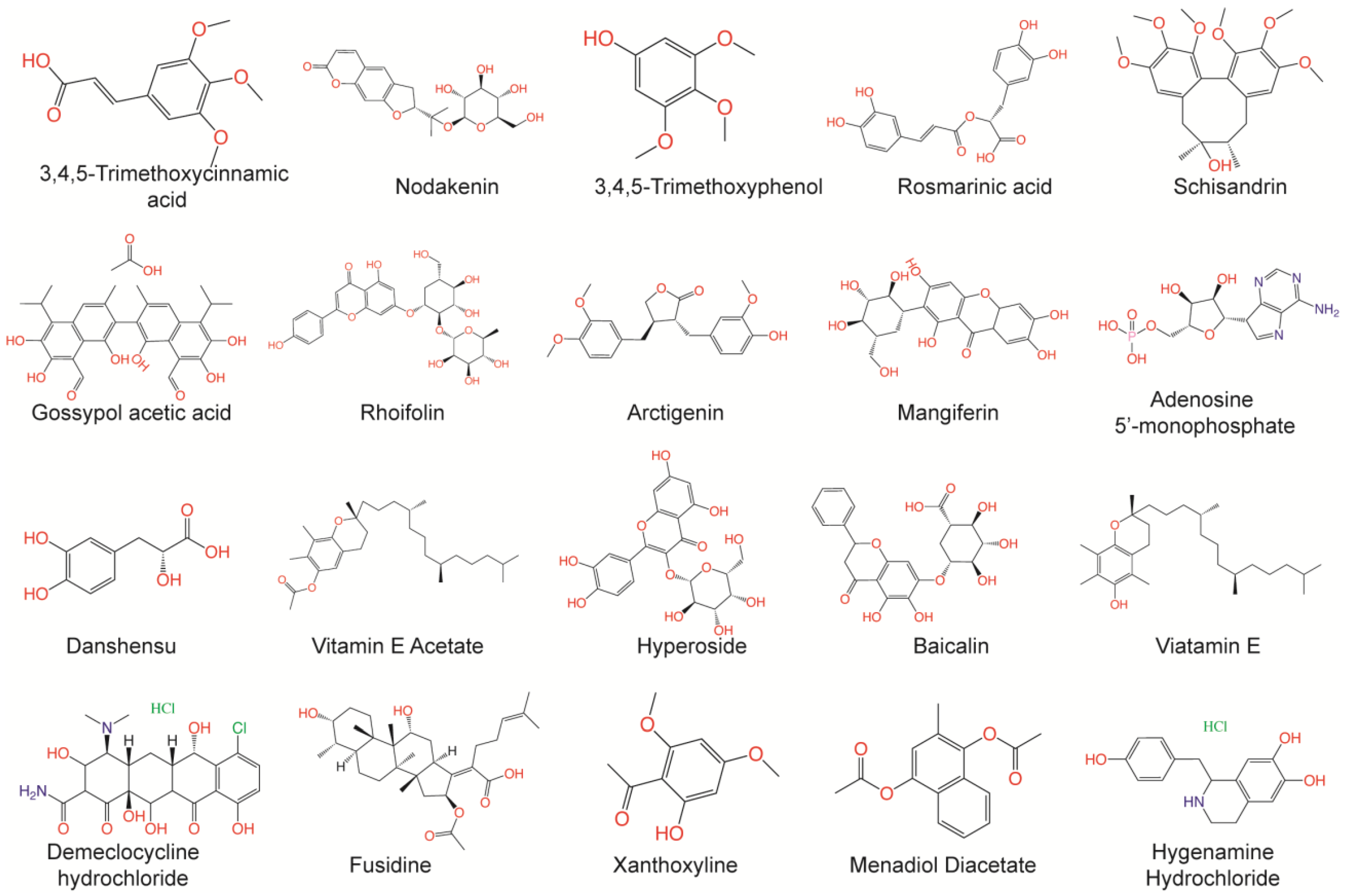
3.3. Prediction of Bioactivities of Natural Compound and De Novo Generated Molecule Libraries
3.4. Molecular Docking Screening
3.5. PAINS Filtering
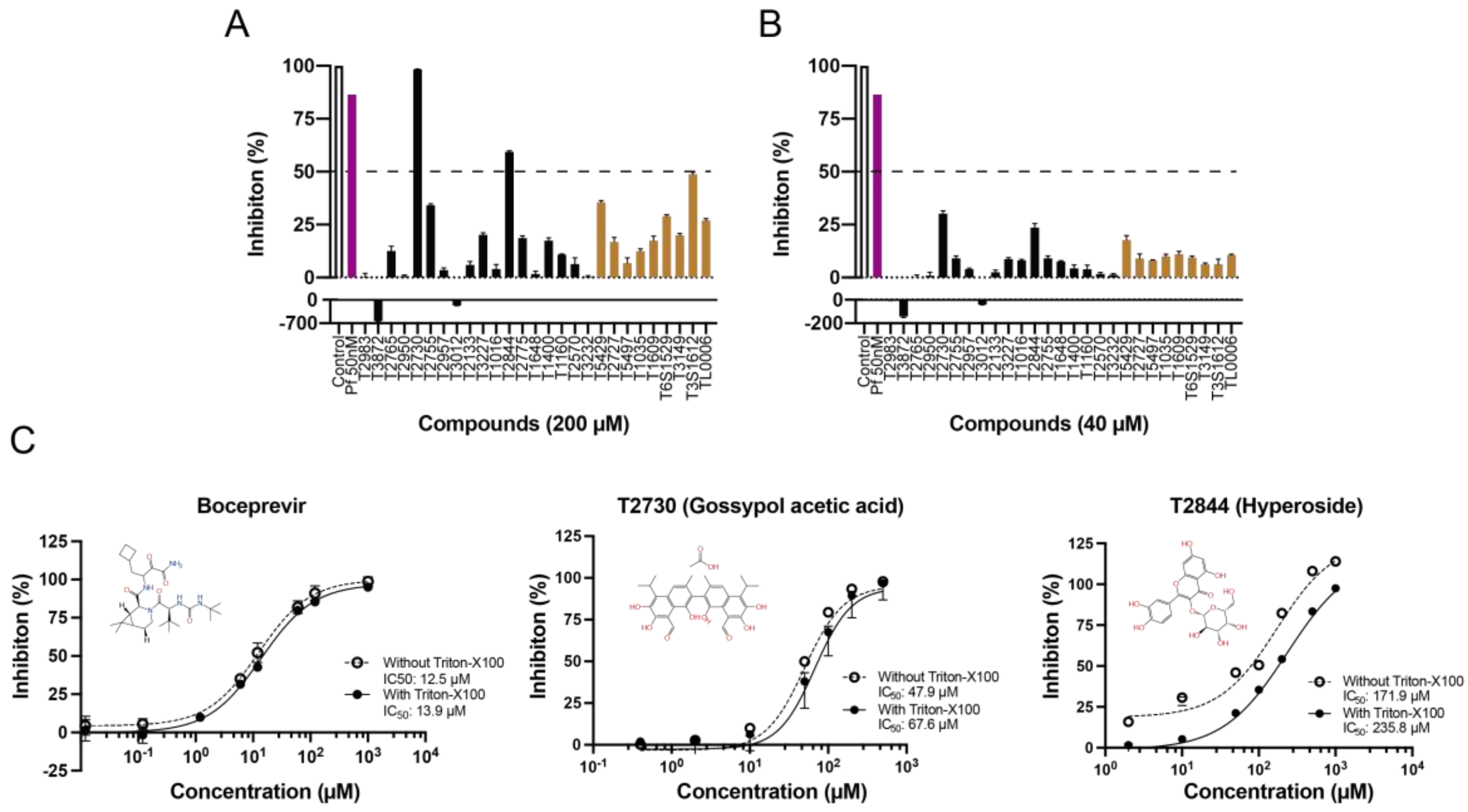
3.6. In Vitro Binding Assay Validation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Worrall, L.J.; Vuckovic, M.; Rosell, F.I.; Gentile, F.; Ton, A.T.; Caveney, N.A.; Ban, F.; Cherkasov, A.; Paetzel, M.; et al. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat. Commun. 2020, 11, 5877. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Dai, W.A.-O.; Zhang, B.A.-O.; Jiang, X.M.; Su, H.A.-O.; Li, J.; Zhao, Y.A.-O.; Xie, X.; Jin, Z.A.-O.X.; Peng, J.; Liu, F.A.-O.X.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar] [CrossRef]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef]
- Muramatsu, T.; Kim, Y.T.; Nishii, W.; Terada, T.; Shirouzu, M.; Yokoyama, S. Autoprocessing mechanism of severe acute respiratory syndrome coronavirus 3C-like protease (SARS-CoV 3CLpro) from its polyproteins. FEBS J. 2013, 280, 2002–2013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 6489. [Google Scholar] [CrossRef] [PubMed]
- Günther, S.A.-O.; Reinke, P.A.-O.; Fernández-García, Y.A.-O.; Lieske, J.A.-O.; Lane, T.A.-O.; Ginn, H.A.-O.; Koua, F.A.-O.; Ehrt, C.A.-O.; Ewert, W.A.-O.; Oberthuer, D.A.-O.; et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 2021, 372, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Shiravi, A.A.; Ardekani, A.; Sheikhbahaei, E.; Heshmat-Ghahdarijani, K. Cardiovascular Complications of SARS-CoV-2 Vaccines: An Overview. Cardiol. Ther. 2022, 11, 13–21. [Google Scholar] [CrossRef]
- Venkadapathi, J.; Govindarajan, V.K.; Sekaran, S.; Venkatapathy, S. A Minireview of the Promising Drugs and Vaccines in Pipeline for the Treatment of COVID-19 and Current Update on Clinical Trials. Front. Mol. Biosci. 2021, 8, 637378. [Google Scholar] [CrossRef]
