
VirClust—A Tool for Hierarchical Clustering, Core Protein Detection and Annotation of (Prokaryotic) Viruses


Abstract
:1. Introduction
2. Materials and Methods
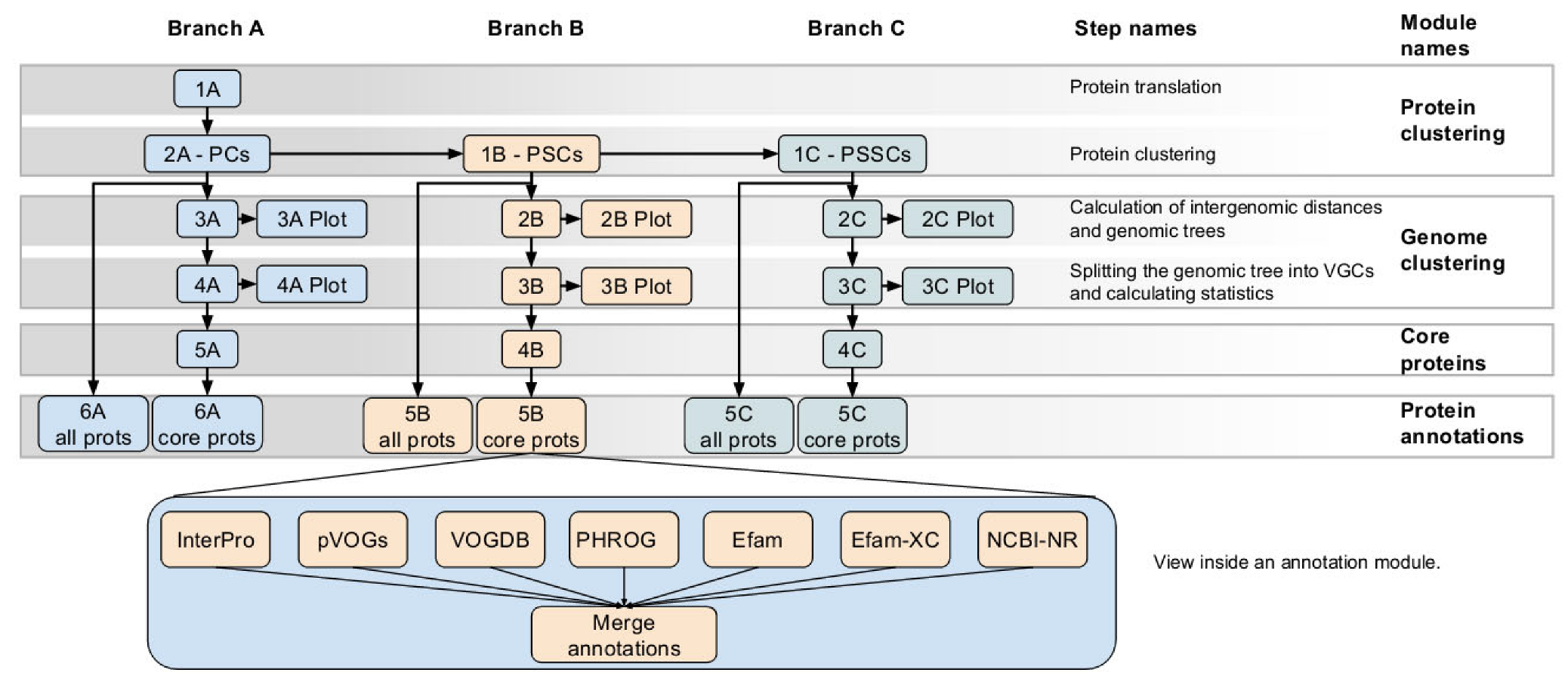
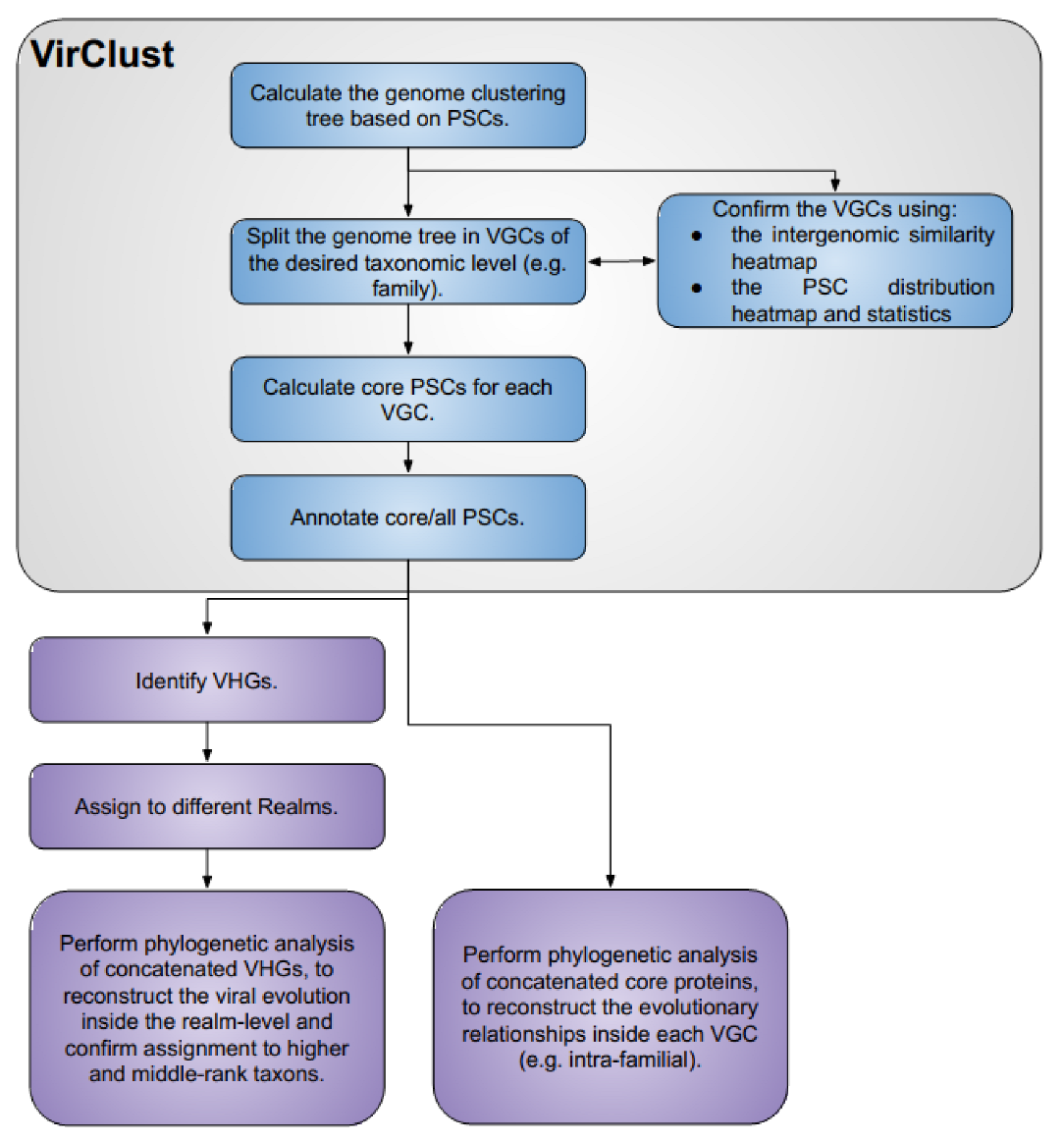
2.1. VirClust—Development and Workflow
2.1.1. Protein Clustering Module from Branch A
Gene Prediction and Protein Prediction—Step 1A
From Proteins to Protein Clusters—Step 2A
2.1.2. Protein Clustering Modules from Branch B and Branch C
From Protein Clusters to Protein Superclusters—Step 1B
From Protein Superclusters to Protein Super-Superclusters—Step 1C
2.1.3. Genome Clustering Modules from Branches A, B, and C
