
Understanding Mutations in Human SARS-CoV-2 Spike Glycoprotein: A Systematic Review & Meta-Analysis
 ,
,  , , , , ,
, , , , ,  , , and
, , and 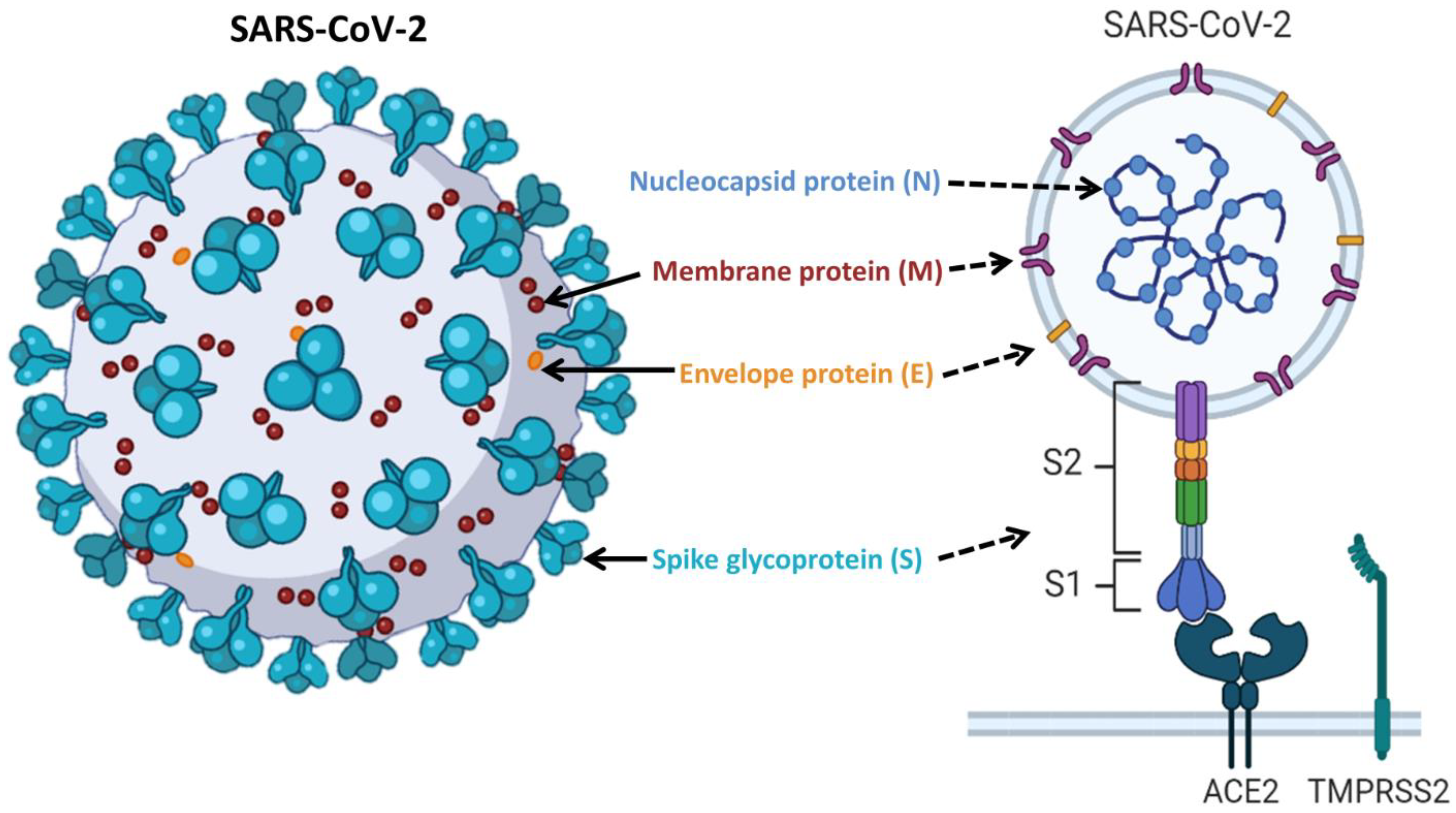{kind=link}
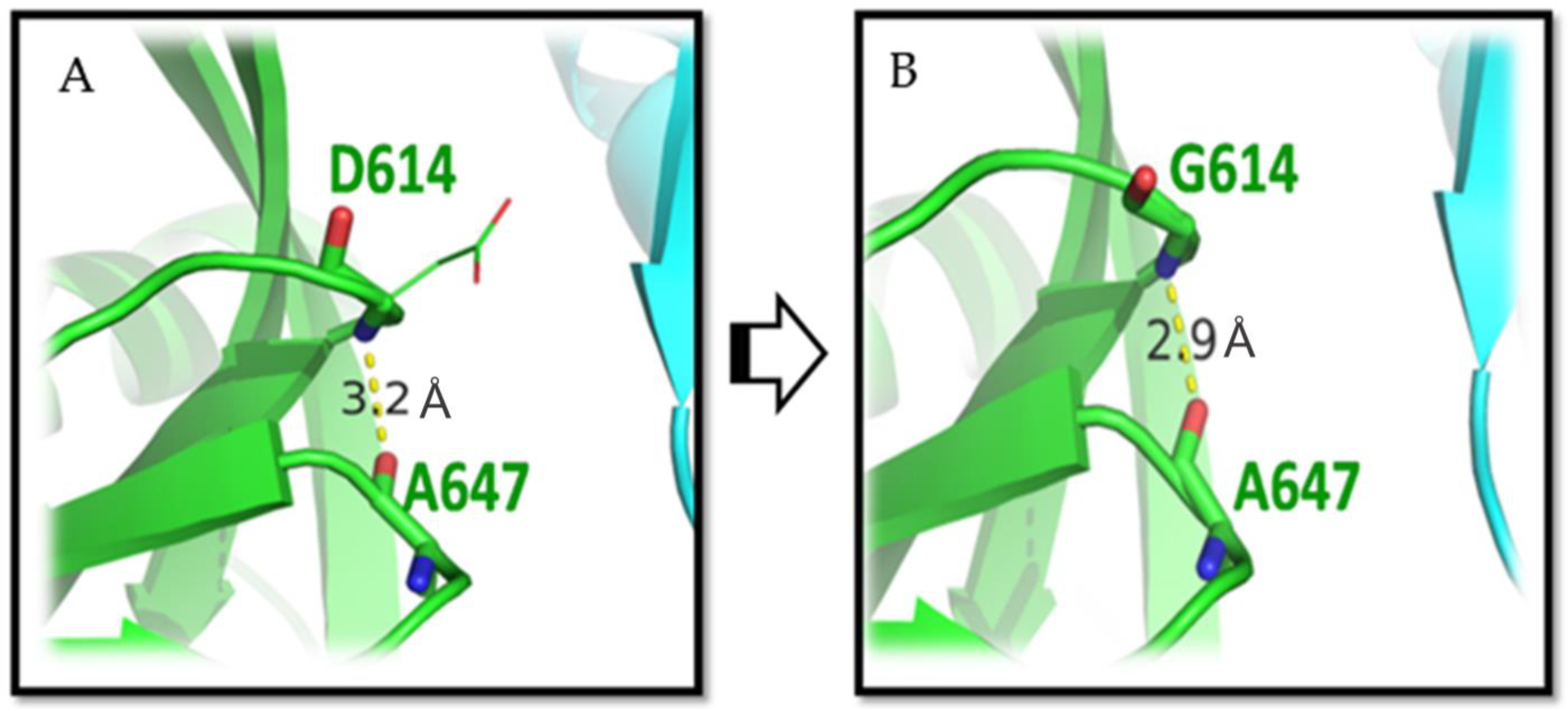{kind=link}
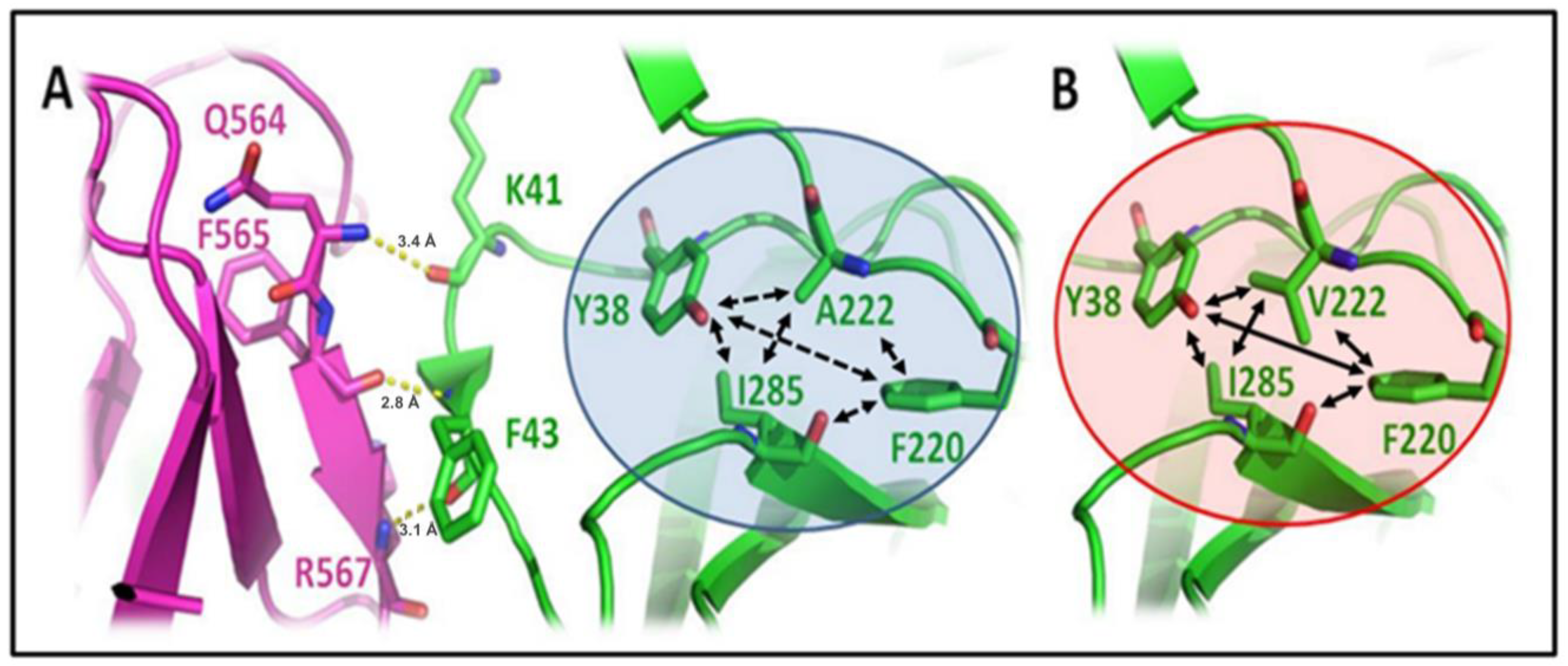{kind=link}
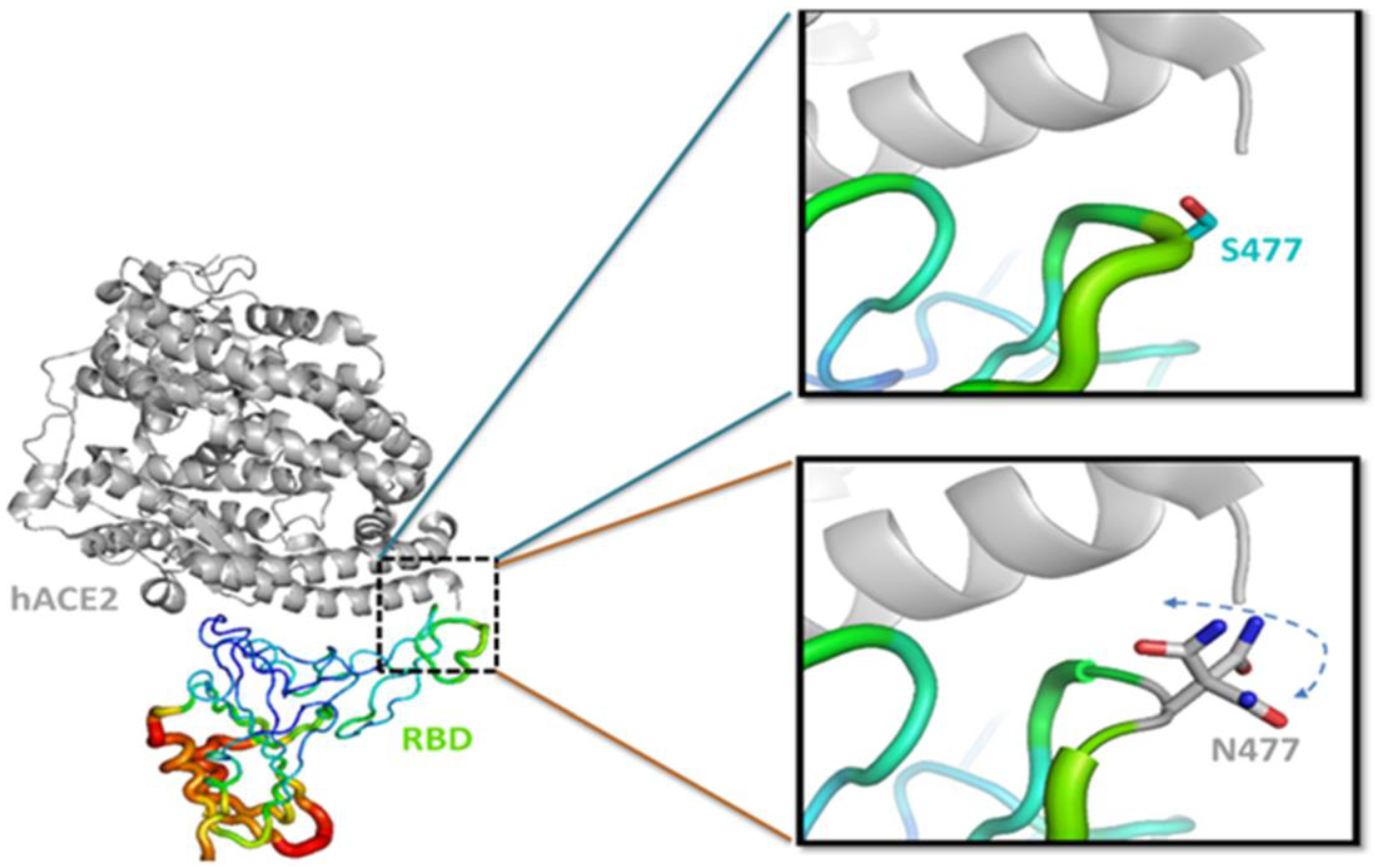{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Modeling of Spike Mutations
2.2. Solvent Accessible Surface Area Calculation
3. Results
3.1. D614G Mutation
3.2. A222V Mutation
3.3. S477N Mutation
3.4. Alpha Variant
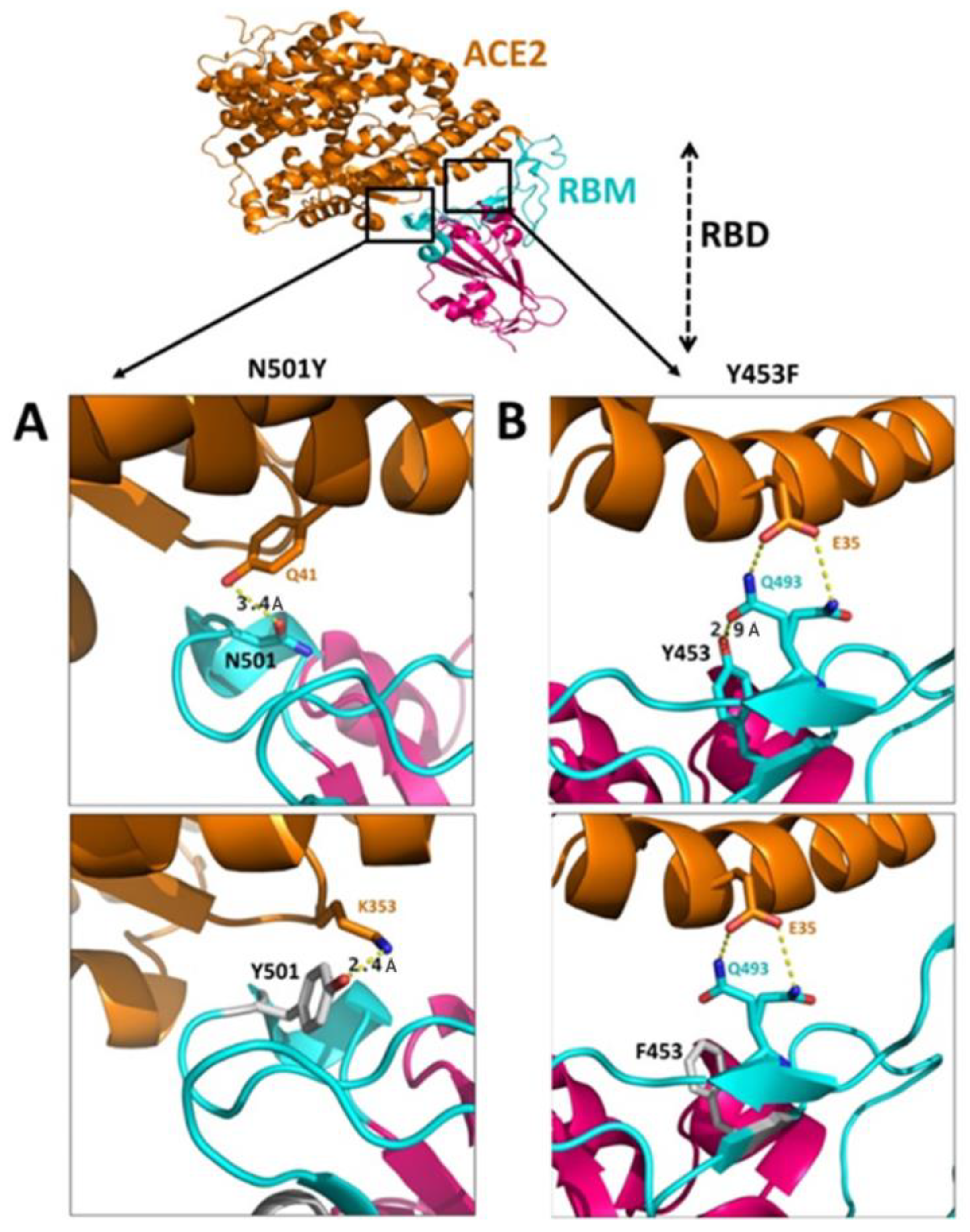
3.4.1. N501Y Mutation
3.4.2. Y453F Mutation
3.4.3. N439K Mutation
3.5. Beta Variant
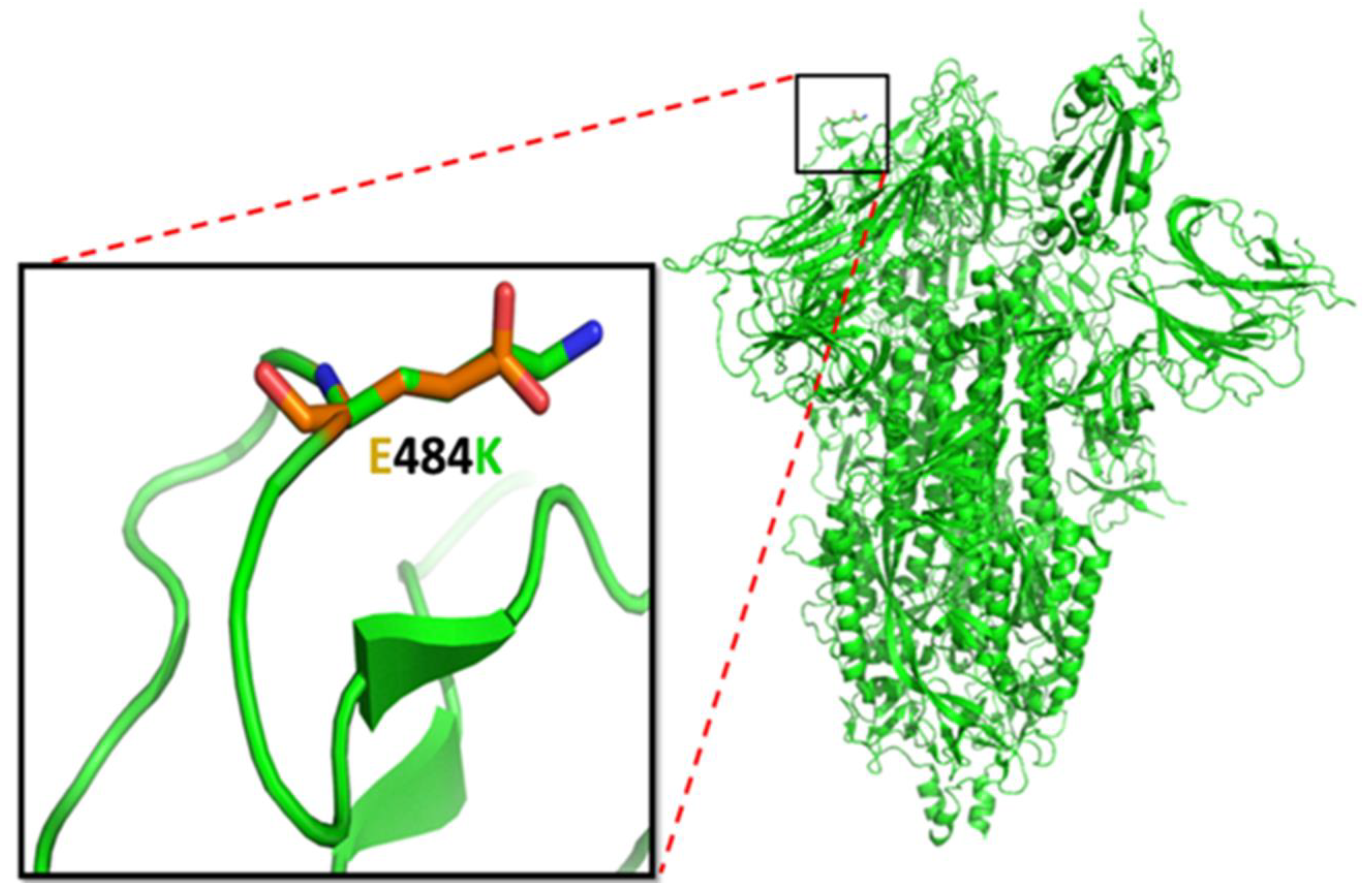
3.5.1. E484K Mutation
3.5.2. K417N Mutation
3.6. Gamma Variant
3.7. Delta Variant
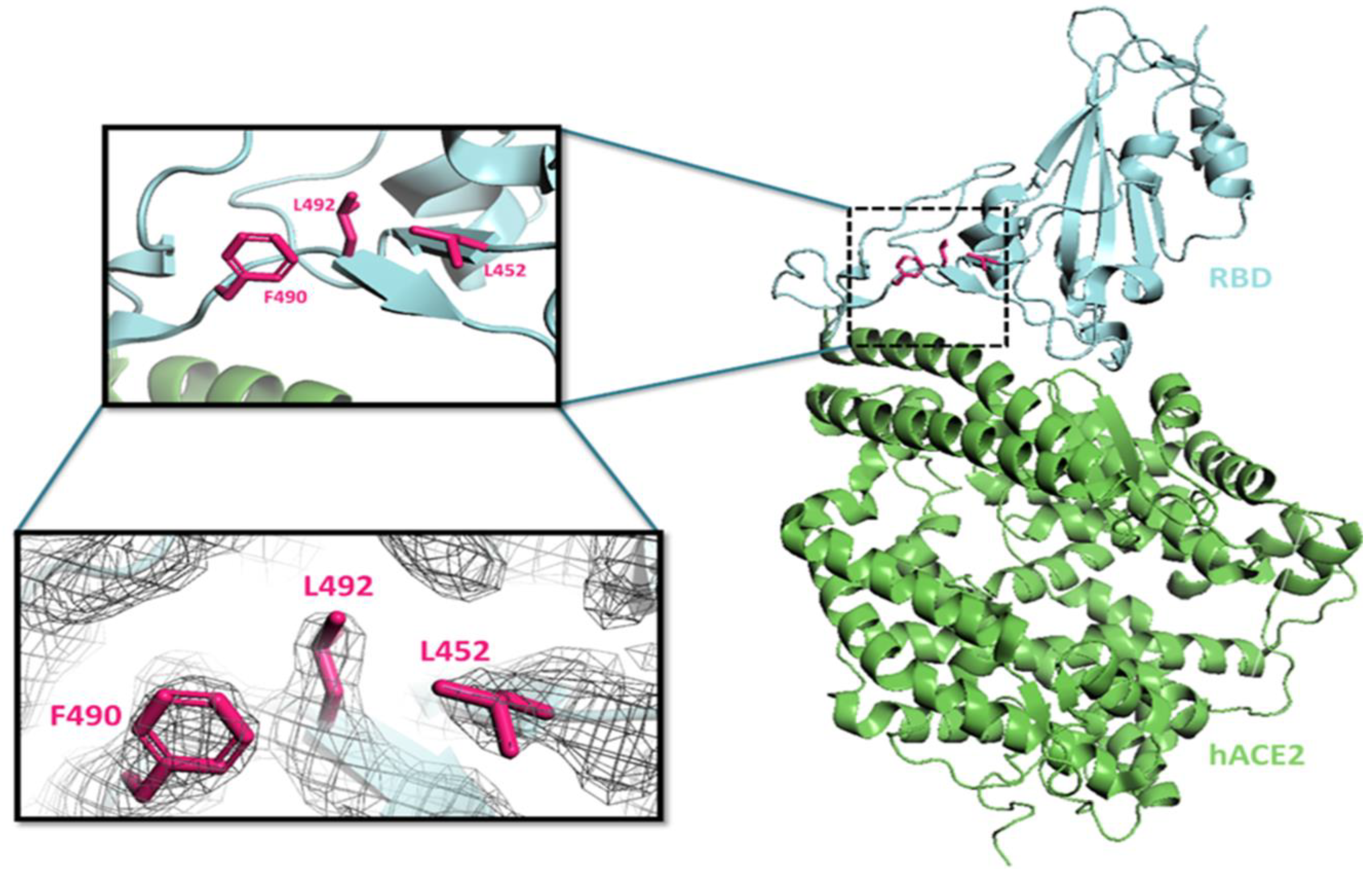
3.7.1. L452R Mutation
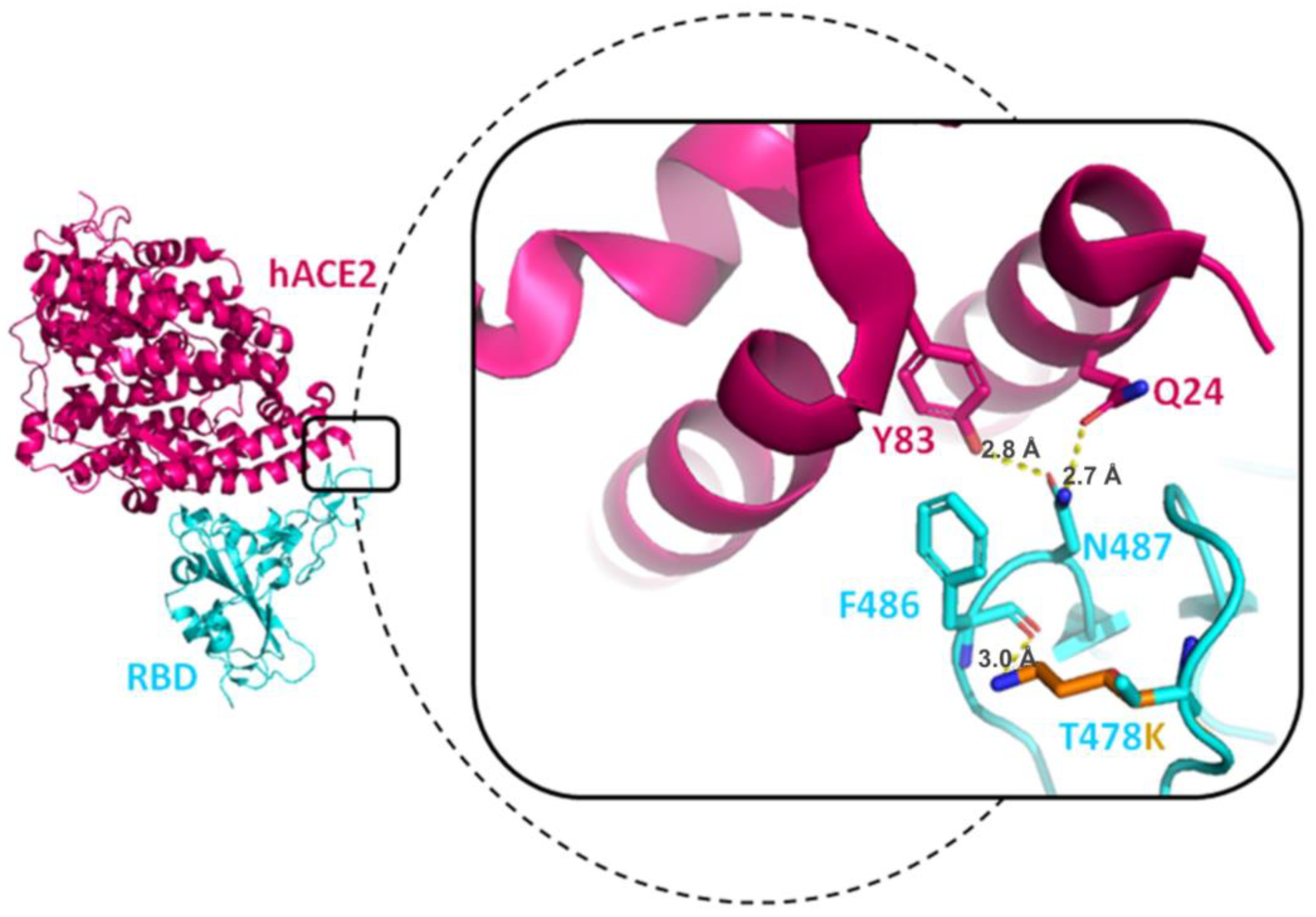
3.7.2. T478K Mutation
3.8. Delta Plus Variant
3.9. Omicron Variant
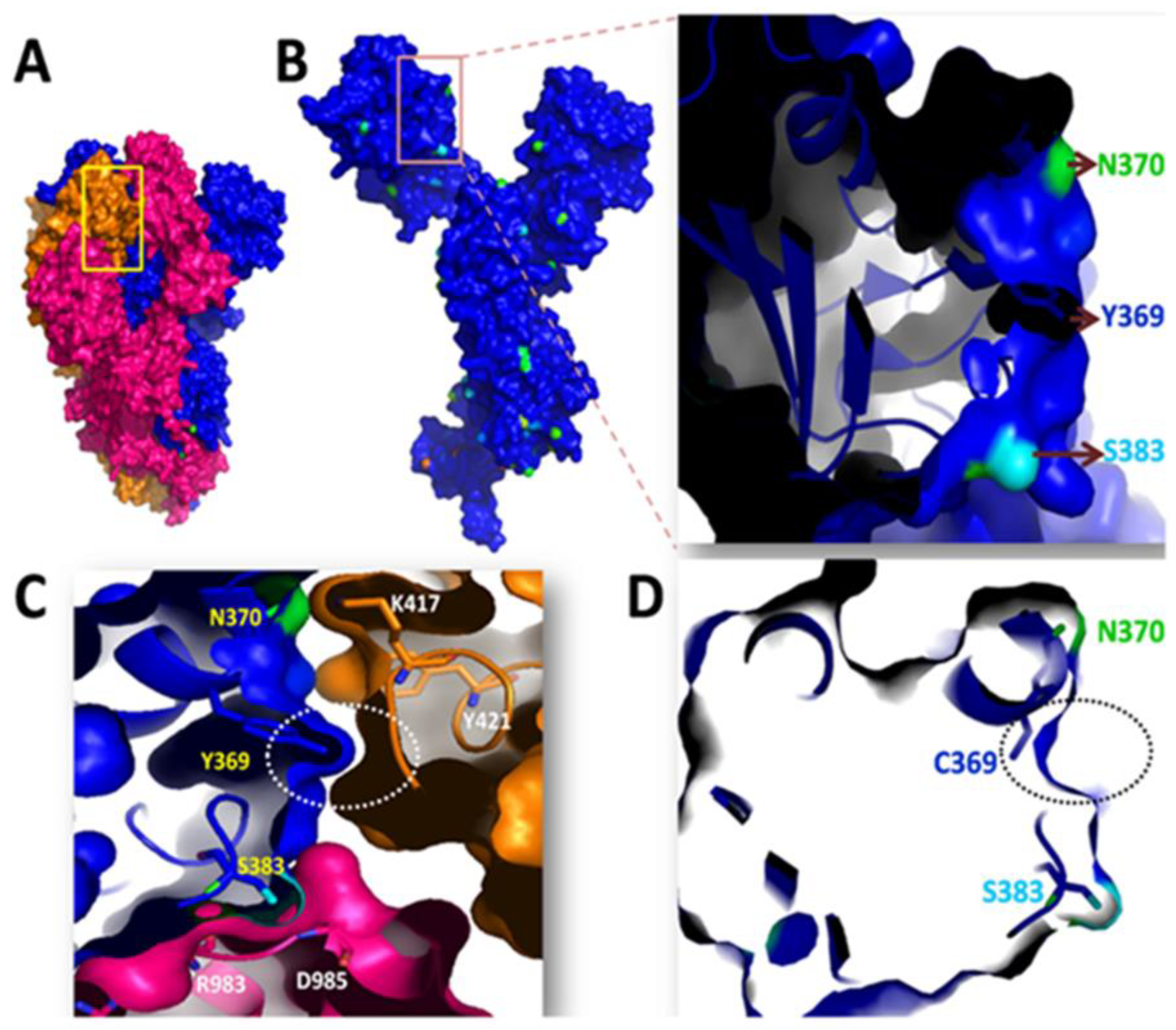
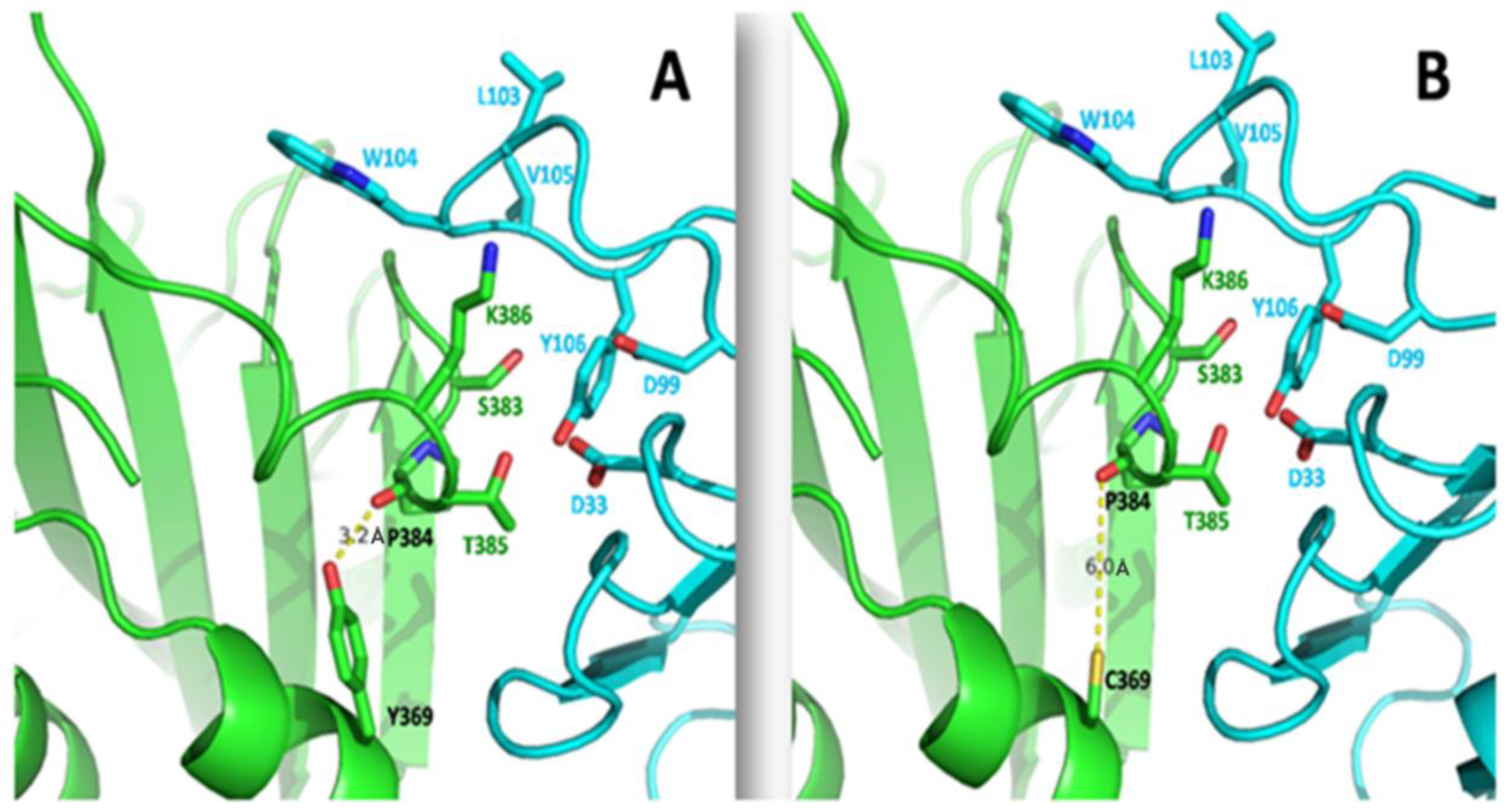
Y369C Mutation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| hACE2 | Human angiotensin converting enzyme 2 |
| mAbs | Monoclonal antibodies |
| NTD | N terminal domain |
| RBD | Receptor binding domain |
| RBM | Receptor binding motif |
| RMSD | Root mean square deviation |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SASA | Solvent accessible surface area |
