
The EBV Gastric Cancer Resource (EBV-GCR): A Suite of Tools for Investigating EBV-Associated Human Gastric Carcinogenesis
 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Implementation and Software
2.2. Sample Collection and Ethics
2.3. Data Sources for Cellular mRNA and miRNA Expression Levels, Patient Cohort Composition, and Analysis Workflow
2.4. Data Sources for Viral mRNA and miRNA Expression Levels, Patient Cohort Composition, and Analysis Workflow
2.5. Data Sources for Immune Landscape Features, Patient Cohort Composition, and Analysis Workflow
2.6. Data Sources for Patient Survival, Patient Cohort Composition, and Analysis Workflow
2.7. Data Sources for DNA Methylation Levels, Patient Cohort Composition, and Analysis Workflow
2.8. Data Sources for Single-Cell Analysis, Patient Cohort Composition, and Analysis Workflow
3. Results
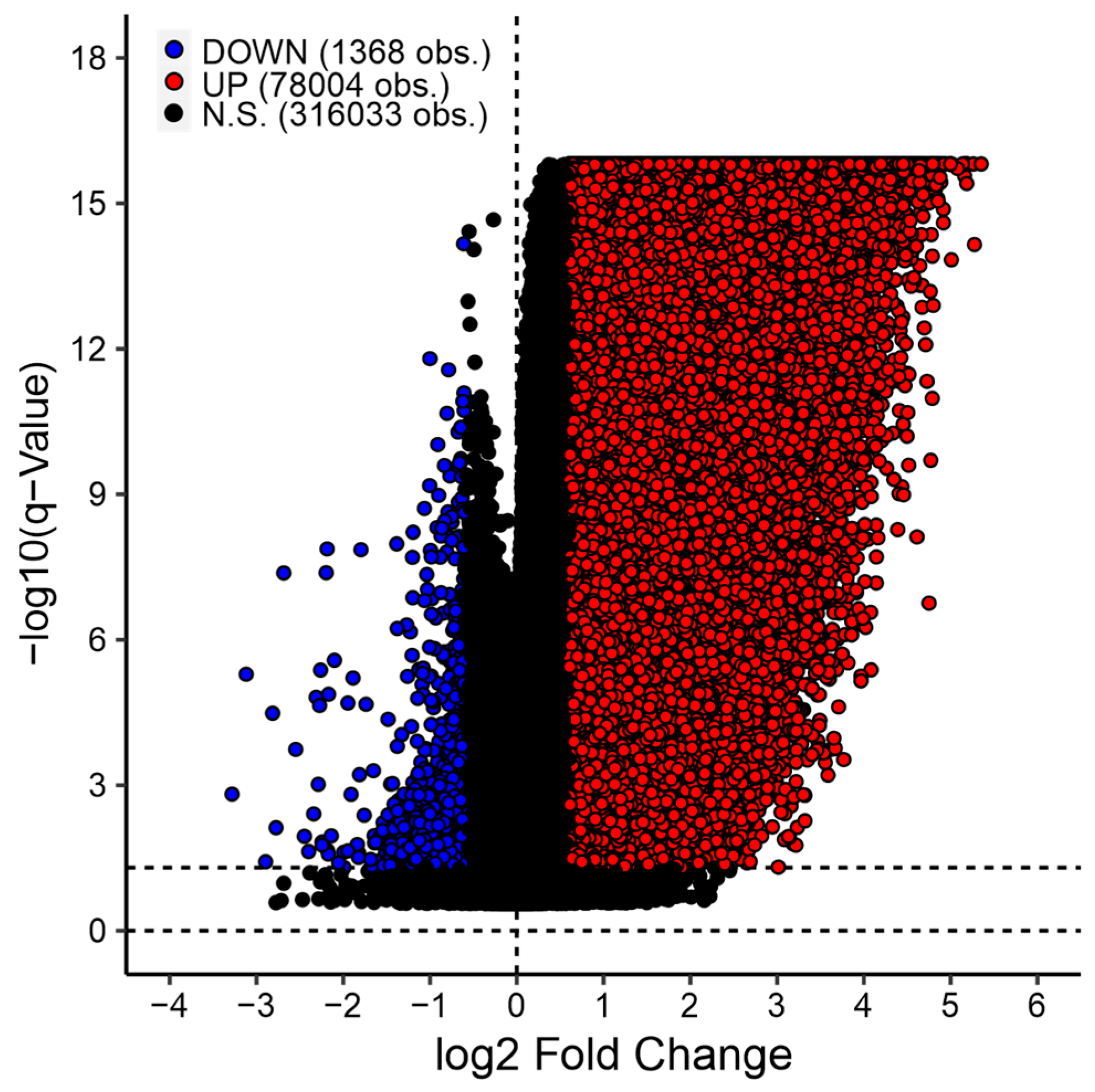
3.1. Differentially Expressed Gene Analysis
3.2. Correlations between Cellular and Viral Gene Expressions
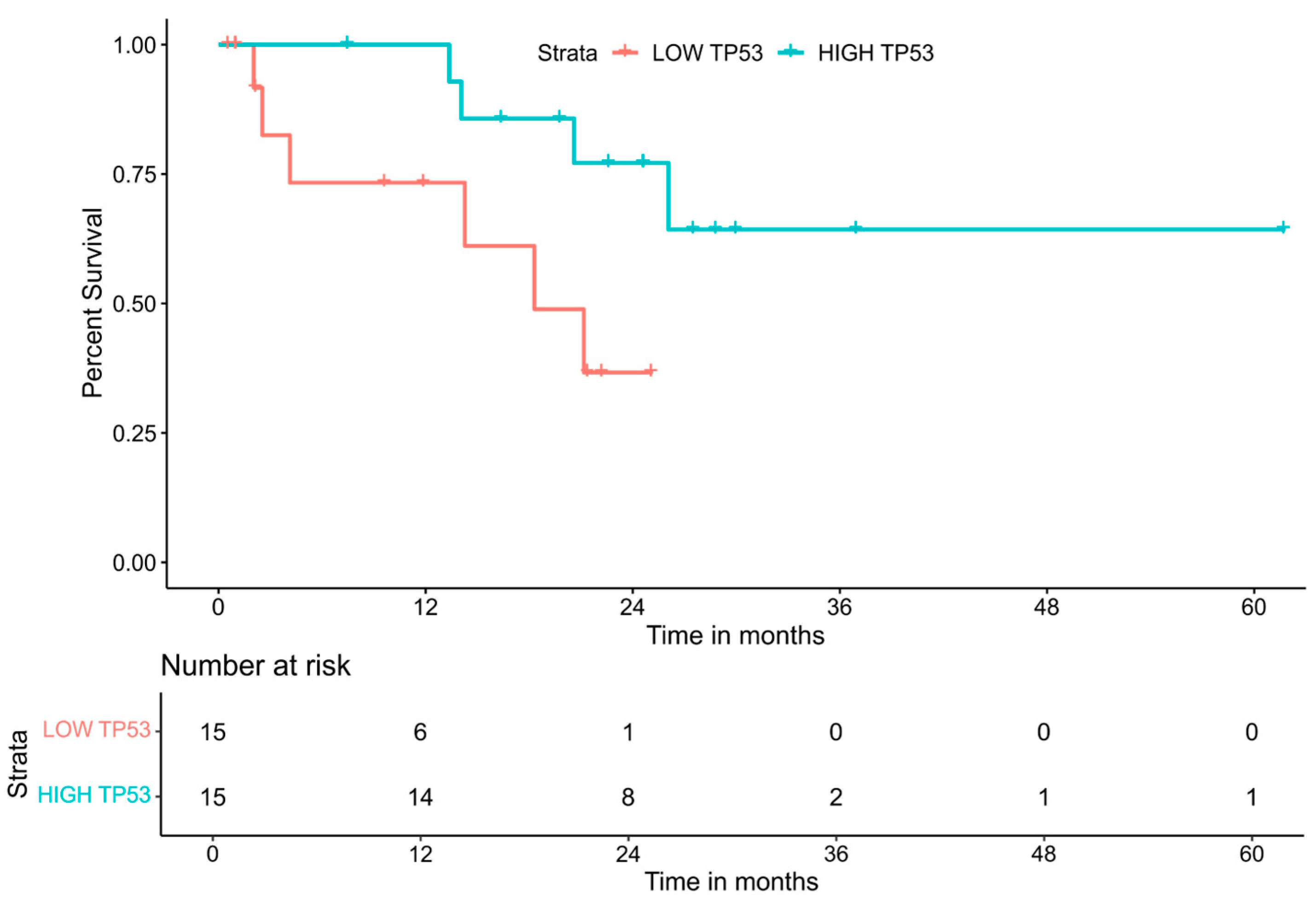
3.3. The Impact of Gene Expression Levels on Overall Survival
3.4. The Correlation of Gene Expression Levels with Immune Landscape Features
3.5. Differential Probe Methylation Analysis
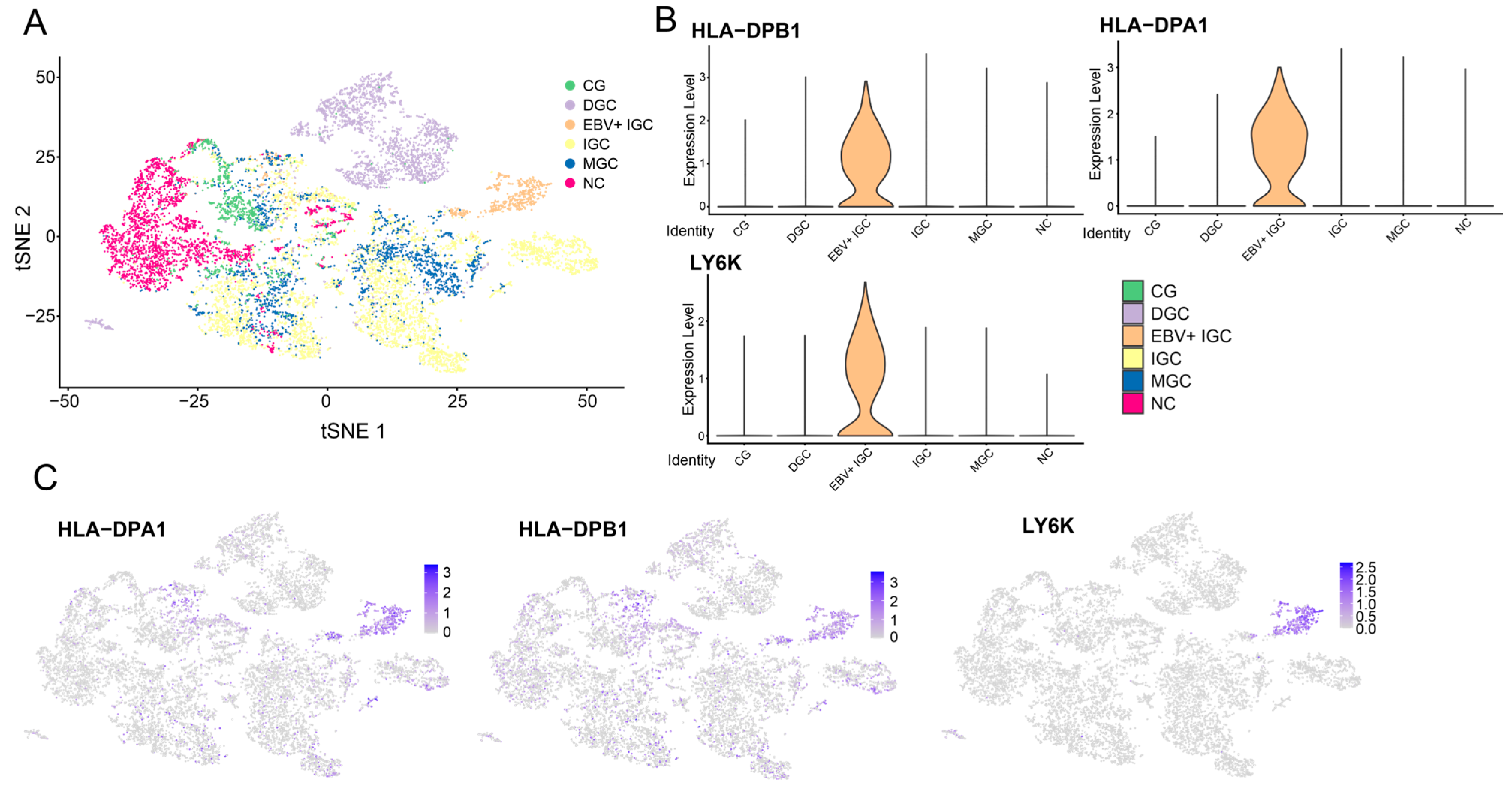
3.6. Single-Cell Analysis
3.7. Example Case Study
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowe, M.; Zuo, J. Immune Responses to Epstein-Barr Virus: Molecular Interactions in the Virus Evasion of CD8+ T Cell Immunity. Microbes Infect. 2010, 12, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Rickinson, A.B. Epstein-Barr Virus: 40 Years On. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef]
- Eichelberg, M.R.; Welch, R.; Guidry, J.T.; Ali, A.; Ohashi, M.; Makielski, K.R.; McChesney, K.; Van Sciver, N.; Lambert, P.F.; Keleș, S.; et al. Epstein-Barr Virus Infection Promotes Epithelial Cell Growth by Attenuating Differentiation-Dependent Exit from the Cell Cycle. mBio 2019, 10, e01332-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, J.; Imai, S.; Oda, T.; Kojima, T.; Okita, K.; Takada, K. Epstein-Barr Virus Promotes Epithelial Cell Growth in the Absence of EBNA2 and LMP1 Expression. J. Virol. 1999, 73, 1286–1292. [Google Scholar] [CrossRef] [Green Version]
- Albanese, M.; Tagawa, T.; Hammerschmidt, W. Strategies of Epstein-Barr Virus to Evade Innate Antiviral Immunity of Its Human Host. Front. Microbiol. 2022, 13, 955603. [Google Scholar] [CrossRef]