- Amoutzias, G.D.; Nikolaidis, M.; Tryfonopoulou, E.; Chlichlia, K.; Markoulatos, P.; Oliver, S.G. The Remarkable Evolutionary Plasticity of Coronaviruses by Mutation and Recombination: Insights for the COVID-19 Pandemic and the Future Evolutionary Paths of SARS-CoV-2. Viruses 2022, 14, 78. [Google Scholar] [CrossRef]
- Malik, J.A.; Ahmed, S.; Mir, A.; Shinde, M.; Bender, O.; Alshammari, F.; Ansari, M.; Anwar, S. The SARS-CoV-2 mutations versus vaccine effectiveness: New opportunities to new challenges. J. Infect. Public Health 2022, 15, 228–240. [Google Scholar] [CrossRef]
- Jang, W.D.; Jeon, S.; Kim, S.; Lee, S.Y. Drugs repurposed for COVID-19 by virtual screening of 6218 drugs and cell-based assay. Proc. Natl. Acad. Sci. USA 2021, 118, e2024302118. [Google Scholar] [CrossRef]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Kumar, Y.; Singh, H.; Patel, C.N. In silico prediction of potential inhibitors for the Main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J. Infect. Public Health 2020, 13, 1210–1223. [Google Scholar] [CrossRef]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.-D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structure basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remedesvir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, K.; Singh, M.K. Drug repurposing in COVID-19: A review with past, present and future. Metab. Open 2021, 12, 100121. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.; Mfone, F.; Shallcross, M.; Pathak, V. Review of Pharmacotherapy Trialed for Management of the Coronavirus Disease-19. Eurasian J. Med. 2021, 53, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.M.; Delaugerre, C.; Le Goff, J.; Mela-Lima, B.; Ponscarme, D.; Goldwirt, L.; de Castro, N. No evidence of rapid antiviral clearance or clinical benefit with the combination of hydroxychloroquine and azithromycin in patients with severe COVID-19 infection. Med. Mal. Infect. 