| VoC | Variant(s) of concern |
| WHO | World Health Organization |
References
- Lotfi, M.; Hamblin, M.R.; Rezaei, N. COVID-19: Transmission, prevention, and potential therapeutic opportunities. Clin. Chim. Acta. 2020, 508, 254–266. [Google Scholar] [CrossRef] [PubMed]
- WHO. Coronavirus Disease 2019 (COVID-19): Situation Report; WHO: Geneva, Switzerland, 2020; Volume 51. [Google Scholar]
- Almehdi, A.M.; Khoder, G.; Alchakee, A.S.; Alsayyid, A.T.; Sarg, N.H.; Soliman, S.S.M. SARS-CoV-2 spike protein: Pathogenesis, vaccines, and potential therapies. Infection 2021, 49, 855–876. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Raghav, S.; Ghosh, A.; Turuk, J.; Kumar, S.; Jha, A.; Madhulika, S.; Priyadarshini, M.; Biswas, V.K.; Shyamli, P.S.; Singh, B.; et al. Analysis of Indian SARS-CoV-2 Genomes Reveals Prevalence of D614G Mutation in Spike Protein Predicting an Increase in Interaction With TMPRSS2 and Virus Infectivity. Front. Microbiol. 2020, 11, 594928. [Google Scholar] [CrossRef]
- Abu-Raddad, L.J.; Chemaitelly, H.; Butt, A.A. Effectiveness of the BNT162b2 COVID-19 Vaccine against the B.1.1.7 and B.1.351 Variants. N. Engl. J. Med. 2021, 385, 187–189. [Google Scholar] [CrossRef]
- Emary, K.R.W.; Golubchik, T.; Aley, P.K.; Ariani, C.V.; Angus, B.; Bibi, S.; Blane, B.; Bonsall, D.; Cicconi, P.; Charlton, S.; et al. Efficacy of ChAdOx1 nCoV-19 (AZD1222) vaccine against SARS-CoV-2 variant of concern 202012/01 (B.1.1.7): An exploratory analysis of a randomised controlled trial. Lancet 2021, 397, 1351–1362. [Google Scholar] [CrossRef]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef]