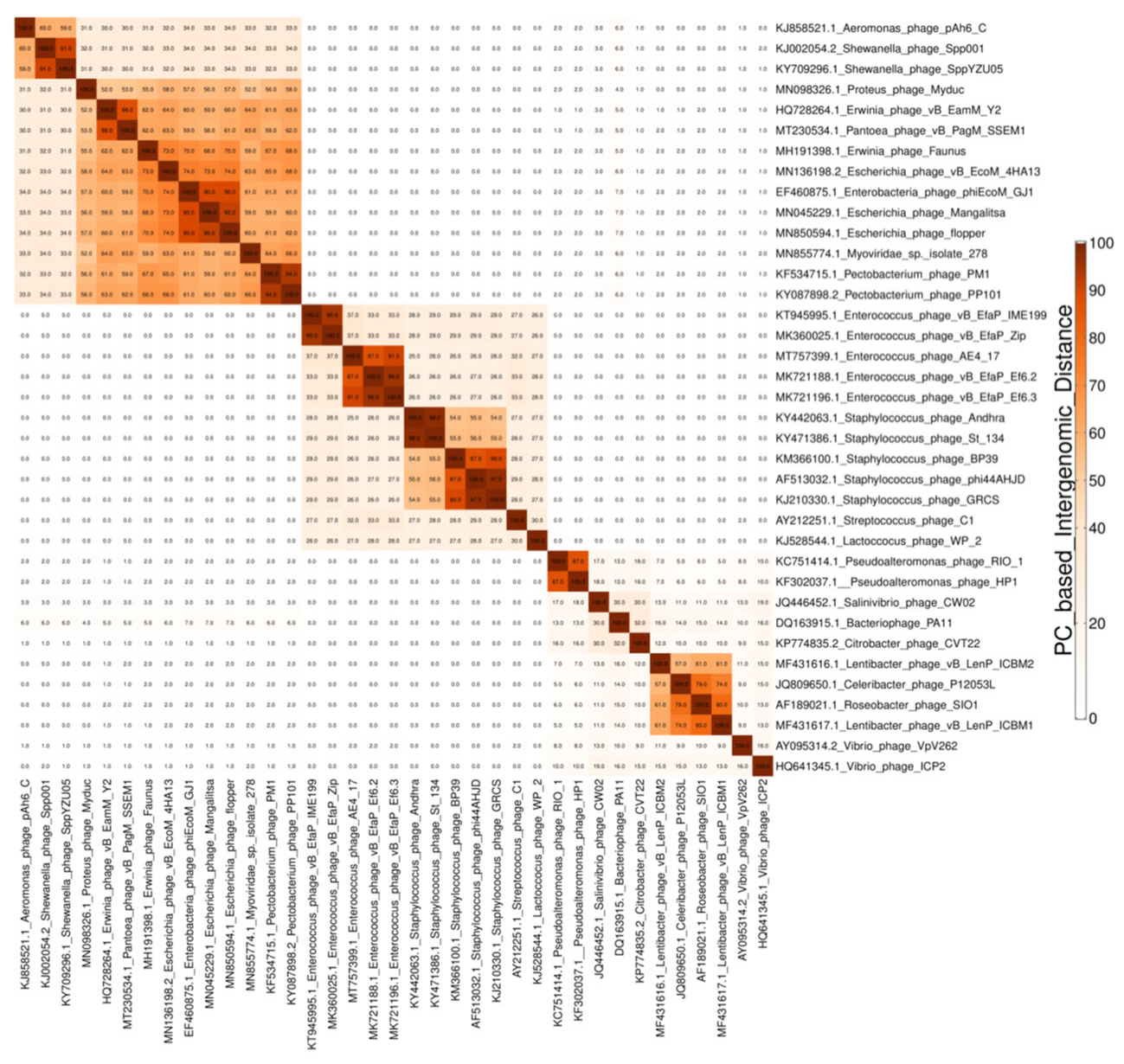
Hierarchical Clustering of the Viral Genomes Based on Their P(SS)C Content—Steps 3A, 2B or 2C
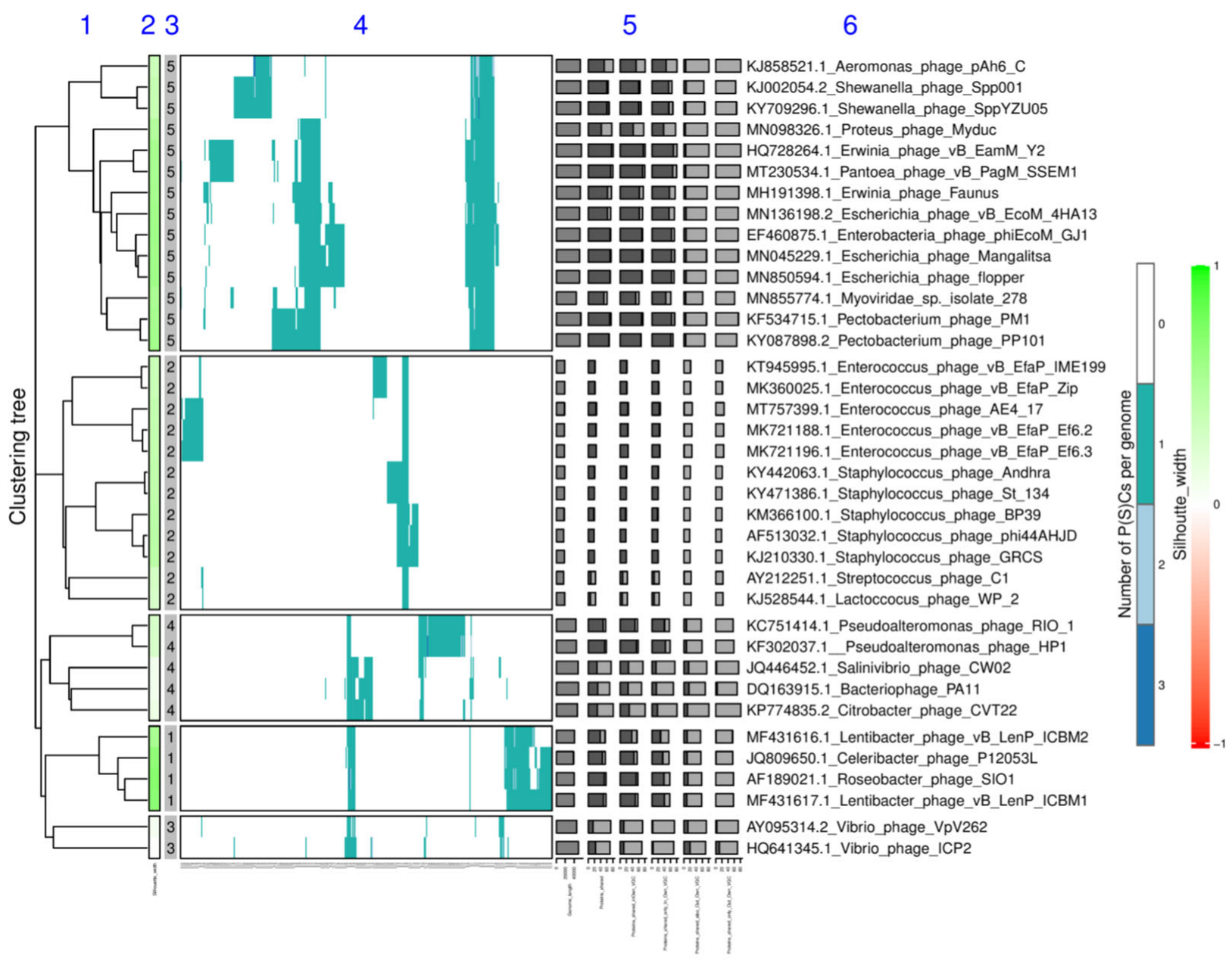
Splitting into Viral Genome Clusters and Related Statistics—Steps 4A, 3B or 3C
2.1.4. Core Proteins Modules from Branches A, B, and C
2.1.5. Protein Annotation Modules from Branches A, B, and C
2.2. Running VirClust on Test Datasets
3. Results and Discussion
3.1. VirClust—A Tool for Viral Genome Clustering, Core Protein Detection, and Protein Annotation
3.2. Availability
3.3. Protein Clustering—Parameters Choice
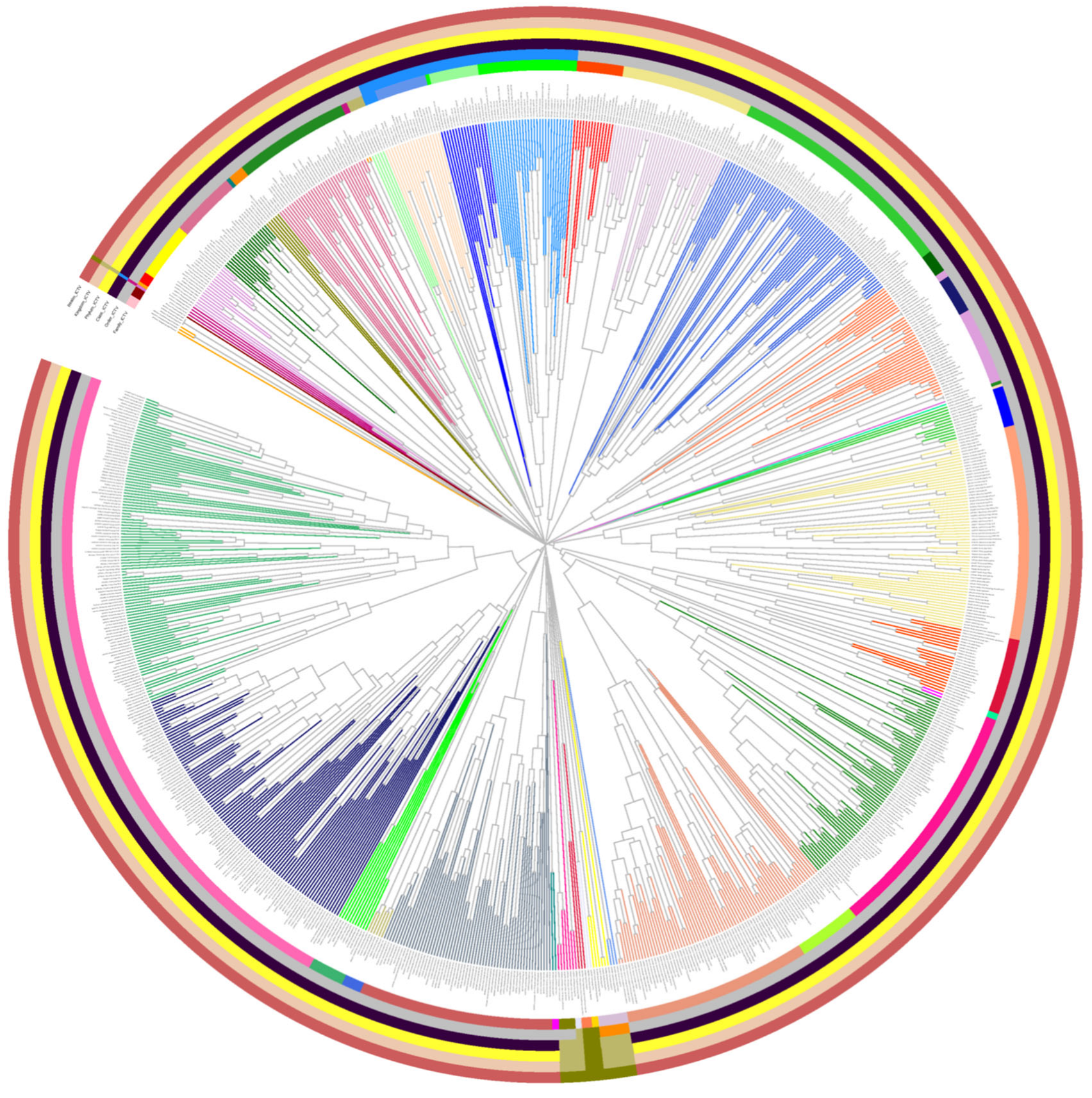
3.4. VirClust Hierarchical Clustering Matches ICTV Virus Classification
