
Immunosuppression as a Hub for SARS-CoV-2 Mutational Drift

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Sources and Collection
2.1.1. Data Sources
2.1.2. Data Collection
2.1.3. Collection, Extraction and Sequencing of RNA Samples
2.2. Processing of Whole-Genome Viral Sequencing Data
2.3. Genomic and Phylogenetic Annotations
Statistical Modeling
2.4. Limitations
2.5. Ethics Declaration
3. Results
3.1. Data Overview
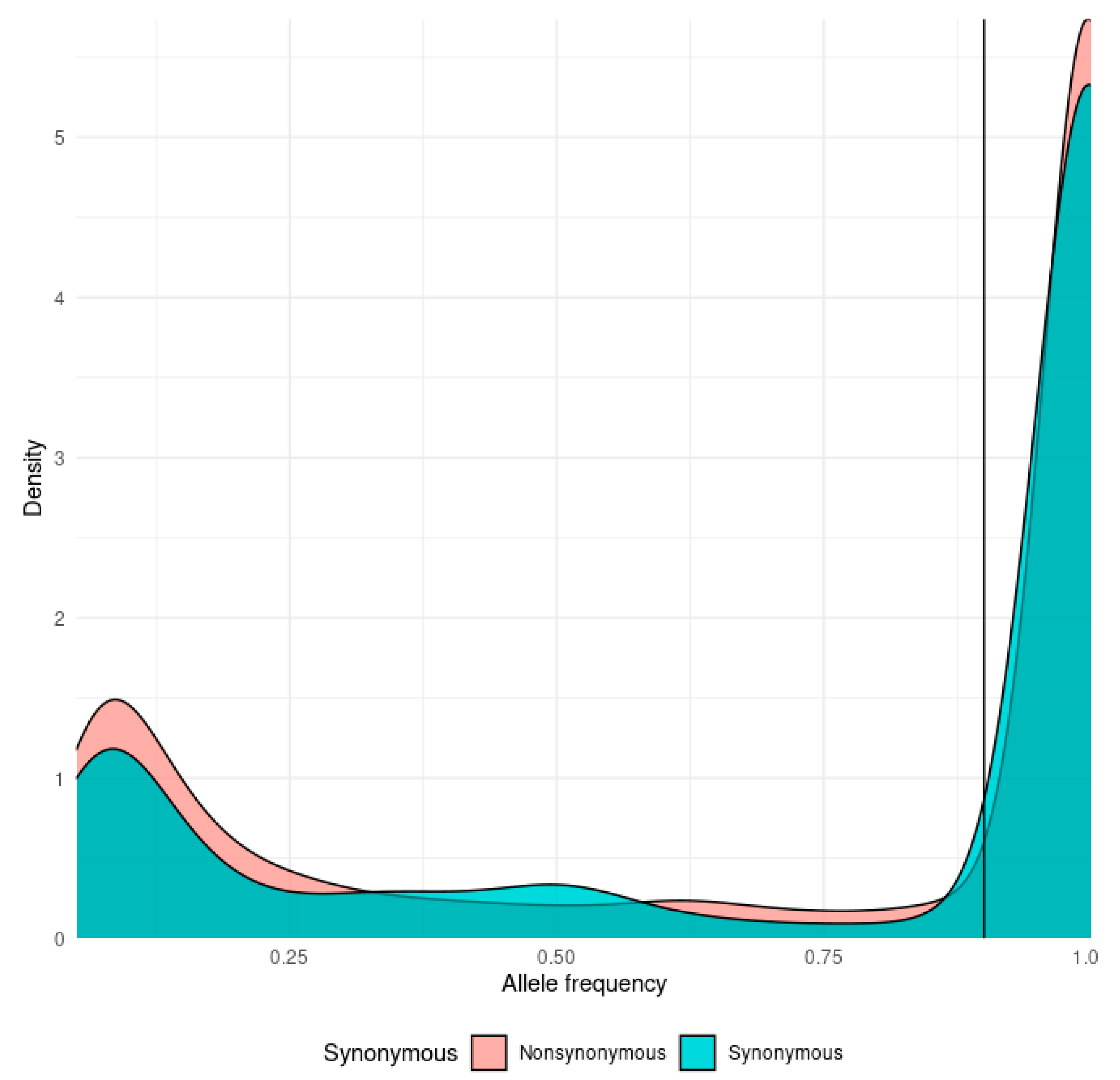
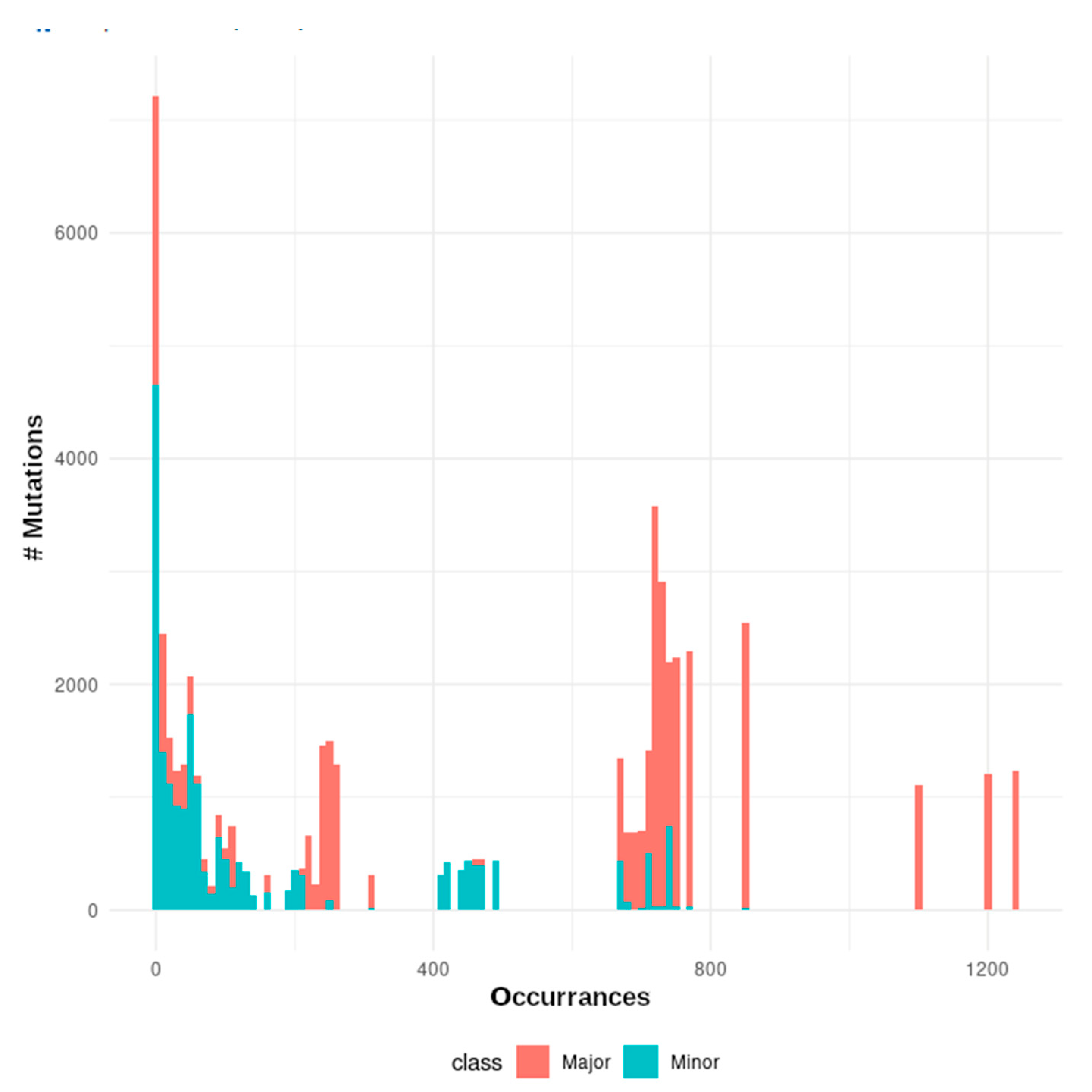
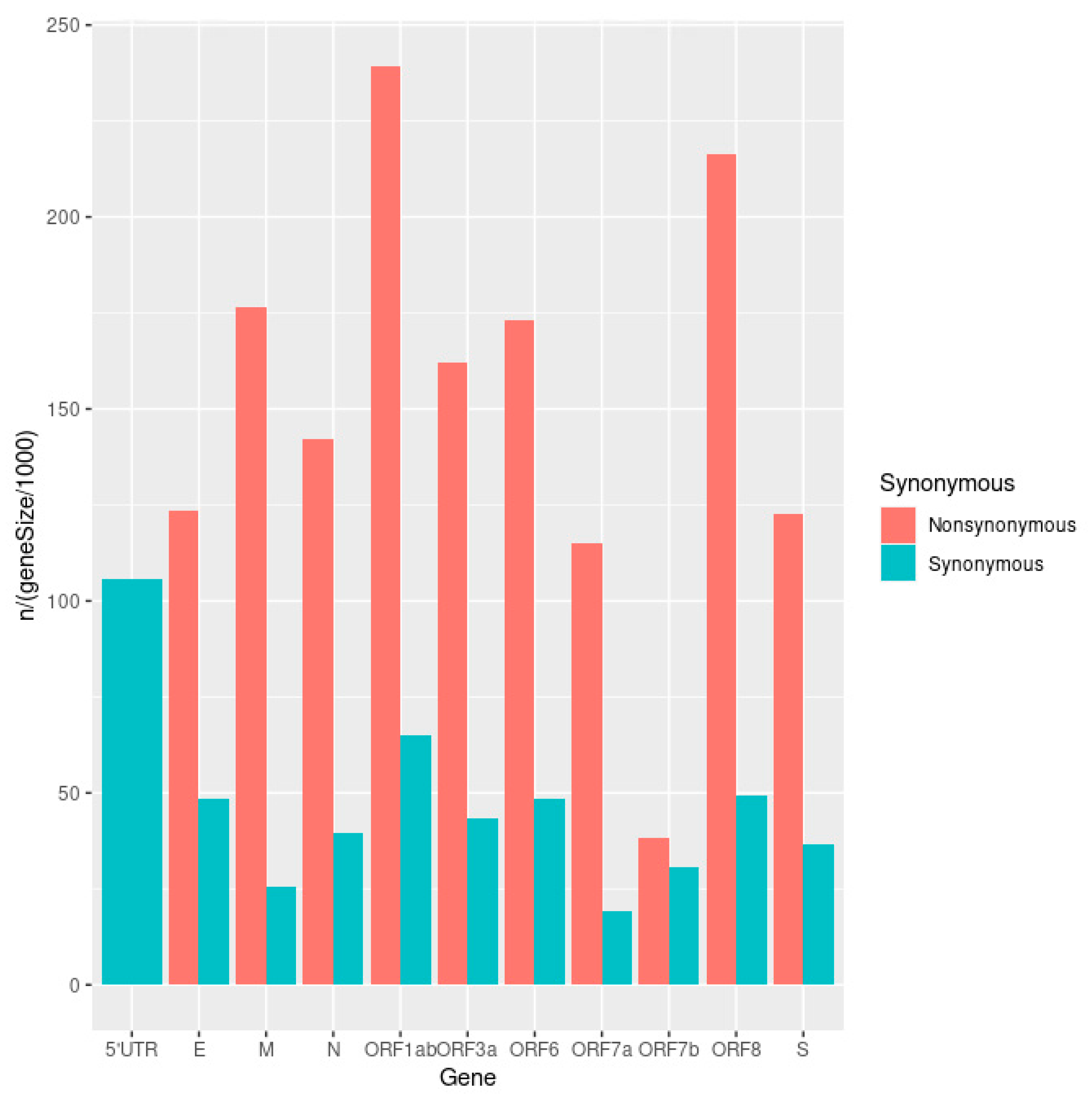
3.2. The mutational Landscape of SARS-CoV-2
3.3. Modelling SARS-CoV-2 Intrahost Diversity
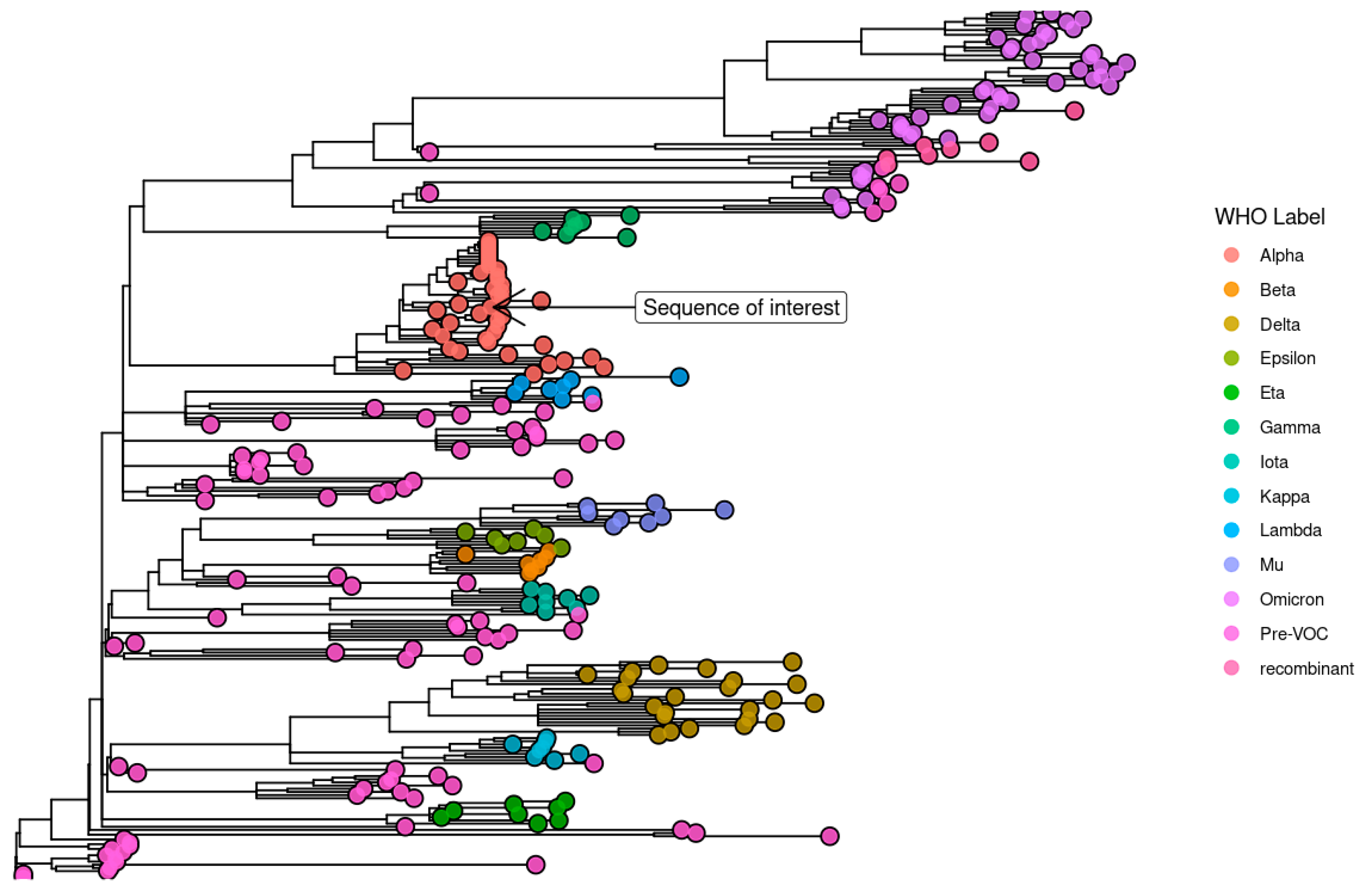
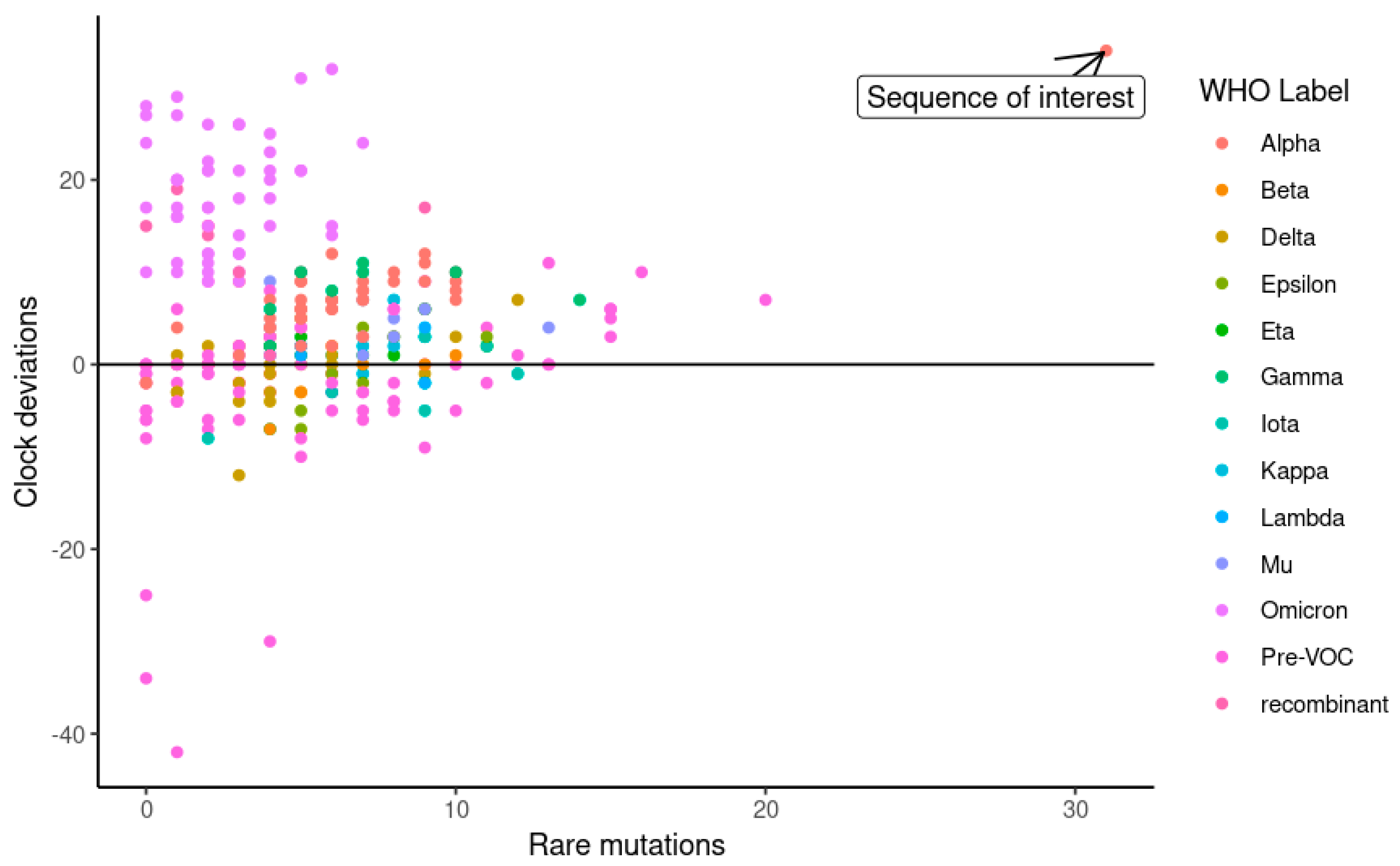
3.4. The Identification of an Unusual Viral Genome from an Immunosuppressed Host
4. Discussion
4.1. A Weak Association between SARS-CoV-2 Intra-Host Diversity and Host Factors