- Mahase, E. COVID-19: Novavax vaccine efficacy is 86% against UK variant and 60% against South African variant. BMJ 2021, 372, n296. [Google Scholar] [CrossRef]
- Taylor, P.C.; Adams, A.C.; Hufford, M.M.; de la Torre, I.; Winthrop, K.; Gottlieb, R.L. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat. Rev. Immunol. 2021, 21, 382–393. [Google Scholar] [CrossRef]
- Lai, C.-C.; Shih, T.-P.; Ko, W.-C.; Tang, H.-J.; Hsueh, P.-R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef]
- Sanjuán, R.; Domingo-Calap, P. Genetic diversity and evolution of viral populations. Encycl. Virol. 2021, 1, 53–61. [Google Scholar]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A Single Mutation in Chikungunya Virus Affects Vector Specificity and Epidemic Potential. PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef]
- Diehl, W.E.; Lin, A.E.; Grubaugh, N.D.; Carvalho, L.M.; Kim, K.; Kyawe, P.P.; McCauley, S.M.; Donnard, E.; Kucukural, A.; McDonel, P.; et al. Ebola Virus Glycoprotein with Increased Infectivity Dominated the 2013-2016 Epidemic. Cell 2016, 167, 1088–1098.e6. [Google Scholar] [CrossRef] [Green Version]