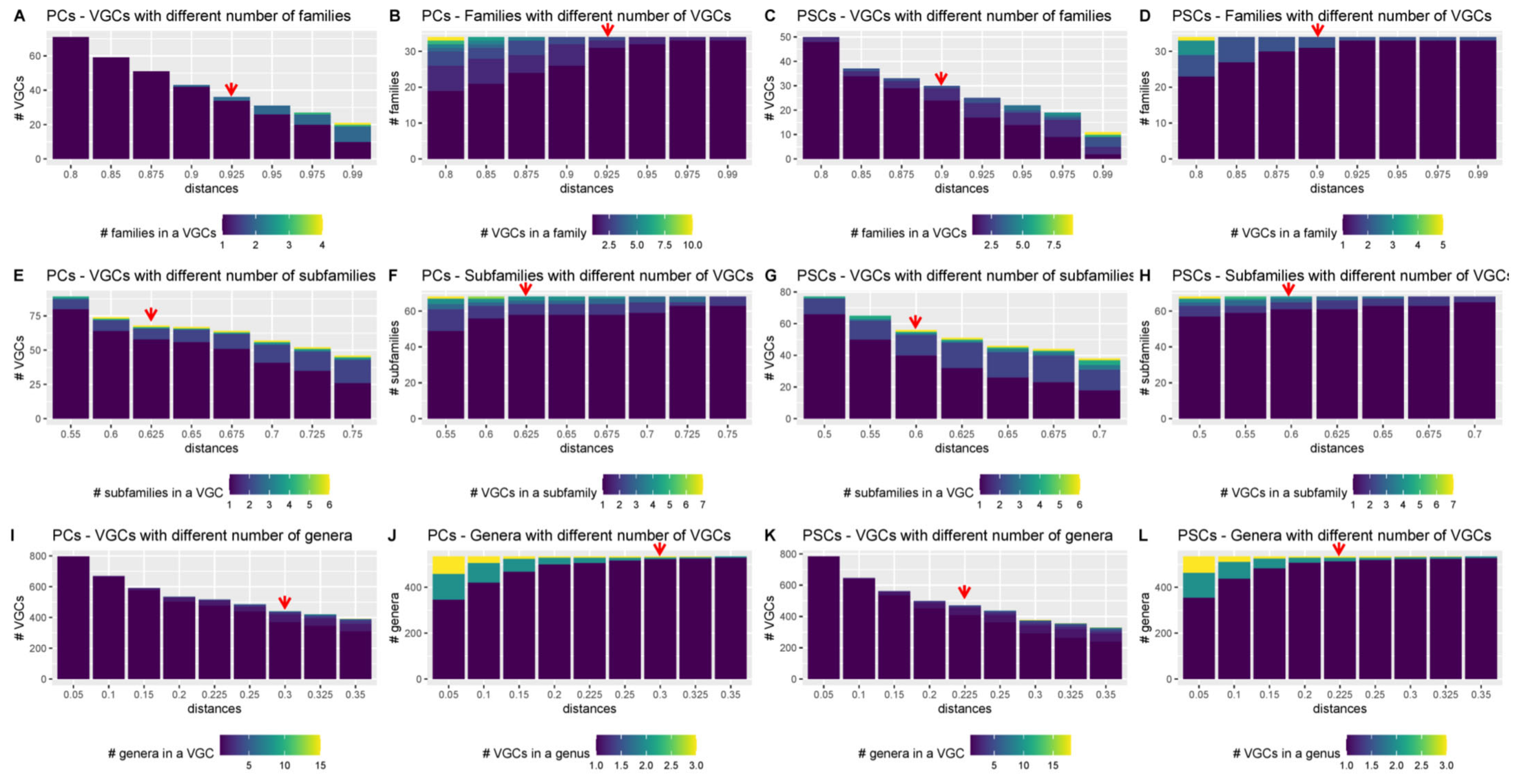
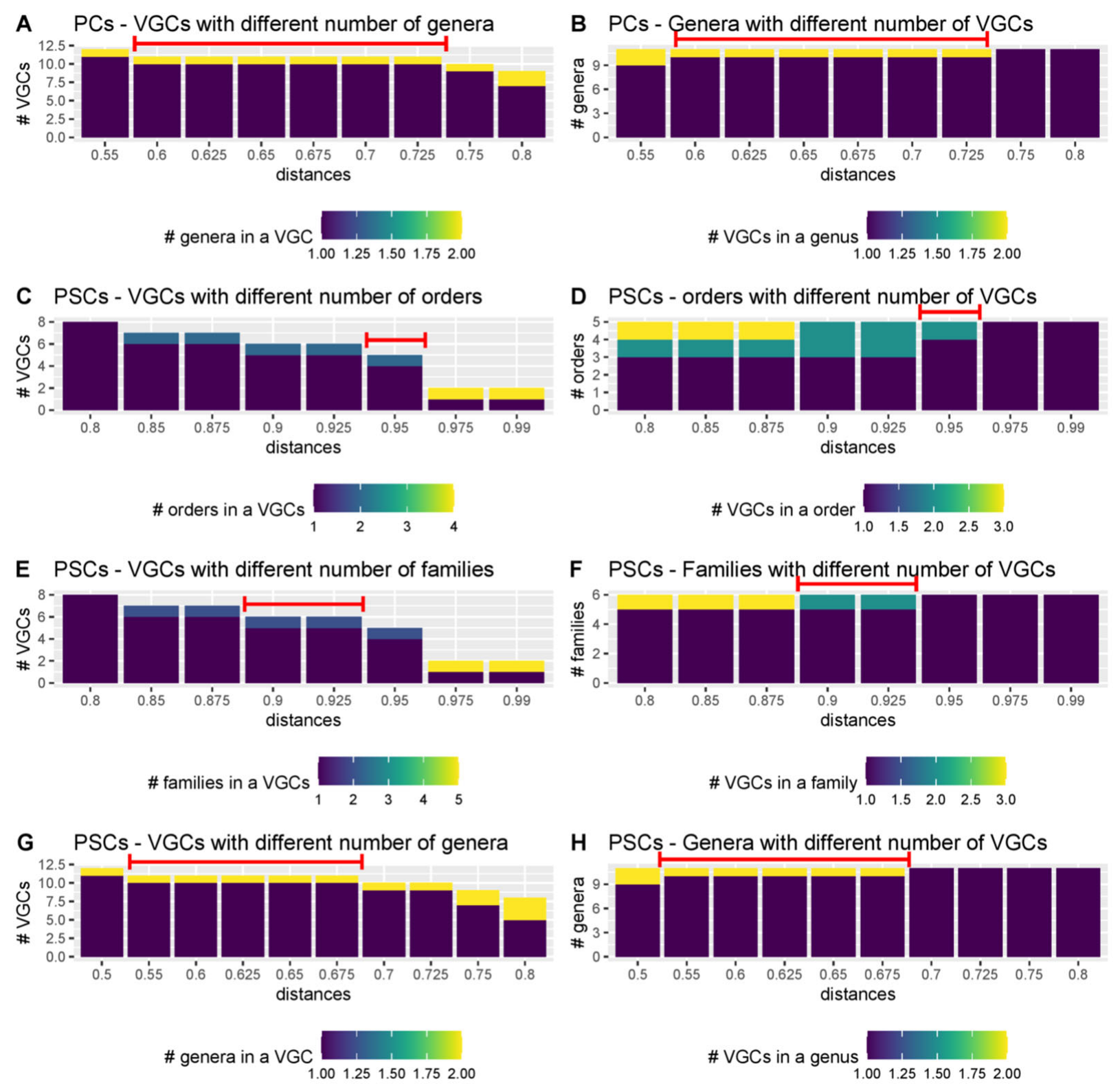
3.5. Distance Thresholds for Different Taxonomic Levels
3.6. Identification and Annotation of Core-Proteins
3.7. A Roadmap for Using VirClust for Virus Taxonomy and Outlook
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global Organization and Proposed Megataxonomy of the Virus World. Microbiol. Mol. Biol. Rev. 2020, 84, e00061-19. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Krupovic, M.; Mushegian, A.; Kropinski, A.M.; Siddell, S.G.; Varsani, A.; Adams, M.J.; Davidson, A.J.; Dulith, B.E.; Harach, B.; et al. The new scope of virus taxonomy: Partitioning the virosphere into 15 hierarchical ranks. Nat. Microbiol. 