4.2. A Case of Chronic SARS-CoV-2 Infection in an Immunocompromised Woman with Hashimoto’s Thyroiditis
4.3. Rare, High Impact Mutations of Nonstructural Proteins and Replication Organelle Constituents in Chronic Infection
4.4. ORF3a Truncation in Chronic Infection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Wang, H.; Paulson, K.R.; Pease, S.A.; Watson, S.; Comfort, H.; Zheng, P. Estimating excess mortality due to the COVID-19 pandemic: A systematic analysis of COVID-19-related mortality, 2020–2021. Lancet 2022, 399, 1513–1536. [Google Scholar] [CrossRef] [PubMed]
- McLean, G.; Kamil, J.; Lee, B.; Moore, P.; Schulz, T.F.; Muik, A. The Impact of Evolving SARS-CoV-2 Mutations and Variants on COVID-19 Vaccines. mBio 2022, 13, e02979-21. [Google Scholar] [CrossRef]
- Harrison, N.L.; Sachs, J.D. Reply to Garry: The origin of SARS-CoV-2 remains unresolved. Proc. Natl. Acad. Sci. USA 2022, 119, e2215826119. [Google Scholar] [CrossRef] [PubMed]
- Burki, T. The origin of SARS-CoV-2 variants of concern. Lancet Infect. Dis. 2022, 22, 174–175. [Google Scholar] [CrossRef]
- Morales, A.C.; Rice, A.M.; Ho, A.T.; Mordstein, C.; Mühlhausen, S.; Watson, S. Causes and Consequences of Purifying Selection on SARS-CoV-2. Genome Biol. Evol. 2021, 13, evab196. [Google Scholar] [CrossRef]
- Li, J.; Du, P.; Yang, L.; Zhang, J.; Song, C.; Chen, D. Two-step fitness selection for intra-host variations in SARS-CoV-2. Cell Rep. 2022, 38, 110205. Available online: https://www.cell.com/cell-reports/abstract/S2211-1247(21)01709-5 (accessed on 14 February 2023). [CrossRef]
- Bendall, E.E.; Callear, A.P.; Getz, A.; Goforth, K.; Edwards, D.; Monto, A.S. Rapid transmission and tight bottlenecks constrain the evolution of highly transmissible SARS-CoV-2 variants. Nat. Commun. 2023, 14, 272. [Google Scholar] [CrossRef] [PubMed]