- Yin, H.; Qu, J.; Peng, Q.; Gan, R. Molecular Mechanisms of EBV-Driven Cell Cycle Progression and Oncogenesis. Med. Microbiol. Immunol. 2019, 208, 573–583. [Google Scholar] [CrossRef] [Green Version]
- Prusinkiewicz, M.A.; Mymryk, J.S. Metabolic Control by DNA Tumor Virus-Encoded Proteins. Pathogens 2021, 10, 560. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rickinson, A. The Global Landscape of EBV-Associated Tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Tay, J.K.; Loh, C.J.L.; Chu, A.J.M.; Yeong, J.P.S.; Lim, C.M.; Toh, H.C. Epstein-Barr Virus Epithelial Cancers-A Comprehensive Understanding to Drive Novel Therapies. Front. Immunol. 2021, 12, 734293. [Google Scholar] [CrossRef]
- Fukayama, M.; Ushiku, T. Epstein-Barr Virus-Associated Gastric Carcinoma. Pathol. Res. Pract. 2011, 207, 529–537. [Google Scholar] [CrossRef]
- Akiba, S.; Koriyama, C.; Herrera-Goepfert, R.; Eizuru, Y. Epstein-Barr Virus Associated Gastric Carcinoma: Epidemiological and Clinicopathological Features. Cancer Sci. 2008, 99, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, F.; Gameiro, S.F.; Tessier, T.M.; Maciver, A.H.; Mymryk, J.S. High Levels of Class I Major Histocompatibility Complex MRNA Are Present in Epstein-Barr Virus-Associated Gastric Adenocarcinomas. Cells 2020, 9, 499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghasemi, F.; Tessier, T.M.; Gameiro, S.F.; Maciver, A.H.; Cecchini, M.J.; Mymryk, J.S. High MHC-II Expression in Epstein-Barr Virus-Associated Gastric Cancers Suggests That Tumor Cells Serve an Important Role in Antigen Presentation. Sci. Rep. 2020, 10, 14786. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Kang, M.-S.; Kim, K.-M. Epstein-Barr Virus-Associated Gastric Carcinoma and Specific Features of the Accompanying Immune Response. J. Gastric Cancer 2016, 16, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Li, J.; Hao, Y.; Nie, Y.; Li, Z.; Qian, M.; Liang, Q.; Yu, J.; Zeng, M.; Wu, K. Differentiated Tumor Immune Microenvironment of Epstein-Barr Virus-Associated and Negative Gastric Cancer: Implication in Prognosis and Immunotherapy. Oncotarget 2017, 8, 67094–67103. [Google Scholar] [CrossRef] [Green Version]
- Salnikov, M.; Prusinkiewicz, M.A.; Lin, S.; Ghasemi, F.; Cecchini, M.J.; Mymryk, J.S. Tumor-Infiltrating T Cells in EBV-Associated Gastric Carcinomas Exhibit High Levels of Multiple Markers of Activation, Effector Gene Expression, and Exhaustion. Viruses 2023, 15, 176. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, T.; Zhou, Y.; Cheng, A.S.L.; Yu, J.; To, K.F.; Kang, W. The Oncogenic Role of Epstein-Barr Virus-Encoded MicroRNAs in Epstein-Barr Virus-Associated Gastric Carcinoma. J. Cell. Mol. Med. 2018, 22, 38–45. [Google Scholar] [CrossRef]