- Deval, J.; Gurard-Levin, Z.A. Opportunities and Challenges in Targeting the Proofreading Activity of SARS-CoV-2 Polymerase Complex. Molecules 2022, 27, 2918. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic. Acids. Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Ramachandran, S.; Kota, P.; Ding, F.; Dokholyan, N.V. Automated minimization of steric clashes in protein structures. Proteins 2011, 79, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Jiang, W.; Zhou, Y. Identifying protein–protein interaction sites in transient complexes with temperature factor, sequence profile and accessible surface area. Amino. Acids. 2010, 38, 263–270. [Google Scholar] [CrossRef]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Webster, R.G.; Webby, R.J. Influenza Virus: Dealing with a Drifting and Shifting Pathogen. Viral Immunol. 2018, 31, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Rothenburg, S.; Brennan, G. Species-Specific Host-Virus Interactions: Implications for Viral Host Range and Virulence. Trends Microbiol. 2020, 28, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Noureddine, F.Y.; Chakkour, M.; El Roz, A.; Reda, J.; Al Sahily, R.; Assi, A.; Joma, M.; Salami, H.; Hashem, S.J.; Harb, B.; et al. The Emergence of SARS-CoV-2 Variant(s) and Its Impact on the Prevalence of COVID-19 Cases in the Nabatieh Region, Lebanon. Med. Sci. 2021, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Salehi-Vaziri, M.; Fazlalipour, M.; Seyed Khorrami, S.M.; Azadmanesh, K.; Pouriayevali, M.H.; Jalali, T.; Shoja, Z.; Maleki, A. The ins and outs of SARS-CoV-2 variants of concern (VOCs). Arch. Virol. 