2020, 5, 668–674. [Google Scholar] [CrossRef]
- Simmonds, P.; Adriaenssens, E.M.; Zerbini, F.M.; Abrescia, N.G.A.; Aiewsakun, P.; Alfenas-Zerbini, P.; Bao, Y.; Barylski, J.; Drosten, C.; Duffy, S.; et al. Four principles to establish a universal virus taxonomy. PLoS Biol. 2023, 21, e3001922. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Senkevich, T.G.; Dolja, V.V. The ancient Virus World and evolution of cells. Biol. Direct 2006, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Koonin, E.V. Multiple origins of viral capsid proteins from cellular ancestors. Proc. Natl. Acad. Sci. USA 2017, 114, E2401–E2410. [Google Scholar] [CrossRef]
- Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origin of viruses: Primordial replicators recruiting capsids from hosts. Nat. Rev. Microbiol. 2019, 17, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, D.; Varsani, A.; Koonin, E.V.; Krupovic, M. Multiple origins of prokaryotic and eukaryotic single-stranded DNA viruses from bacterial and archaeal plasmids. Nat. Commun. 2019, 10, 3425. [Google Scholar] [CrossRef]
- Iranzo, J.; Krupovic, M.; Koonin, E.V. The Double-Stranded DNA Virosphere as a Modular Hierarchical Network of Gene Sharing. mBio 2016, 7, e00978-16. [Google Scholar] [CrossRef]
- Krupovic, M.; Kuhn, J.H.; Wang, F.; Baquero, D.P.; Dolja, V.V.; Egelman, E.H.; Prangishvili, D.; Koonin, E.V. Adnaviria: A new realm for archaeal filamentous viruses with linear A-form double-stranded DNA genomes. J. Virol. 2021, 95, e00673-21. [Google Scholar] [CrossRef]
- Hepojoki, J.; Hetzel, U.; Paraskevopoulou, S.; Drosten, C.; Harrach, B.; Zerbini, M.; Koonin, E.; Krupovic, M.; Dolja, V.; Kuhn, J. ICTV Taxonomy Proposal: Create One New Realm (Ribozyviria) including One New Family (Kolmioviridae) Including Genus Deltavirus and Seven New Genera for a Total of 15 Species. Available online: https://ictv.global/ictv/proposals/2020.012D.R.Ribozyviria.zip (accessed on 20 March 2023).