- Morales-Sanchez, A.; Fuentes-Panana, E.M. Epstein-Barr Virus-Associated Gastric Cancer and Potential Mechanisms of Oncogenesis. Curr. Cancer Drug Targets 2017, 17, 534–554. [Google Scholar] [CrossRef]
- Tavakoli, A.; Monavari, S.H.; Solaymani Mohammadi, F.; Kiani, S.J.; Armat, S.; Farahmand, M. Association between Epstein-Barr Virus Infection and Gastric Cancer: A Systematic Review and Meta-Analysis. BMC Cancer 2020, 20, 493. [Google Scholar] [CrossRef]
- Shinozaki-Ushiku, A.; Kunita, A.; Fukayama, M. Update on Epstein-Barr Virus and Gastric Cancer (Review). Int. J. Oncol. 2015, 46, 1421–1434. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Snapper, C.M. Epstein Barr Virus: Development of Vaccines and Immune Cell Therapy for EBV-Associated Diseases. Front. Immunol. 2021, 12, 734471. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.J.; Kim, J.Y.; Long, N.P.; Min, J.E.; Kim, H.M.; Yoon, J.H.; Anh, N.H.; Park, M.C.; Kwon, S.W.; Lee, S.K. Comprehensive Multi-Omics Analysis Reveals Aberrant Metabolism of Epstein-Barr-Virus-Associated Gastric Carcinoma. Cells 2019, 8, 1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, E.M.; Gewurz, B.E. Epstein-Barr Virus Oncoprotein-Driven B Cell Metabolism Remodeling. PLoS Pathog. 2022, 18, e1010254. [Google Scholar] [CrossRef]
- Hassani, A.; Khan, G. What Do Animal Models Tell Us about the Role of EBV in the Pathogenesis of Multiple Sclerosis? Front. Immunol. 2022, 13, 1036155. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.H.; Baiocchi, R.A. Murine Models of Epstein-Barr Virus-Associated Lymphomagenesis. ILAR J. 2016, 57, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Cherniack, A.D.; Sanchez-Vega, F.; Seoane, J.A.; Farshidfar, F.; Bowlby, R.; Islam, M.; et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Cancer Cell 2018, 33, 721–735.e8. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Li, H.; Jiang, S.; Li, R.; Li, W.; Chen, H.; Bo, X. A Survey and Evaluation of Web-Based Tools/Databases for Variant Analysis of TCGA Data. Brief. Bioinform. 2019, 20, 1524–1541. [Google Scholar] [CrossRef]