2022, 167, 327–344. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Malik, J.A.; Mulla, A.H.; Farooqi, T.; Pottoo, F.H.; Anwar, S.; Rengasamy, K.R.R. Targets and strategies for vaccine development against SARS-CoV-2. Biomed. Pharmacother. 2021, 137, 111254. [Google Scholar] [CrossRef]
- Dai, L.; Gao, G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021, 21, 73–82. [Google Scholar] [CrossRef]
- Sadoff, J.; Gray, G.; Vandebosch, A.; Cárdenas, V.; Shukarev, G.; Grinsztejn, B.; Goepfert, P.A.; Truyers, C.; Fennema, H.; Spiessens, B.; et al. Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against COVID-19. N. Engl. J. Med. 2021, 384, 2187–2201. [Google Scholar] [CrossRef]
- Bernal, J.L.; Andrews, N.; Gower, C.; Robertson, C.; Stowe, J.; Tessier, E.; Simmons, R.; Cottrell, S.; Roberts, R.; O’Doherty, M.; et al. Effectiveness of the Pfizer-BioNTech and Oxford-AstraZeneca vaccines on COVID-19 related symptoms, hospital admissions, and mortality in older adults in England: Test negative case-control study. BMJ 2021, 373, n1088. [Google Scholar] [CrossRef]
- Arora, P.; Pöhlmann, S.; Hoffmann, M. Mutation D614G increases SARS-CoV-2 transmission. Signal Transduct. Target. Ther. 2021, 6, 101. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Hanage, W.P.; Rasmussen, A.L. Making Sense of Mutation: What D614G Means for the COVID-19 Pandemic Remains Unclear. Cell 2020, 182, 794–795. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef]