2020, 50, 384. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H. Big Data and Artificial Intelligence Modeling for Drug Discovery. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 573–589. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, Y.; Li, J.; Wang, M.; Saravanan, K.M.; Wei, J.; Tze-Yang Ng, J.; Tofazzal Hossain, M.; Liu, M.; Zhang, H.; et al. A novel virtual screening procedure identifies Pralatrexate as inhibitor of SARS-CoV-2 RdRp and it reduces viral replication in vitro. PLoS Comput. Biol. 2020, 16, e1008489. [Google Scholar] [CrossRef]
- Santana, M.V.S.; Silva-Jr, F.P. De novo design and bioactivity prediction of SARS-CoV-2 main protease inhibitors using recurrent neural network-based transfer learning. BMC Chem. 2021, 15, 8. [Google Scholar] [CrossRef]
- Zhang, H.; Saravanan, K.M.; Yang, Y.; Hossain, M.T.; Li, J.; Ren, X.; Pan, Y.; Wei, Y. Deep Learning Based Drug Screening for Novel Coronavirus 2019-nCov. Interdiscip. Sci. 2020, 12, 368–376. [Google Scholar] [CrossRef]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, 2000028. [Google Scholar] [CrossRef] [PubMed]
- Tahir Ul Qamar, M.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.L. Structural basis of SARS-CoV-2 3CL(pro) and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Nand, M.; Maiti, P.; Joshi, T.; Chandra, S.; Pande, V.; Kuniyal, J.C.; Ramakrishnan, M.A. Virtual screening of anti-HIV1 compounds against SARS-CoV-2: Machine learning modeling, chemoinformatics and molecular dynamics simulation based analysis. Sci. Rep. 2020, 10, 20397. [Google Scholar] [CrossRef] [PubMed]
- Joshi, T.; Joshi, T.; Pundir, H.; Sharma, P.; Mathpal, S.; Chandra, S. Predictive modeling by deep learning, virtual screening and molecular dynamics study of natural compounds against SARS-CoV-2 main protease. J. Biomol. Struct. Dyn. 2021, 39, 6728–6746. [Google Scholar] [CrossRef]