- Salnikov, M.; Gameiro, S.F.; Zeng, P.Y.F.; Barrett, J.W.; Nichols, A.C.; Mymryk, J.S. The HPV Induced Cancer Resource (THInCR): A Suite of Tools for Investigating HPV-Dependent Human Carcinogenesis. mSphere 2022, 7, e0031722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hu, S.; Min, M.; Ni, Y.; Lu, Z.; Sun, X.; Wu, J.; Liu, B.; Ying, X.; Liu, Y. Dissecting Transcriptional Heterogeneity in Primary Gastric Adenocarcinoma by Single Cell RNA Sequencing. Gut 2021, 70, 464–475. [Google Scholar] [CrossRef]
- Chakravorty, S.; Yan, B.; Wang, C.; Wang, L.; Quaid, J.T.; Lin, C.F.; Briggs, S.D.; Majumder, J.; Canaria, D.A.; Chauss, D.; et al. Integrated Pan-Cancer Map of EBV-Associated Neoplasms Reveals Functional Host-Virus Interactions. Cancer Res. 2019, 79, 6010–6023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungerleider, N.; Bullard, W.; Kara, M.; Wang, X.; Roberts, C.; Renne, R.; Tibbetts, S.; Flemington, E.K. EBV MiRNAs Are Potent Effectors of Tumor Cell Transcriptome Remodeling in Promoting Immune Escape. PLoS Pathog. 2021, 17, e1009217. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.S. Epstein-Barr Virus: A Master Epigenetic Manipulator. Curr. Opin. Virol. 2017, 26, 74–80. [Google Scholar] [CrossRef] [PubMed]
- De Re, V.; Caggiari, L.; De Zorzi, M.; Fanotto, V.; Miolo, G.; Puglisi, F.; Cannizzaro, R.; Canzonieri, V.; Steffan, A.; Farruggia, P.; et al. Epstein-Barr Virus BART MicroRNAs in EBV- Associated Hodgkin Lymphoma and Gastric Cancer. Infect. Agents Cancer 2020, 15, 42. [Google Scholar] [CrossRef]
- Polakovicova, I.; Jerez, S.; Wichmann, I.A.; Sandoval-Bórquez, A.; Carrasco-Véliz, N.; Corvalán, A.H. Role of MicroRNAs and Exosomes in Helicobacter Pylori and Epstein-Barr Virus Associated Gastric Cancers. Front. Microbiol. 2018, 9, 636. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, J.; Iizasa, H.; Yoshiyama, H.; Nakamura, M.; Saito, M.; Sasaki, S.; Shimokuri, K.; Yanagihara, M.; Sakai, K.; Suehiro, Y.; et al. The Role of Epigenetic Regulation in Epstein-Barr Virus-Associated Gastric Cancer. Int. J. Mol. Sci. 2017, 18, 1606. [Google Scholar] [CrossRef] [Green Version]
- Nagy, Á.; Munkácsy, G.; Győrffy, B. Pancancer Survival Analysis of Cancer Hallmark Genes. Sci. Rep. 2021, 11, 6047. [Google Scholar] [CrossRef]
- Chen, X.; Pan, C.; Xu, C.; Sun, Y.; Geng, Y.; Kong, L.; Xiao, X.; Zhao, Z.; Zhou, W.; Huang, L.; et al. Identification of Survival-associated Key Genes and Long Non-coding RNAs in Glioblastoma Multiforme by Weighted Gene Co-expression Network Analysis. Int. J. Mol. Med. 2019, 43, 1709–1722. [Google Scholar] [CrossRef]