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC-A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [PubMed]
- Aiewsakun, P.; Simmonds, P. The genomic underpinnings of eukaryotic virus taxonomy: Creating a sequence-based framework for family-level virus classification. Microbiome 2018, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Aiewsakun, P.; Adriaenssens, E.M.; Lavigne, R.; Kropinski, A.M.; Simmonds, P. Evaluation of the genomic diversity of viruses infecting bacteria, archaea and eukaryotes using a common bioinformatic platform: Steps towards a unified taxonomy. J. Gen. Virol. 2018, 99, 1331–1343. [Google Scholar] [CrossRef]
- Bolduc, B.; Jang, H.B.; Doulcier, G.; You, Z.-Q.; Roux, S.; Sullivan, M.B. vConTACT: An iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ 2017, 5, e3243. [Google Scholar] [CrossRef] [PubMed]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 5 May 2022).
- Noguchi, H.; Taniguchi, T.; Itoh, T. MetaGeneAnnotator: Detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2008, 15, 387–396. [Google Scholar] [CrossRef]
- Charif, D.; Seqin, R. 1.0-2: A contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis. In Structural Approaches to Sequence Evolution: Molecules, Networks, Populations; Bastolla, U., Porto, M., Roman, H.E., Vendruscolo, M., Eds.; Springer: New York, NY, USA, 2007; pp. 207–232. ISBN 978-3-540-35305-8. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef]
- Suzuki, R.; Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006, 22, 1540–1542. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H.; Terada, Y. Selective Inference for Testing Trees and Edges in Phylogenetics. Front. Ecol. Evol. 2019, 7, 459. [Google Scholar] [CrossRef]
- Maechler, M.; Rousseeuw, P.; Struyf, A.; Hubert, M.; Hornik, K. Cluster: Cluster Analysis Basics and Extensions. Available online: https://CRAN.R-project.org/package=cluster (accessed on 12 May 2020).
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic virus orthologous groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017, 45, D491–D498. [Google Scholar] [CrossRef]
- Kiening, M.; Ochsenreiter, R.; Hellinger, H.-J.; Rattei, T.; Hofacker, I.; Frishman, D. Conserved Secondary Structures in Viral mRNAs. Viruses 2019, 11, 401. [Google Scholar] [CrossRef]
- Terzian, P.; Olo Ndela, E.; Galiez, C.; Lossouarn, J.; Pérez Bucio, R.E.; Mom, R.; Toussaint, A.; Petit, M.-A.; Enault, F. PHROG: Families of prokaryotic virus proteins clustered using remote homology. NAR Genom. Bioinform. 2021, 3, lqab067. [Google Scholar] [CrossRef]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 2019, 20, 473. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.-Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Zucker, F.; Bischoff, V.; Olo Ndela, E.; Heyerhoff, B.; Poehlein, A.; Freese, H.M.; Roux, S.; Simon, M.; Enault, F.; Moraru, C. New Microviridae isolated from Sulfitobacter reveals two cosmopolitan subfamilies of single-stranded DNA phages infecting marine and terrestrial Alphaproteobacteria. Virus Evol. 2022, 8, veac070. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef] [PubMed]
- Zayed, A.A.; Lücking, D.; Mohssen, M.; Cronin, D.; Bolduc, B.; Gregory, A.C.; Hargreaves, K.R.; Piehowski, P.D.; White, R.A.; Huang, E.L.; et al. efam: An expanded, metaproteome-supported HMM profile database of viral protein families. Bioinformatics 2021, 37, 4202–4208. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.X.; Mahbob, M.; Ragan, M.A. Clustering evolving proteins into homologous families. BMC Bioinform. 2013, 14, 120. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters Step2A | E-Value = 10−5, | E-Value = 10−5, | E-Value = 10−5, | |
|---|---|---|---|---|
| Bitscore = 50, | Bitscore = 50, | Bitscore = 50, | ||
| Coverage = 100, | Coverage = 80, | Coverage = 0, | ||
| %id = 100 | %id = 50 | %id = 0 | ||
| Number of PCs | 1982 | 1124 | 805 | |