- Beck, B.R.; Shin, B.; Choi, Y.; Park, S.; Kang, K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput. Struct. Biotechnol. J. 2020, 18, 784–790. [Google Scholar] [CrossRef]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. BERT: Pre-training of deep bidirectional transformers for language understanding. In Proceedings of the NAACL-HLT, Minneapolis, MN, USA, 2–7 June 2019; pp. 4171–4186. [Google Scholar]
- Liu, Y.; Ott, M.; Goyal, N.; Du, J.; Joshi, M.; Chen, D.; Levy, O.; Lewis, M.; Zettlemoyer, L.; Stoyanov, V. RoBERTa: A Robustly Optimized BERT Pretraining Approach. arXiv 2019, arXiv:1907.11692. [Google Scholar]
- Rogers, A.; Kovaleva, O.; Rumshisky, A. A Primer in BERTology: What We Know About How BERT Works. Trans. Assoc. Comput. Linguist. 2020, 8, 842–866. [Google Scholar] [CrossRef]
- Chithrananda, S.; Grand, G.; Bharath, R. ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction. arXiv 2020, arXiv:2010.09885. [Google Scholar]
- Wolf, T.; Debut, L.; Sanh, V.; Chaumond, J.; Delangue, C.; Moi, A.; Cistac, P.; Rault, T.; Louf, R.; Funtowicz, M.; et al. Transformers: State-of-the-Art Natural Language Processing. In Proceedings of the 2020 EMNLP, Online, 16–20 November 2020; pp. 38–45. [Google Scholar]
- Raffel, C.; Shazeer, N.; Roberts, A.; Lee, K.; Narang, S.; Matena, M.; Zhou, Y.; Li, W.; Liu, P.J. Exploring the limits of transfer learning with a unified text-to-text transformer. J. Mach. Learn. Res. 2020, 21, 5485–5551. [Google Scholar]
- Wang, S.; Guo, Y.; Wang, Y.; Sun, H.; Huang, J. Smiles-Bert: Large Scale Unsupervised Pre-Training for Molecular Property Prediction. In Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Niagara Falls, NY, USA, 7–10 September 2019; pp. 429–436. [Google Scholar]
- Honda, S.; Shi, S.; Ueda, H.R. SMILES transformer: Pre-trained molecular fingerprint for low data drug discovery. arXiv 2019, arXiv:1911.04738. [Google Scholar]