- Lythgoe, K.A.; Hall, M.; Ferretti, L.; de Cesare, M.; MacIntyre-Cockett, G.; Trebes, A. SARS-CoV-2 within-host diversity and transmission. Science 2021, 372, eabg0821. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, Y.; Sun, W.; Zhang, L.; Ji, J.; Zhang, Z. Population Bottlenecks and Intra-Host Evolution during Human-to-Human Transmission of SARS-CoV-2. Front. Med. 2021, 8, 585358. Available online: https://www.frontiersin.org/articles/10.3389/fmed.2021.585358 (accessed on 14 February 2023). [CrossRef]
- Tai, J.H.; Sun, H.Y.; Tseng, Y.C.; Li, G.; Chang, S.Y.; Yeh, S.H. Contrasting Patterns in the Early Stage of SARS-CoV-2 Evolution between Humans and Minks. Mol. Biol. Evol. 2022, 39, msac156. [Google Scholar] [CrossRef]
- MacLean, O.A.; Lytras, S.; Weaver, S.; Singer, J.B.; Boni, M.F.; Lemey, P. Natural selection in the evolution of SARS-CoV-2 in bats created a generalist virus and highly capable human pathogen. PLoS Biol. 2021, 19, e3001115. [Google Scholar] [CrossRef] [PubMed]