- Machnik, M.; Cylwa, R.; Kiełczewski, K.; Biecek, P.; Liloglou, T.; Mackiewicz, A.; Oleksiewicz, U. The Expression Signature of Cancer-Associated KRAB-ZNF Factors Identified in TCGA Pan-Cancer Transcriptomic Data. Mol. Oncol. 2019, 13, 701–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusinkiewicz, M.A.; Gameiro, S.F.; Ghasemi, F.; Dodge, M.J.; Zeng, P.Y.F.; Maekebay, H.; Barrett, J.W.; Nichols, A.C.; Mymryk, J.S. Survival-Associated Metabolic Genes in Human Papillomavirus-Positive Head and Neck Cancers. Cancers 2020, 12, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, Z. The History and Advances in Cancer Immunotherapy: Understanding the Characteristics of Tumor-Infiltrating Immune Cells and Their Therapeutic Implications. Cell Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Tumor Immunology and Tumor Evolution: Intertwined Histories. Immunity 2020, 52, 55–81. [Google Scholar] [CrossRef]
- Zhang, S.; Lv, M.; Cheng, Y.; Wang, S.; Li, C.; Qu, X. Immune Landscape of Advanced Gastric Cancer Tumor Microenvironment Identifies Immunotherapeutic Relevant Gene Signature. BMC Cancer 2021, 21, 1324. [Google Scholar] [CrossRef]
- Zhang, C.; Li, D.; Yu, R.; Li, C.; Song, Y.; Chen, X.; Fan, Y.; Liu, Y.; Qu, X. Immune Landscape of Gastric Carcinoma Tumor Microenvironment Identifies a Peritoneal Relapse Relevant Immune Signature. Front. Immunol. 2021, 12, 651033. [Google Scholar] [CrossRef]
- Deng, S.-Z.; Wang, X.-X.; Zhao, X.-Y.; Bai, Y.-M.; Zhang, H.-M. Exploration of the Tumor Immune Landscape and Identification of Two Novel Immunotherapy-Related Genes for Epstein-Barr Virus-Associated Gastric Carcinoma via Integrated Bioinformatics Analysis. Front. Surg. 2022, 9, 898733. [Google Scholar] [CrossRef]
- Stanland, L.J.; Luftig, M.A. The Role of EBV-Induced Hypermethylation in Gastric Cancer Tumorigenesis. Viruses 2020, 12, 1222. [Google Scholar] [CrossRef]
- Jing, J.-J.; Li, H.; Wang, Z.-Y.; Zhou, H.; Sun, L.-P.; Yuan, Y. Aberrantly Methylated-Differentially Expressed Genes and Pathways in Epstein-Barr Virus-Associated Gastric Cancer. Future Oncol. 2020, 16, 187–197. [Google Scholar] [CrossRef]
- Zhu, M.; Liang, Q.; Chen, T.; Kong, Q.; Ye, G.; Yu, S.; Li, X.; He, Q.; Liu, H.; Hu, Y.; et al. Identification and Validation of Methylated Differentially Expressed MiRNAs and Immune Infiltrate Profile in EBV-Associated Gastric Cancer. Clin. Epigenetics 2021, 13, 22. [Google Scholar] [CrossRef]