- Schwaller, P.; Laino, T.; Gaudin, T.; Bolgar, P.; Hunter, C.A.; Bekas, C.; Lee, A.A. Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction. ACS Cent. Sci. 2019, 5, 1572–1583. [Google Scholar] [CrossRef] [PubMed]
- Maziarka, Ł.D.; Tomasz Mucha, S.; Rataj, K.; Tabor, J.; Jastrzębski, S. Molecule attention transformer. arXiv 2020, arXiv:2002.08264. [Google Scholar]
- Bjerrum, E.J. SMILES Enumeration as Data Augmentation for Neural Network Modeling of Molecules. arxiv 2017, arXiv:1703.07076. [Google Scholar]
- Douangamath, A.; Fearon, D.; Gehrtz, P.; Krojer, T.; Lukacik, P.; Owen, C.D.; Resnick, E.; Strain-Damerell, C.; Aimon, A.; Abranyi-Balogh, P.; et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020, 11, 5047. [Google Scholar] [CrossRef]
- Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M.; et al. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388. [Google Scholar] [CrossRef]
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: Free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics 2017, 33, 3658–3660. [Google Scholar] [CrossRef]
- Tang, B.; He, F.; Liu, D.; Fang, M.; Wu, Z.; Xu, D. AI-aided design of novel targeted covalent inhibitors against SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Mok, N.Y.; Maxe, S.; Brenk, R. Locating sweet spots for screening hits and evaluating pan-assay interference filters from the performance analysis of two lead-like libraries. J. Chem. Inf. Model. 2013, 53, 534–544. [Google Scholar] [CrossRef]
- Feng, B.Y.; Shoichet, B.K. A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006, 1, 550–553. [Google Scholar] [CrossRef]
- O’Donnell, H.R.; Tummino, T.A.; Bardine, C.; Craik, C.S.; Shoichet, B.K. Colloidal Aggregators in Biochemical SARS-CoV-2 Repurposing Screens. J. Med. Chem. 2021, 64, 17530–17539. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cheng, W.; Hua, B.; Wang, S.; Yang, D. Effects of Gossypol Acetate on Proliferation and Apoptosis in Lymphoblastoid Cell Line and Primary ALL and CLL Cells. Blood 2005, 106, 4405. [Google Scholar] [CrossRef]