- Konings, F.; Perkins, M.D.; Kuhn, J.H.; Pallen, M.J.; Alm, E.J.; Archer, B.N. SARS-CoV-2 Variants of Interest and Concern naming scheme conducive for global discourse. Nat. Microbiol. 2021, 6, 821–823. [Google Scholar] [CrossRef]
- Tay, J.H.; Porter, A.F.; Wirth, W.; Duchene, S. The Emergence of SARS-CoV-2 Variants of Concern Is Driven by Acceleration of the Substitution Rate. Mol. Biol. Evol. 2022, 39, msac013. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hannon, W.W.; Loes, A.N.; Hauser, K.; Dillen, J.R. Shifting mutational constraints in the SARS-CoV-2 receptor-binding domain during viral evolution. Science 2022, 377, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Chaguza, C.; Hahn, A.M.; Petrone, M.E.; Zhou, S.; Ferguson, D.; Breban, M.I. Accelerated SARS-CoV-2 intrahost evolution leading to distinct genotypes during chronic infection. Cell Rep. Med. 2023, 4, 100943. Available online: https://www.cell.com/cell-reports-medicine/abstract/S2666-379100035-6 (accessed on 16 February 2023). [CrossRef]
- Corey, L.; Beyrer, C.; Cohen, M.S.; Michael, N.L.; Bedford, T.; Rolland, M. SARS-CoV-2 Variants in Patients with Immunosuppression. N. Engl. J. Med. 2021, 385, 562–566. [Google Scholar] [CrossRef]
- Harari, S.; Tahor, M.; Rutsinsky, N.; Meijer, S.; Miller, D.; Henig, O. Drivers of adaptive evolution during chronic SARS-CoV-2 infections. Nat. Med. 2022, 28, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Rockett, R.; Basile, K.; Maddocks, S.; Fong, W.; Agius, J.E.; Johnson-Mackinnon, J. Resistance Mutations in SARS-CoV-2 Delta Variant after Sotrovimab Use. N. Engl. J. Med. 2022, 386, 1477–1479. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. In Proceedings of the 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), Rio de Janeiro, Brazil, 20–24 May 2019; Available online: https://ieeexplore.ieee.org/document/8820962 (accessed on 14 March 2023).