| Intergenomic distances | ||||
| 0 pctl | 0.00 | 0.00 | 0.00 | |
| 25th pctl | 1.00 | 0.97 | 0.74 | |
| 50th pctl | 1.00 | 1.00 | 0.99 | |
| 75th pctl | 1.00 | 1.00 | 1.00 | |
| 100th pctl | 1.00 | 1.00 | 1.00 | |
| VGC | Family | Number of Core PSCs | Genome Length (kbps) Range | Gene Count Range | Phage Count | Minimum Intergenomic Similarity (%) | MCP | TerL | Por | DNApol | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | Unknown Function | ||||||||||
| Duplodnaviria | |||||||||||
| 1 | Peduoviridae | 7 | 0 | 28.8–40.6 | 37–56 | 59 | 16 | + | + | + | - |
| 3 | Autographiviridae | 9 | 0 | 30.8–47.7 | 29–65 | 106 | 17 | + | + | - | + |
| 4 | Autographiviridae | 2 | 0 | 10.4–47.8 | 15–69 | 99 | 18 | - | - | - | - |
| 5 | Kyanoviridae + Ackermanviridae | 32 | 0 | 144.4–252.5 | 178–324 | 76 | 17 | + | + | + | + |
| 6 | Herelleviridae | 25 | 3 | 106.1–167.5 | 126–294 | 65 | 14 | + | + | + | + |
| 8 | Zobellviridae | 10 | 3 | 38.9–49.7 | 55–82 | 11 | 16 | + | + | - | + |
| 9 | Salamasviridae + Rountreeviridae + Guelinviridae | 5 | 0 | 16.7–29 | 19–51 | 44 | 13 | + | + | - | + |
| 10 | Drexlerviridae | 23 | 5 | 44.3–51.9 | 62–87 | 40 | 47 | + | + | - | - |
| 11 | Straboviridae | 34 | 3 | 121.5–248.1 | 191–421 | 73 | 19 | - | - | - | + |
| 12 | Steigviridae | 12 | 4 | 93.6–104.6 | 65–119 | 16 | 14 | + | + | + | - |
| 15 | Casjensviridae | 17 | 1 | 54.5–64 | 62–88 | 35 | 26 | + | + | + | - |
| 16 | Mesyanzhinovviridae | 13 | 0 | 56.6–64.1 | 77–93 | 12 | 22 | - | + | + | + |
| 17 | Demerecviridae | 31 | 0 | 104.4–128.7 | 137–192 | 23 | 26 | + | - | + | + |
| 18 | Vilmaviridae | 25 | 0 | 70.2–84.4 | 113–151 | 17 | 22 | + | + | + | + |
| 19 | Zierdtviridae | 23 | 1 | 64.2–70.6 | 86–94 | 20 | 25 | + | + | + | + |
| 20 | Schitoviridae | 13 | 1 | 59.1–104 | 71–127 | 70 | 13 | + | + | + | + |
| 21 | Chaseviridae | 27 | 4 | 44.7–56.7 | 62–82 | 14 | 39 | + | + | + | + |
| 22 | Kwiatkowskiviridae | 156 | 92 | 146.4–149.9 | 243–274 | 6 | 69 | + | + | - | + |
| 23 | Aggregaviridae + Assiduviridae | 6 | 4 | 43.2–57.5 | 80–110 | 4 | 12 | - | - | - | - |
| 24 | Pachyviridae | 12 | 3 | 71.5–78.9 | 105–119 | 5 | 12 | - | + | + | - |
| 25 | Pervagoviridae | 80 | 56 | 72.6–73 | 84–86 | 2 | 99 | - | + | + | + |
| 27 | Duneviridae + Helgolandviridae | 5 | 3 | 37.6–46.6 | 48–63 | 6 | 12 | - | - | - | - |
| 30 | Crevaviridae + Intestiviridae | 23 | 10 | 83.5–98.1 | 77–95 | 20 | 27 | + | + | + | + |
| 31 | Orlajensenviridae | 24 | 4 | 17.4–17.5 | 24–24 | 3 | 100 | + | + | + | - |
| 33 | Winoviridae | 10 | 8 | 34.8–39.7 | 49–62 | 3 | 18 | - | - | - | - |
| 34 | Molycolviridae | 183 | 149 | 124.2–124.7 | 193–200 | 2 | 95 | + | - | + | + |
| 35 | Forsetiviridae | 55 | 37 | 44–47.2 | 66–76 | 2 | 77 | + | + | + | - |
| 36 | Suoliviridae | 20 | 10 | 92.6–104.1 | 94–172 | 30 | 19 | + | - | + | + |
| Varidnaviria | |||||||||||
| 2 | Matshushitaviridae | 28 | 23 | 17.1–19.7 | 35–40 | 2 | 75 | - | + | - | - |
| 7 | Corticoviridae + Autolykiviridae | 3 | 0 | 10.1–10.7 | 16–21 | 7 | 15 | + | + | - | - |
| 13 | Simuloviridae | 12 | 11 | 16.4–19 | 29–31 | 3 | 47 | - | + | - | - |
| 14 | Tectiviridae | 3 | 0 | 14.4–16.6 | 22–30 | 6 | 11 | - | + | - | + |
| 28 | Tectiviridae | 8 | 3 | 17.3–18.3 | 30–34 | 3 | 27 | - | + | - | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moraru, C. VirClust—A Tool for Hierarchical Clustering, Core Protein Detection and Annotation of (Prokaryotic) Viruses. Viruses 2023, 15, 1007. https://doi.org/10.3390/v15041007
Moraru C. VirClust—A Tool for Hierarchical Clustering, Core Protein Detection and Annotation of (Prokaryotic) Viruses. Viruses. 2023; 15(4):1007. https://doi.org/10.3390/v15041007
Chicago/Turabian StyleMoraru, Cristina. 2023. "VirClust—A Tool for Hierarchical Clustering, Core Protein Detection and Annotation of (Prokaryotic) Viruses" Viruses 15, no. 4: 1007. https://doi.org/10.3390/v15041007