- Ferenczyova, K.; Kalocayova, B.A.-O.; Bartekova, M.A.-O. Potential Implications of Quercetin and its Derivatives in Cardioprotection. Int. J. Mol. Sci. 2020, 21, 1585. [Google Scholar] [CrossRef] [PubMed]
- Khaerunnisa, S.; Kurniawan, H.; Awaluddin, R.; Suhartati, S.; Soetjipto, S. Potential inhibitor of COVID-19 main protease (Mpro) from several medicinal plant compounds by molecular docking study. Preprints 2020, 2020030226. [Google Scholar] [CrossRef]
- Chaves, O.A.; Fintelman-Rodrigues, N.; Wang, X.; Sacramento, C.Q.; Temerozo, J.R.; Ferreira, A.C.; Mattos, M.; Pereira-Dutra, F.; Bozza, P.T.; Castro-Faria-Neto, H.C.; et al. Commercially Available Flavonols Are Better SARS-CoV-2 Inhibitors than Isoflavone and Flavones. Viruses 2022, 14, 1458. [Google Scholar] [CrossRef]
- Chaves, O.A.; Lima, C.R.; Fintelman-Rodrigues, N.; Sacramento, C.Q.; de Freitas, C.S.; Vazquez, L.; Temerozo, J.R.; Rocha, M.E.N.; Dias, S.S.G.; Carels, N.; et al. Agathisflavone, a natural biflavonoid that inhibits SARS-CoV-2 replication by targeting its proteases. Int. J. Biol. Macromol. 2022, 222, 1015–1026. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fold 1 | Fold 2 | Fold 3 | Fold 4 | Fold 5 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Label | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 |
| Original | 142 | 57,771 | 75 | 57,838 | 168 | 57,754 | 164 | 57,749 | 80 | 57,833 |
| Augmentation_10 | 1420 | 57,771 | 750 | 57,838 | 1680 | 57,754 | 1640 | 57,749 | 800 | 57,833 |
| Augmentation_20 | 2840 | 57,771 | 1500 | 57,838 | 3260 | 57,754 | 3280 | 57,749 | 1600 | 57,833 |
| Augmentation_80 | 11,360 | 57,771 | 6000 | 57,838 | 13,440 | 57,754 | 13,120 | 57,749 | 6400 | 57,833 |
| Model | Transfer Learning | GCNN | Random Forest | |||||
|---|---|---|---|---|---|---|---|---|
| Dataset | Original | 10 | 20 | 80 | 20 | 80 | 20 | 80 |