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [Green Version]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Zhao, L.; Illingworth, C.J.R. Measurements of intrahost viral diversity require an unbiased diversity metric. Virus Evol. 2019, 5, vey041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Ma, L.; Zou, D.; Tian, D.; Li, C.; Zhu, J. The Global Landscape of SARS-CoV-2 Genomes, Variants, and Haplotypes in 2019nCoVR. Genom. Proteom. Bioinform. 2020, 18, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díez-Fuertes, F.; Iglesias-Caballero, M.; García-Pérez, J.; Monzón, S.; Jiménez, P.; Varona, S. A Founder Effect Led Early SARS-CoV-2 Transmission in Spain. J. Virol. 2021, 95, e01583-20. [Google Scholar] [CrossRef]
- Cele, S.; Karim, F.; Lustig, G.; San, J.E.; Hermanus, T.; Tegally, H. SARS-CoV-2 prolonged infection during advanced HIV disease evolves extensive immune escape. Cell Host Microbe 2022, 30, 154–162. [Google Scholar] [CrossRef]
- Iketani, S.; Mohri, H.; Culbertson, B.; Hong, S.J.; Duan, Y.; Luck, M.I. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature 2023, 613, 558–564. [Google Scholar] [CrossRef]
- Ricciardi, S.; Guarino, A.M.; Giaquinto, L.; Polishchuk, E.V.; Santoro, M.; Di Tullio, G. The role of NSP6 in the biogenesis of the SARS-CoV-2 replication organelle. Nature 2022, 606, 761–768. [Google Scholar] [CrossRef]
- Li, Y.; Pustovalova, Y.; Shi, W.; Gorbatyuk, O.; Sreeramulu, S.; Schwalbe, H. Crystal structure of the CoV-Y domain of SARS-CoV-2 nonstructural protein 3. Sci. Rep. 2023, 13, 2890. [Google Scholar] [CrossRef] [PubMed]
- Faizan, M.I.; Chaudhuri, R.; Sagar, S.; Albogami, S.; Chaudhary, N.; Azmi, I. NSP4 and ORF9b of SARS-CoV-2 Induce Pro-Inflammatory Mitochondrial DNA Release in Inner Membrane-Derived Vesicles. Cells 2022, 11, 2969. [Google Scholar] [CrossRef] [PubMed]
- Nemudryi, A.; Nemudraia, A.; Wiegand, T.; Nichols, J.; Snyder, D.T.; Hedges, J.F. SARS-CoV-2 genomic surveillance identifies naturally occurring truncation of ORF7a that limits immune suppression. Cell Rep. 2021, 35, 109197. [Google Scholar] [CrossRef]
- Valcarcel, A.; Bensussen, A.; Álvarez-Buylla, E.R.; Díaz, J. Structural Analysis of SARS-CoV-2 ORF8 Protein: Pathogenic and Therapeutic Implications. Front. Genet. 2021, 12, 693227. Available online: https://www.frontiersin.org/articles/10.3389/fgene.2021.693227 (accessed on 15 March 2023). [CrossRef] [PubMed]
- Firth, A.E. A putative new SARS-CoV protein, 3c, encoded in an ORF overlapping ORF3a. J. Gen. Virol. 2020, 101, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Finkel, Y.; Mizrahi, O.; Nachshon, A.; Weingarten-Gabbay, S.; Morgenstern, D.; Yahalom-Ronen, Y. The coding capacity of SARS-CoV-2. Nature 2021, 589, 125–130. [Google Scholar] [CrossRef]