- Zou, J.; Xie, X.; Fontes-Garfias, C.R.; Swanson, K.A.; Kanevsky, I.; Tompkins, K.; Cutler, M.; Cooper, D.; Dormitzer, P.R.; Shi, P.-Y. The effect of SARS-CoV-2 D614G mutation on BNT162b2 vaccine-elicited neutralization. NPJ Vaccines 2021, 6, 44. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef]
- Singh, A.; Steinkellner, G.; Köchl, K.; Gruber, K.; Gruber, C.C. Serine 477 plays a crucial role in the interaction of the SARS-CoV-2 spike protein with the human receptor ACE2. Sci. Rep. 2021, 11, 4320. [Google Scholar] [CrossRef] [PubMed]
- Nelson-Sathi, S.; Umasankar, P.K.; Sreekumar, E.; Nair, R.R.; Joseph, I.; Nori, S.R.C.; Philip, J.S.; Prasad, R.; Navyasree, K.V.; Ramesh, S.; et al. Mutational landscape and in silico structure models of SARS-CoV-2 spike receptor binding domain reveal key molecular determinants for virus-host interaction. BMC Mol. Cell Biol. 2022, 23, 2. [Google Scholar] [CrossRef] [PubMed]
- Vasireddy, D.; Vanaparthy, R.; Mohan, G.; Malayala, S.V.; Atluri, P. Review of COVID-19 Variants and COVID-19 Vaccine Efficacy: What the Clinician Should Know? J. Clin. Med. Res. 2021, 13, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.-M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 2021, 29, 477–488.e4. [Google Scholar] [CrossRef]
- Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-SARS-CoV-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 17 March 2023).