| mcc | 0.06931 | 0.88577 | 0.77580 | 0.97618 | 0.17995 | 0.26405 | 0.47148 | 0.46608 |
| tp | 1.4 | 1021.4 | 2294.6 | 9712 | 192 | 2160 | 948 | 3792 |
| tn | 57787.4 | 57757.8 | 57761.6 | 57755 | 57489.2 | 54236.6 | 57784.2 | 57783.8 |
| fp | 0 | 29.8 | 26 | 32.6 | 298.4 | 3551 | 3.4 | 3.8 |
| fn | 124.4 | 236.6 | 219.6 | 352 | 232.4 | 7904 | 1568 | 6272 |
| auroc | 0.52137 | 0.96239 | 0.98226 | 0.99226 | 0.76543 | 0.76389 | 0.77730 | 0.77897 |
| auprc | 0.03054 | 0.88366 | 0.95221 | 0.98621 | 0.3119 | 0.50636 | 0.52265 | 0.65047 |
| recall | 0.01532 | 0.80871 | 0.90753 | 0.96350 | 0.08885 | 0.19786 | 0.31199 | 0.31759 |
| accuracy | 0.99785 | 0.99550 | 0.98794 | 0.99436 | 0.95663 | 0.83353 | 0.97394 | 0.90750 |
| precision | 0.36667 | 0.97514 | 0.98818 | 0.99568 | 0.59492 | 0.84163 | 0.97671 | 0.99222 |
| f1 | 0.02881 | 0.88391 | 0.94605 | 0.97931 | 0.11763 | 0.20417 | 0.38709 | 0.39592 |
| Model | Transfer Learning | GCNN | Random Forest | |||||
|---|---|---|---|---|---|---|---|---|
| Dataset | Original | 10 | 20 | 80 | 20 | 80 | 20 | 80 |
| mcc | 0 | 0.30798 | 0.30973 | 0.26022 | 0.05526 | 0.08691 | 0.08652 | 0.08652 |
| tp | 0 | 16.6 | 22.6 | 26.2 | 4.8 | 11.8 | 0.8 | 0.8 |
| tn | 802 | 789.8 | 774 | 746.2 | 787.8 | 754.2 | 802 | 802 |
| fp | 0 | 12.2 | 28 | 55.8 | 14.2 | 47.8 | 0 | 0 |
| fn | 78 | 61.4 | 55.4 | 51.8 | 73.2 | 66.2 | 77.2 | 77.2 |
| auroc | 0.50025 | 0.66905 | 0.67788 | 0.68109 | 0.66972 | 0.68249 | 0.68298 | 0.71836 |
| auprc | 0.08868 | 0.28671 | 0.28427 | 0.23152 | 0.20616 | 0.23784 | 0.35956 | 0.40239 |
| recall | 0 | 0.21282 | 0.28974 | 0.39990 | 0.06154 | 0.15128 | 0.01026 | 0.01026 |
| accuracy | 0.72909 | 0.91636 | 0.90523 | 0.87773 | 0.90068 | 0.87045 | 0.91227 | 0.91227 |
| precision | 0 | 0.58623 | 0.44416 | 0.31989 | 0.13992 | 0.23694 | 0.8 | 0.8 |
| f1 | 0 | 0.29778 | 0.34973 | 0.32647 | 0.07880 | 0.10446 | 0.02025 | 0.02025 |