- Gupta, S.; Mallick, D.; Banerjee, K.; Mukherjee, S.; Sarkar, S.; Lee, S.T. D155Y substitution of SARS-CoV-2 ORF3a weakens binding with Caveolin-1. Comput. Struct. Biotechnol. J. 2022, 20, 766–778. [Google Scholar] [CrossRef]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef]
- Zhang, J.; Ejikemeuwa, A.; Gerzanich, V.; Nasr, M.; Tang, Q.; Simard, J.M. Understanding the Role of SARS-CoV-2 ORF3a in Viral Pathogenesis and COVID-19. Front. Microbiol. 2022, 13, 854567. Available online: https://www.frontiersin.org/articles/10.3389/fmicb.2022.854567 (accessed on 15 March 2023). [CrossRef]
- Wang, R.; Yang, X.; Chang, M.; Xue, Z.; Wang, W.; Bai, L. ORF3a Protein of Severe Acute Respiratory Syndrome Coronavirus 2 Inhibits Interferon-Activated Janus Kinase/Signal Transducer and Activator of Transcription Signaling via Elevating Suppressor of Cytokine Signaling 1. Front. Microbiol. 2021, 12, 752597. Available online: https://www.frontiersin.org/articles/10.3389/fmicb.2021.752597 (accessed on 15 March 2023). [CrossRef]
- Su, W.Q.; Yu, X.J.; Zhou, C.M. SARS-CoV-2 ORF3a Induces Incomplete Autophagy via the Unfolded Protein Response. Viruses 2021, 13, 2467. [Google Scholar] [CrossRef]
- Miao, G.; Zhao, H.; Li, Y.; Ji, M.; Chen, Y.; Shi, Y. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 2021, 56, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Redondo, N.; Zaldívar-López, S.; Garrido, J.J.; Montoya, M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front. Immunol. 2021, 12, 708264. Available online: https://www.frontiersin.org/articles/10.3389/fimmu.2021.708264 (accessed on 15 March 2023). [CrossRef] [PubMed]
- Weigang, S.; Fuchs, J.; Zimmer, G.; Schnepf, D.; Kern, L.; Beer, J. Within-host evolution of SARS-CoV-2 in an immunosuppressed COVID-19 patient as a source of immune escape variants. Nat. Commun. 2021, 12, 6405. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Immunocompetent (N = 1165) | Immunosuppressed ᵃ (N = 30) |
|---|---|---|
| Males (fraction) | 692/1165 | 20/31 |
| Age, mean ± SE | 35.6 ± 0.5 | 60.8 ± 3.1 |
| Diabetes mellitus ᵃ, % | 7.20% | 41.90% |
| Vaccinated ᵃ, % | 90.40% | 96.70% |
| Smoker ᵃ, % (N.A. %) | 10.4% (24%) | 6.4% (0%) |
| Alpha variant (fraction) | 693/1165 | 22/30 |
| Delta variant (fraction) | 236/1165 | 4/30 |
| Other VOC (fraction) | 7/1165 | 0/30 |
| Pre-VOC (fraction) | 199/1165 | 4/30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shapira, G.; Patalon, T.; Gazit, S.; Shomron, N. Immunosuppression as a Hub for SARS-CoV-2 Mutational Drift. Viruses 2023, 15, 855. https://doi.org/10.3390/v15040855
Shapira G, Patalon T, Gazit S, Shomron N. Immunosuppression as a Hub for SARS-CoV-2 Mutational Drift. Viruses. 2023; 15(4):855. https://doi.org/10.3390/v15040855
Chicago/Turabian StyleShapira, Guy, Tal Patalon, Sivan Gazit, and Noam Shomron. 2023. "Immunosuppression as a Hub for SARS-CoV-2 Mutational Drift" Viruses 15, no. 4: 855. https://doi.org/10.3390/v15040855