- He, D.; Zhang, Y.; Zhang, N.; Zhou, L.; Chen, J.; Jiang, Y.; Shao, C. Aberrant Gene Promoter Methylation of P16, FHIT, CRBP1, WWOX, and DLC-1 in Epstein-Barr Virus-Associated Gastric Carcinomas. Med. Oncol. 2015, 32, 92. [Google Scholar] [CrossRef]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lönnberg, T. A Practical Guide to Single-Cell RNA-Sequencing for Biomedical Research and Clinical Applications. Genome Med. 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.L.; Jones, R.J.; Kenney, S.C.; Rivenbark, A.G.; Tang, W.; Knight, E.R.; Coleman, W.B.; Gulley, M.L. Epstein-Barr Virus-Specific Methylation of Human Genes in Gastric Cancer Cells. Infect. Agents Cancer 2010, 5, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, R.H.; Dekroon, R.; Raab-Traub, N. Alterations in Cellular Expression in EBV Infected Epithelial Cell Lines and Tumors. PLoS Pathog. 2019, 15, e1008071. [Google Scholar] [CrossRef]
- Okabe, A.; Funata, S.; Matsusaka, K.; Namba, H.; Fukuyo, M.; Rahmutulla, B.; Oshima, M.; Iwama, A.; Fukayama, M.; Kaneda, A. Regulation of Tumour Related Genes by Dynamic Epigenetic Alteration at Enhancer Regions in Gastric Epithelial Cells Infected by Epstein-Barr Virus. Sci. Rep. 2017, 7, 7924. [Google Scholar] [CrossRef] [Green Version]
- Qian, Z.; Jiang, Y.; Shou, C.; Yu, J.; Huang, D.; Xie, H.; Zhou, L.; Chen, D.; Zheng, S. Validation of the DNA Methylation Landscape of TFF1/TFF2 in Gastric Cancer. Cancers 2022, 14, 5474. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ge, D.; Lu, C. The SMART App: An Interactive Web Application for Comprehensive DNA Methylation Analysis and Visualization. Epigenetics Chromatin 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, M.; Brägelmann, J.; Schultze, J.L.; Perner, S. Web-TCGA: An Online Platform for Integrated Analysis of Molecular Cancer Data Sets. BMC Bioinform. 2016, 17, 72. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # of Patient Samples Analyzed | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient Subset | mRNA-seq | miRNA-seq | Cellular mRNA vs. Viral mRNA | Cellular miRNA vs. Viral mRNA | Cellular mRNA vs. Viral miRNA | Cellular miRNA vs. Viral miRNA | Immune Comparisons—Cellular mRNA | Immune Comparisons—Cellular miRNA | Immune Comparisons—Viral mRNA/miRNA | Overall Survival—Cellular mRNA | Overall Survival—Cellular miRNA | Methylation |
| EBV | 30 | 29 | 26 | 25 | 29 | 30 | 30 | 29 | 26 (mRNA) 29 (miRNA) | 30 | 29 | 29 |
| MSI | 73 | 59 | NA | NA | NA | NA | 73 | 59 | NA | 72 | 58 | 58 |
| CIN | 223 | 203 | 223 | 201 | 220 | 199 | 202 | |||||
| GS | 60 | 46 | 60 | 46 | 49 | 45 | 46 | |||||
| POLE | 7 | 7 | 7 | 7 | 7 | 7 | 7 | |||||
| Normal | 35 | 41 | NA | NA | NA | NA | 18 | |||||