| Model | Input | Mcc | Auroc | Auprc | Recall | Accuracy | Precision | f1 |
|---|---|---|---|---|---|---|---|---|
| Transfer Learning | Active 20 | 0.37804 | 0.68186 | 0.34433 | 0.34359 | 0.91341 | 0.51978 | 0.41321 |
| Active 80 | 0.29978 | 0.68306 | 0.26118 | 0.34359 | 0.89091 | 0.37632 | 0.35833 | |
| Chemprop | original | 0.17636 | 0.68152 | 0.19321 | 0.12821 | 0.90341 | 0.37037 | 0.19048 |
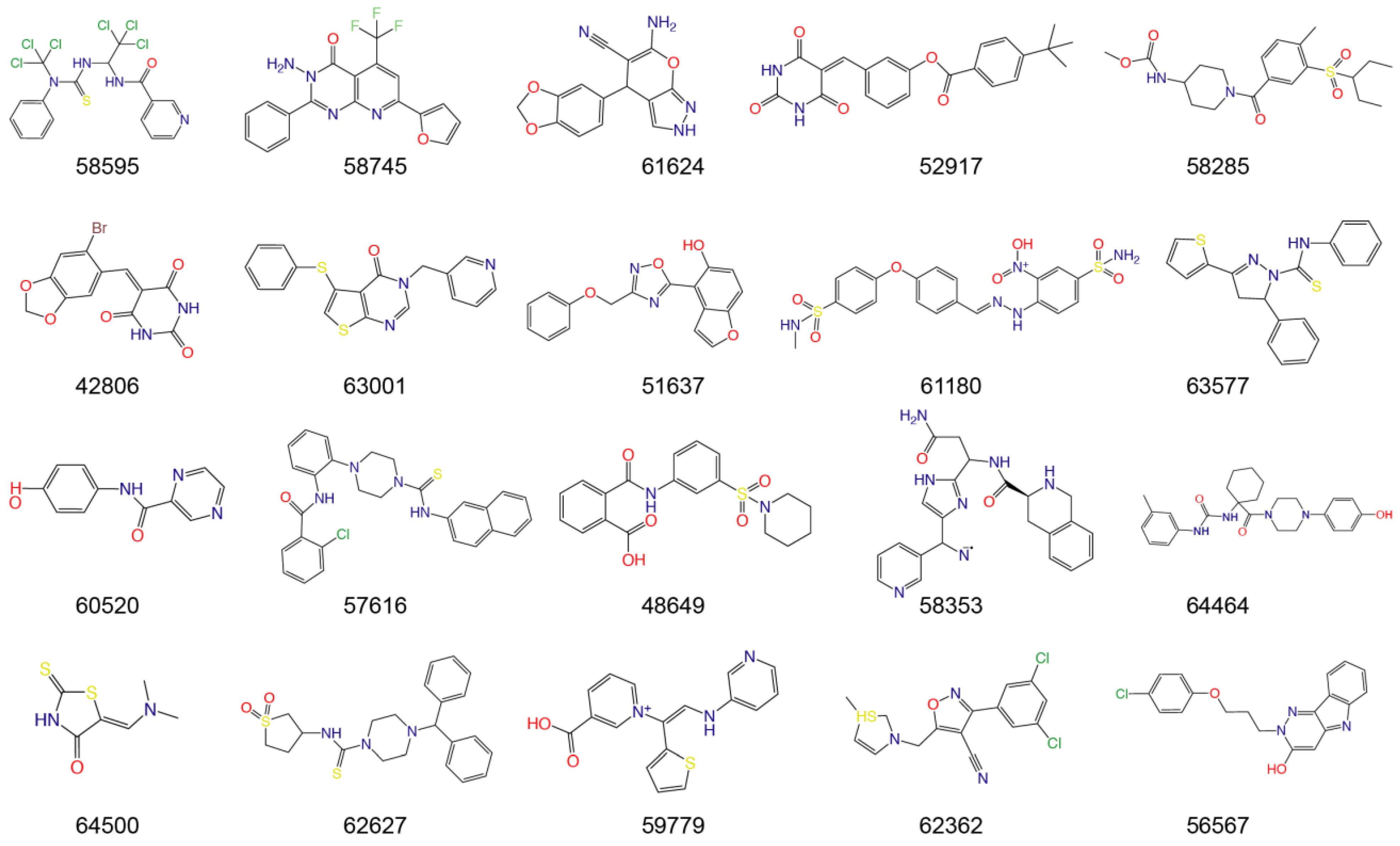
| IDs | Name | Source | Structure | Docking Score (kcal/mol) | Mpro Residues Interacting with Molecules through H-Bond and Other Types |
|---|---|---|---|---|---|
| PF-07321332 | - | - |  | −9.2 |  |
| T5429 | Theaflavin 3,3′-digallate | Black tea |  | −10.4 |  |
| T2727 | Salvianolic acid B | Slvia miltiorrhiza |  | −9.2 |  |
| T5497 | AMAROGENTIN | Gentiana scabra |  | −8.9 |  |
| T1035 | Hesperidin | Citrus sinensis |  | −8.8 |  |
| T1609 | NAD+ | Punica granatum |  | −8.8 |  |
| T6S1529 | 1,5-Dicaffeoylquinic acid | Lonicera japonica |  | −8.8 |  |
| T3149 | Salvianolic Acid C | Slvia miltiorrhiza |  | −8.7 |  |
| T3S1612 | Kuwanon G | Morus alba |  | −8.7 |  |
| TL0006 | Chicoric Acid | Cichorium intybus |  | −8.6 |  |
| T3242 | Breviscapin | Erigeron |  | −8.5 |  |
| 58353 | - | - |  | −8.7 |  |
| 52917 | - | - |  | −8.6 |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Liang, B.; Sang, X.; An, J.; Huang, Z. Discovery of Potential Inhibitors of SARS-CoV-2 Main Protease by a Transfer Learning Method. Viruses 2023, 15, 891. https://doi.org/10.3390/v15040891
Zhang H, Liang B, Sang X, An J, Huang Z. Discovery of Potential Inhibitors of SARS-CoV-2 Main Protease by a Transfer Learning Method. Viruses. 2023; 15(4):891. https://doi.org/10.3390/v15040891
Chicago/Turabian StyleZhang, Huijun, Boqiang Liang, Xiaohong Sang, Jing An, and Ziwei Huang. 2023. "Discovery of Potential Inhibitors of SARS-CoV-2 Main Protease by a Transfer Learning Method" Viruses 15, no. 4: 891. https://doi.org/10.3390/v15040891