- Rathnasinghe, R.; Jangra, S.; Cupic, A.; Martínez-Romero, C.; Mulder, L.C.F.; Kehrer, T.; Yildiz, S.; Choi, A.; Mena, I.; De Vrieze, J.; et al. The N501Y mutation in SARS-CoV-2 spike leads to morbidity in obese and aged mice and is neutralized by convalescent and post-vaccination human sera. medRxiv 2021. [Google Scholar] [CrossRef]
- Shahhosseini, N.; Babuadze, G.; Wong, G.; Kobinger, G.P. Mutation Signatures and In Silico Docking of Novel SARS-CoV-2 Variants of Concern. Microorganisms 2021, 9, 926. [Google Scholar] [CrossRef]
- Tian, F.; Tong, B.; Sun, L.; Shi, S.; Zheng, B.; Wang, Z.; Dong, X.; Zheng, P. Mutation N501Y in RBD of Spike Protein Strengthens the Interaction between COVID-19 and its Receptor ACE2. bioRxiv 2021. [Google Scholar] [CrossRef]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.T.M.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature 2022, 602, 294–299. [Google Scholar] [CrossRef]
- Xie, X.; Liu, Y.; Liu, J.; Zhang, X.; Zou, J.; Fontes-Garfias, C.R.; Xia, H.; Swanson, K.A.; Cutler, M.; Cooper, D.; et al. Neutralization of SARS-CoV-2 spike 69/70 deletion, E484K and N501Y variants by BNT162b2 vaccine-elicited sera. Nat. Med. 2021, 27, 620–621. [Google Scholar] [CrossRef]
- Focosi, D.; Maggi, F. Neutralising antibody escape of SARS-CoV-2 spike protein: Risk assessment for antibody-based COVID-19 therapeutics and vaccines. Rev. Med. Virol. 2021, 31, e2231. [Google Scholar] [CrossRef]
- Hayashi, T.; Yaegashi, N.; Konishi, I. Effect of RBD mutations in spike glycoprotein of SARS-CoV-2 on neutralizing IgG affinity. medRxiv 2021. [Google Scholar] [CrossRef]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018. [Google Scholar] [CrossRef]
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; da Silva Filipe, A.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell 2021, 184, 1171–1187.e20. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021, 371, 850–854. [Google Scholar] [CrossRef]
- Shen, X.; Tang, H.; McDanal, C.; Wagh, K.; Fischer, W.; Theiler, J.; Yoon, H.; Li, D.; Haynes, B.F.; Sanders, K.O.; et al. SARS-CoV-2 variant B.1.1.7 is susceptible to neutralizing antibodies elicited by ancestral spike vaccines. Cell Host Microbe. 2021, 29, 529–539.e3. [Google Scholar] [CrossRef]
- Aleem, A.; Akbar Samad, A.B.; Slenker, A.K. Emerging Variants of SARS-CoV-2 and Novel Therapeutics against Coronavirus (COVID-19); StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Andreano, E.; Piccini, G.; Licastro, D.; Casalino, L.; Johnson, N.V.; Paciello, I.; Monego, S.D.; Pantano, E.; Manganaro, N.; Manenti, A.; et al. SARS-CoV-2 escape from a highly neutralizing COVID-19 convalescent plasma. Proc. Natl. Acad. Sci. USA 2021, 118, e2103154118. [Google Scholar] [CrossRef]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe. 2021, 29, 463–476.e6. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Dingens, A.S.; Bloom, J.D. Complete map of SARS-CoV-2 RBD mutations that escape the monoclonal antibody LY-CoV555 and its cocktail with LY-CoV016. Cell Rep. Med. 2021, 2, 100255. [Google Scholar] [CrossRef] [PubMed]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.-H.; Michailidis, E.; et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife 2020, 9, e61312. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Buzko, O.; Spilman, P.R.; Niazi, K.; Rabizadeh, S.; Soon-Shiong, P.R. Molecular dynamic simulation reveals E484K mutation enhances spike RBD-ACE2 affinity and the combination of E484K, K417N and N501Y mutations (501Y. V2 variant) induces conformational change greater than N501Y mutant alone, potentially resulting in an escape mutant. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhou, D.; Dejnirattisai, W.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Evidence of escape of SARS-CoV-2 variant B.1.351 from natural and vaccine-induced sera. Cell 2021, 184, 2348–2361.e6. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Huang, D.; Lee, C.-C.D.; Wu, N.C.; Jackson, A.M.; Zhu, X.; Liu, H.; Peng, L.; van Gils, M.J.; Sanders, R.W.; et al. Structural and functional ramifications of antigenic drift in recent SARS-CoV-2 variants. Science 2021, 373, 818–823. [Google Scholar] [CrossRef]
- Pondé, R.A.A. Physicochemical effect of the N501Y, E484K/Q, K417N/T, L452R and T478K mutations on the SARS-CoV-2 spike protein RBD and its influence on agent fitness and on attributes developed by emerging variants of concern. Virology 2022, 572, 44–54. [Google Scholar] [CrossRef]
- Khan, A.; Zia, T.; Suleman, M.; Khan, T.; Ali, S.S.; Abbasi, A.A.; Mohammad, A.; Wei, D.-Q. Higher infectivity of the SARS-CoV-2 new variants is associated with K417N/T, E484K, and N501Y mutants: An insight from structural data. J. Cell Physiol. 2021, 236, 7045–7057. [Google Scholar] [CrossRef]
- Li, Q.; Nie, J.; Wu, J.; Zhang, L.; Ding, R.; Wang, H.; Zhang, Y.; Li, T.; Liu, S.; Zhang, M.; et al. SARS-CoV-2 501Y.V2 variants lack higher infectivity but do have immune escape. Cell 2021, 184, 2362–2371.e9. [Google Scholar] [CrossRef]
- Cheng, M.H.; Krieger, J.M.; Banerjee, A.; Xiang, Y.; Kaynak, B.; Shi, Y.; Arditi, M.; Bahar, I. Impact of new variants on SARS-CoV-2 infectivity and neutralization: A molecular assessment of the alterations in the spike-host protein interactions. iScience 2022, 25, 103939. [Google Scholar] [CrossRef]
- Barton, M.I.; MacGowan, S.A.; Kutuzov, M.A.; Dushek, O.; Barton, G.J.; van der Merwe, P.A. Effects of common mutations in the SARS-CoV-2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. Elife 2021, 10, e70658. [Google Scholar] [CrossRef]
- Liu, H.; Wei, P.; Kappler, J.W.; Marrack, P.; Zhang, G. SARS-CoV-2 Variants of Concern and Variants of Interest Receptor Binding Domain Mutations and Virus Infectivity. Front. Immunol. 2022, 13, 825256. [Google Scholar] [CrossRef]
- Banho, C.A.; Sacchetto, L.; Campos, G.R.F.; Bittar, C.; Possebon, F.S.; Ullmann, L.S.; Marques, B.D.C.; da Silva, G.C.D.; Moraes, M.M.; Parra, M.C.P.; et al. Impact of SARS-CoV-2 Gamma lineage introduction and COVID-19 vaccination on the epidemiological landscape of a Brazilian city. Commun. Med. 2022, 2, 41. [Google Scholar] [CrossRef]
- Zhan, Y.; Yin, H.; Yin, J.Y.B. 1.617.2 (Delta) Variant of SARS-CoV-2: Features, transmission and potential strategies. Int. J. Biol. Sci. 2022, 18, 1844–1851. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Singh, J.; Rahman, S.A.; Ehtesham, N.Z.; Hira, S.; Hasnain, S.E. SARS-CoV-2 variants of concern are emerging in India. Nat. Med. 2021, 27, 1131–1133. [Google Scholar] [CrossRef] [PubMed]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 2020, 584, 115–119. [Google Scholar] [CrossRef]
- Tchesnokova, V.; Kulasekara, H.; Larson, L.; Bowers, V.; Rechkina, E.; Kisiela, D.; Sledneva, Y.; Choudhury, D.; Maslova, I.; Deng, K.; et al. Acquisition of the L452R Mutation in the ACE2-Binding Interface of Spike Protein Triggers Recent Massive Expansion of SARS-CoV-2 Variants. J. Clin. Microbiol. 2021, 59, e0092121. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, S.; Mercatelli, D.; Rakhimov, A.; Giorgi, F.M. Preliminary report on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Spike mutation T478K. J. Med. Virol. 2021, 93, 5638–5643. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, J.; Gao, K.; Wei, G.-W. Vaccine-escape and fast-growing mutations in the United Kingdom, the United States, Singapore, Spain, India, and other COVID-19-devastated countries. Genomics 2021, 113, 2158–2170. [Google Scholar] [CrossRef]