| Immune Landscape Features * | |
|---|---|
| Aneuploidy score | Monocytes |
| B cells memory | Neutrophils |
| B cells naive | NK cells activated |
| BCR evenness | NK cells resting |
| BCR richness | Nonsilent mutation rate |
| BCR Shannon | No. of segments |
| CTA score | Plasma cells |
| Dendritic cells | Proliferation |
| Dendritic cells activated | Silent mutation rate |
| Dendritic cells resting | SNV neoantigens |
| Eosinophils | Stromal fraction |
| Fraction altered | T cells CD4 memory activated |
| Homologous recombination defects | T cells CD4 memory resting |
| IFN gamma response | T cells CD4 Naive |
| Indel neoantigens | T cells CD8 |
| Intratumor heterogeneity | T cells follicular helper |
| Leukocyte fraction | T cells gamma delta |
| Lymphocyte infiltration signature score | T cells regulatory Tregs |
| Lymphocytes | TCR evenness |
| Macrophage regulation | TCR richness |
| Macrophages | TCR Shannon |
| Macrophages M0 | TGF beta response |
| Macrophages M1 | Th1 cells |
| Macrophages M2 | Th17 cells |
| Mast cells | Th2 cells |
| Mast cells activated | Wound healing |
| Mast cells resting | |
| Cellular mRNAs | Cellular miRNAs | |||||
|---|---|---|---|---|---|---|
| Viral mRNA | # of Positive Correlations | # of Negative Correlations | # of Non-Significant Correlations | # of Positive Correlations | # of Negative Correlations | # of Non-Significant Correlations |
| A73 | 0 | 4 | 20,527 | 0 | 0 | 1046 |
| BALF1 | 2 | 0 | 20,529 | 0 | 0 | 1046 |
| BALF2 | 2 | 1 | 20,528 | 5 | 63 | 978 |
| BALF3 | 0 | 0 | 20,531 | 0 | 6 | 1040 |
| BALF4 | 1105 | 1077 | 18,349 | 0 | 7 | 1039 |
| BALF5 | 852 | 952 | 18,727 | 2 | 50 | 994 |
| BARF0 | 1 | 0 | 20,530 | 0 | 0 | 1046 |
| BARF1 | 1 | 0 | 20,530 | 0 | 1 | 1045 |
| BNRF1 | 1 | 0 | 20,530 | 0 | 0 | 1046 |
| EBER1 | 1008 | 1140 | 18,383 | 36 | 4 | 1006 |
| EBNA-1 | 4 | 7 | 20,520 | 0 | 0 | 1046 |
| LF2 | 109 | 114 | 20,308 | 0 | 0 | 1046 |
| LMP2B | 0 | 0 | 20,531 | 0 | 2 | 1044 |
| RPMS1 | 740 | 526 | 19,265 | 5 | 16 | 1025 |
| Cellular mRNAs | Cellular miRNAs | |||||
|---|---|---|---|---|---|---|
| Viral mRNA | # of Positive Correlations | # of Negative Correlations | # of Non-Significant Correlations | # of Positive Correlations | # of Negative Correlations | # of Non-Significant Correlations |
| ebv-miR-BART10-3p | 1351 | 4371 | 14,809 | 166 | 19 | 861 |
| ebv-miR-BART11-5p | 6 | 19 | 20,506 | 5 | 2 | 1039 |
| ebv-miR-BART12 | 0 | 0 | 20,531 | 77 | 0 | 969 |
| ebv-miR-BART13-3p | 99 | 239 | 20,193 | 20 | 0 | 1026 |