- Thakur, V.; Bhola, S.; Thakur, P.; Patel, S.K.S.; Kulshrestha, S.; Ratho, R.K.; Kumar, P. Waves and variants of SARS-CoV-2: Understanding the causes and effect of the COVID-19 catastrophe. Infection 2022, 50, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, M.; Sharma, A.; Priyanka; Thakur, N.; Rajkhowa, T.K.; Choudhary, O.P. Delta variant (B.1.617.2) of SARS-CoV-2: Mutations, impact, challenges and possible solutions. Hum. Vaccin. Immunother. 2022, 18, 2068883. [Google Scholar] [CrossRef]
- Davis, C.; Logan, N.; Tyson, G.; Orton, R.; Harvey, W.T.; Perkins, J.S.; Mollett, G.; Blacow, R.M.; COVID-19 Genomics UK (COG-UK) Consortium; Peacock, T.P.; et al. Reduced neutralisation of the Delta (B.1.617.2) SARS-CoV-2 variant of concern following vaccination. PLoS Pathog. 2021, 17, e1010022. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Roy, H. The Delta Plus variant of COVID-19: Will it be the worst nightmare in the SARS-CoV-2 pandemic? J. Biomed. Sci. 2021, 8, 1–2. [Google Scholar] [CrossRef]
- Salvatore, M.; Bhattacharyya, R.; Purkayastha, S.; Zimmermann, L.; Ray, D.; Hazra, A.; Kleinsasser, M.; Mellan, T.A.; Whittaker, C.; Flaxman, S.; et al. Resurgence of SARS-CoV-2 in India: Potential role of the B. 1.617. 2 (Delta) variant and delayed interventions. medRxiv 2021. [Google Scholar] [CrossRef]
- Ren, S.-Y.; Wang, W.-B.; Gao, R.-D.; Zhou, A.-M. Omicron variant (B.1.1.529) of SARS-CoV-2: Mutation, infectivity, transmission, and vaccine resistance. World J. Clin. Cases 2022, 10, 1–11. [Google Scholar] [CrossRef]
- Dhawan, M.; Saied, A.A.; Mitra, S.; Alhumaydhi, F.A.; Emran, T.B.; Wilairatana, P. Omicron variant (B.1.1.529) and its sublineages: What do we know so far amid the emergence of recombinant variants of SARS-CoV-2? Biomed. Pharm. 2022, 154, 113522. [Google Scholar] [CrossRef]
- Quarleri, J.; Galvan, V.; Delpino, M.V. Omicron variant of the SARS-CoV-2: A quest to define the consequences of its high mutational load. Geroscience 2022, 44, 53–56. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- VanBlargan, L.A.; Errico, J.M.; Halfmann, P.J.; Zost, S.J.; Crowe, J.E.; Purcell, L.A.; Kawaoka, Y.; Corti, D.; Fremont, D.H.; Diamond, M.S. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat. Med. 2022, 28, 490–495. [Google Scholar] [CrossRef]
- Cele, S.; Jackson, L.; Khoury, D.S.; Khan, K.; Moyo-Gwete, T.; Tegally, H.; San, J.E.; Cromer, D.; Scheepers, C.; Amoako, D.G.; et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature 2022, 602, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Haslwanter, D.; Dieterle, M.E.; Wec, A.Z.; O’Brien, C.M.; Sakharkar, M.; Florez, C.; Tong, K.; Rappazzo, C.G.; Lasso, G.; Vergnolle, O.; et al. A Combination of Receptor-Binding Domain and N-Terminal Domain Neutralizing Antibodies Limits the Generation of SARS-CoV-2 Spike Neutralization-Escape Mutants. Mbio 2021, 12, e0247321. [Google Scholar] [CrossRef] [PubMed]
- Javanmardi, K.; Segall-Shapiro, T.H.; Chou, C.-W.; Boutz, D.R.; Olsen, R.J.; Xie, X.; Xia, H.; Shi, P.-Y.; Johnson, C.D.; Annapareddy, A.; et al. Antibody escape and cryptic cross-domain stabilization in the SARS-CoV-2 Omicron spike protein. Cell Host Microbe 2022, 30, 1242–1254.e6. [Google Scholar] [CrossRef] [PubMed]
- Ni, D.; Lau, K.; Turelli, P.; Raclot, C.; Beckert, B.; Nazarov, S.; Pojer, F.; Myasnikov, A.; Stahlberg, H.; Trono, D. Structural analysis of the Spike of the Omicron SARS-CoV-2 variant by cryo-EM and implications for immune evasion. bioRxiv 2021. [Google Scholar] [CrossRef]
- Asif, A.; Ilyas, I.; Abdullah, M.; Sarfraz, S.; Mustafa, M.; Mahmood, A. The Comparison of Mutational Progression in SARS-CoV-2: A Short Updated Overview. J. Mol. Pathol. 2022, 3, 201–218. [Google Scholar] [CrossRef]
- Rophina, M.; Pandhare, K.; Shamnath, A.; Imran, M.; Jolly, B.; Scaria, V. ESC: A comprehensive resource for SARS-CoV-2 immune escape variants. Nucleic Acids Res. 2022, 50, D771–D776. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Yuan, S.; Kok, K.-H.; To, K.K.-W.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.-Y.; Poon, R.W.-S.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Kyriakidis, N.C.; López-Cortés, A.; González, E.V.; Grimaldos, A.B.; Prado, E.O. SARS-CoV-2 vaccines strategies: A comprehensive review of phase 3 candidates. NPJ Vaccines 2021, 6, 28. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [Green Version]
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2-What Do They Mean? JAMA 2021, 325, 529–531. [Google Scholar] [CrossRef]
- Abdool Karim, S.S.; de Oliveira, T. New SARS-CoV-2 Variants-Clinical, Public Health, and Vaccine Implications. N. Engl. J. Med. 2021, 384, 1866–1868. [Google Scholar] [CrossRef]
- Chakkour, M.; Salami, A.; Olleik, D.; Kamal, I.; Noureddine, F.Y.; Roz, A.E.; Ghssein, G. Risk Markers of COVID-19, a Study from South-Lebanon. COVID 2022, 2, 867–876. [Google Scholar] [CrossRef]
- Wang, Z.; Schmidt, F.; Weisblum, Y.; Muecksch, F.; Barnes, C.O.; Finkin, S.; Schaefer-Babajew, D.; Cipolla, M.; Gaebler, C.; Lieberman, J.A.; et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature 2021, 592, 616–622. [Google Scholar] [CrossRef]
- Pouwels, K.B.; Pritchard, E.; Matthews, P.C.; Stoesser, N.; Eyre, D.W.; Vihta, K.-D.; House, T.; Hay, J.; Bell, J.I.; Newton, J.N.; et al. Effect of Delta variant on viral burden and vaccine effectiveness against new SARS-CoV-2 infections in the UK. Nat. Med. 2021, 27, 2127–2135. [Google Scholar] [CrossRef]
- Yadav, P.D.; Sapkal, G.N.; Ella, R.; Sahay, R.R.; Nyayanit, D.A.; Patil, D.Y.; Deshpande, G.; Shete, A.M.; Gupta, N.; Mohan, V.K.; et al. Neutralization of Beta and Delta variant with sera of COVID-19 recovered cases and vaccinees of inactivated COVID-19 vaccine BBV152/Covaxin. J. Travel Med. 2021, 28, taab104. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, R.; Srivastava, Y.; Muthuramalingam, P.; Singh, S.K.; Verma, G.; Tiwari, S.; Tandel, N.; Beura, S.K.; Panigrahi, A.R.; Maji, S.; et al. Understanding Mutations in Human SARS-CoV-2 Spike Glycoprotein: A Systematic Review & Meta-Analysis. Viruses 2023, 15, 856. https://doi.org/10.3390/v15040856
Kumar R, Srivastava Y, Muthuramalingam P, Singh SK, Verma G, Tiwari S, Tandel N, Beura SK, Panigrahi AR, Maji S, et al. Understanding Mutations in Human SARS-CoV-2 Spike Glycoprotein: A Systematic Review & Meta-Analysis. Viruses. 2023; 15(4):856. https://doi.org/10.3390/v15040856
Chicago/Turabian StyleKumar, Reetesh, Yogesh Srivastava, Pandiyan Muthuramalingam, Sunil Kumar Singh, Geetika Verma, Savitri Tiwari, Nikunj Tandel, Samir Kumar Beura, Abhishek Ramachandra Panigrahi, Somnath Maji, and et al. 2023. "Understanding Mutations in Human SARS-CoV-2 Spike Glycoprotein: A Systematic Review & Meta-Analysis" Viruses 15, no. 4: 856. https://doi.org/10.3390/v15040856