| ebv-miR-BART13-5p | 328 | 169 | 20,034 | 0 | 3 | 1043 |
| ebv-miR-BART14-3p | 31 | 467 | 20,033 | 140 | 0 | 906 |
| ebv-miR-BART14-5p | 391 | 1864 | 18,276 | 237 | 5 | 804 |
| ebv-miR-BART15 | 939 | 2977 | 16,615 | 209 | 6 | 831 |
| ebv-miR-BART1-5p | 64 | 988 | 19,479 | 13 | 0 | 1033 |
| ebv-miR-BART17-3p | 519 | 2971 | 17,041 | 124 | 6 | 916 |
| ebv-miR-BART17-5p | 0 | 0 | 20,531 | 1 | 0 | 1045 |
| ebv-miR-BART18-3p | 315 | 212 | 20,004 | 0 | 0 | 1046 |
| ebv-miR-BART19-3p | 0 | 9 | 20,522 | 23 | 0 | 1023 |
| ebv-miR-BART19-5p | 706 | 3171 | 16,654 | 223 | 8 | 815 |
| ebv-miR-BART20-5p | 420 | 1538 | 18,573 | 43 | 0 | 1003 |
| ebv-miR-BART21-3p | 0 | 0 | 20,531 | 117 | 4 | 925 |
| ebv-miR-BART22 | 891 | 3928 | 15,712 | 123 | 17 | 906 |
| ebv-miR-BART2-3p | 233 | 1094 | 19,204 | 217 | 6 | 823 |
| ebv-miR-BART2-5p | 506 | 3218 | 16,807 | 155 | 5 | 886 |
| ebv-miR-BART3-3p | 147 | 524 | 19,860 | 71 | 0 | 975 |
| ebv-miR-BART4-3p | 116 | 388 | 20,027 | 121 | 4 | 921 |
| ebv-miR-BART4-5p | 241 | 1301 | 18,989 | 221 | 9 | 816 |
| ebv-miR-BART5-5p | 815 | 3885 | 15,831 | 162 | 16 | 868 |
| ebv-miR-BART6-3p | 606 | 2812 | 17,113 | 191 | 6 | 849 |
| ebv-miR-BART6-5p | 402 | 1066 | 19,063 | 16 | 0 | 1030 |
| ebv-miR-BART7-3p | 173 | 696 | 19,662 | 121 | 0 | 925 |
| ebv-miR-BART7-5p | 275 | 672 | 19,584 | 166 | 2 | 878 |
| ebv-miR-BART8-3p | 858 | 3550 | 16,123 | 129 | 16 | 901 |
| ebv-miR-BART8-5p | 570 | 2792 | 17,169 | 187 | 7 | 852 |
| ebv-miR-BART9-3p | 432 | 1766 | 18,333 | 29 | 6 | 1011 |
| ebv-miR-BART9-5p | 959 | 3215 | 16,357 | 148 | 4 | 894 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salnikov, M.Y.; Wang, E.; Christensen, E.; Prusinkiewicz, M.A.; Shooshtari, P.; Mymryk, J.S. The EBV Gastric Cancer Resource (EBV-GCR): A Suite of Tools for Investigating EBV-Associated Human Gastric Carcinogenesis. Viruses 2023, 15, 853. https://doi.org/10.3390/v15040853
Salnikov MY, Wang E, Christensen E, Prusinkiewicz MA, Shooshtari P, Mymryk JS. The EBV Gastric Cancer Resource (EBV-GCR): A Suite of Tools for Investigating EBV-Associated Human Gastric Carcinogenesis. Viruses. 2023; 15(4):853. https://doi.org/10.3390/v15040853
Chicago/Turabian StyleSalnikov, Mikhail Y., Eric Wang, Erik Christensen, Martin A. Prusinkiewicz, Parisa Shooshtari, and Joe S. Mymryk. 2023. "The EBV Gastric Cancer Resource (EBV-GCR): A Suite of Tools for Investigating EBV-Associated Human Gastric Carcinogenesis" Viruses 15, no. 4: 853. https://doi.org/10.3390/v15040853