
RAPIDprep: A Simple, Fast Protocol for RNA Metagenomic Sequencing of Clinical Samples

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimens
2.2. RAPIDprep Assay
2.3. Development of Final Assay Conditions
2.4. Severe Acute Respiratory Infections in Children Cohort
2.5. Bioinformatic Analysis of RNA-mNGS Data
3. Results and Discussion
3.1. Development of the RAPIDprep Assay
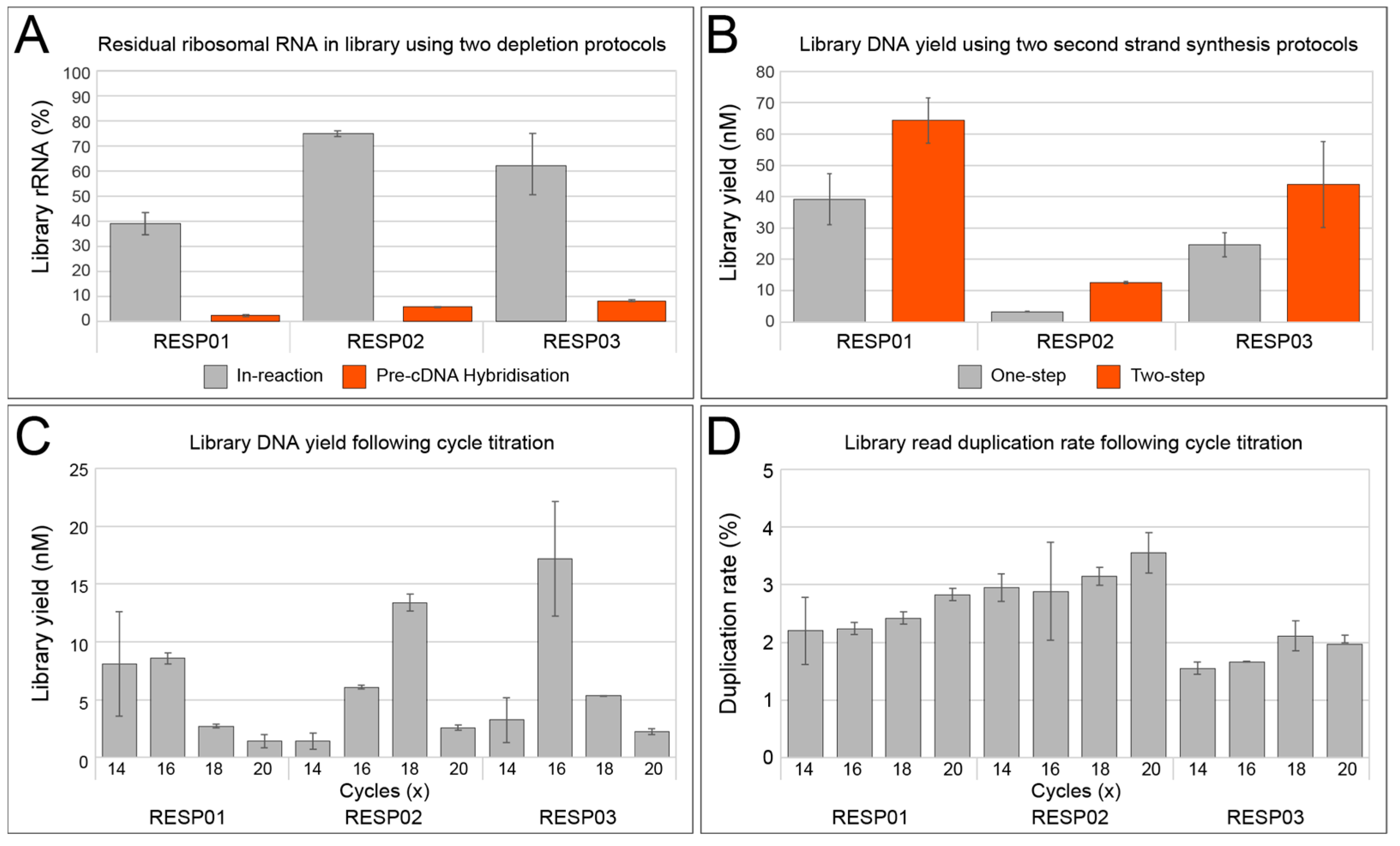
3.1.1. rRNA Depletion
3.1.2. Double-Stranded cDNA Synthesis
3.1.3. Library Amplification
3.2. Application of the RAPIDprep Assay to a Panel of Respiratory Samples
3.3. Viral Sequence Identification, Genome Recovery, and Quantitative Performace
3.4. Comparison of RAPIDprep to Commercial Assay
3.5. Study Limitations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schmitz, J.E.; Stratton, C.W.; Persing, D.H.; Tang, Y.W. Forty Years of Molecular Diagnostics for Infectious Diseases. J. Clin. Microbiol. 2022, 60, e0244621. [Google Scholar] [CrossRef] [PubMed]
- Annand, E.J.; Horsburgh, B.A.; Xu, K.; Reid, P.A.; Poole, B.; de Kantzow, M.C.; Brown, N.; Tweedie, A.; Michie, M.; Grewar, J.D.; et al. Novel Hendra Virus Variant Detected by Sentinel Surveillance of Horses in Australia. Emerg. Infect. Dis. 2022, 28, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Zhang, D.; Lou, X.; Yan, H.; Pan, J.; Mao, H.; Tang, H.; Shu, Y.; Zhao, Y.; Liu, L.; Li, J.; et al. Metagenomic analysis of viral nucleic acid extraction methods in respiratory clinical samples. BMC Genom. 2018, 19, 773. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhao, S.; Yu, B.; Wu, W.-C.; Hu, Y.; Tian, J.-H.; Yin, W.; Ni, F.; Hu, H.-L.; Geng, S.; et al. Total infectome characterization of respiratory infections in pre-COVID-19 Wuhan, China. PLoS Pathog. 2022, 18, e1010259. [Google Scholar] [CrossRef] [PubMed]
- Serpa, P.H.; Deng, X.; Abdelghany, M.; Crawford, E.; Malcolm, K.; Caldera, S.; Fung, M.; McGeever, A.; Kalantar, K.L.; Lyden, A.; et al. Metagenomic prediction of antimicrobial resistance in critically ill patients with lower respiratory tract infections. Genome Med. 2022, 14, 1–74. [Google Scholar] [CrossRef]
- Li, Y.; Deng, X.; Hu, F.; Wang, J.; Liu, Y.; Huang, H.; Ma, J.; Zhang, J.; Zhang, F.; Zhang, C. Metagenomic analysis identified co-infection with human rhinovirus C and bocavirus 1 in an adult suffering from severe pneumonia. J. Infect. 2018, 76, 311–313. [Google Scholar] [CrossRef]
- Subramoney, K.; Mtileni, N.; Bharuthram, A.; Davis, A.; Kalenga, B.; Rikhotso, M.; Maphahlele, M.; Giandhari, J.; Naidoo, Y.; Pillay, S.; et al. Identification of SARS-CoV-2 Omicron variant using spike gene target failure and genotyping assays, Gauteng, South Africa, 2021. J. Med. Virol. 2022, 94, 3676–3684. [Google Scholar] [CrossRef]
- Itokawa, K.; Sekizuka, T.; Hashino, M.; Tanaka, R.; Kuroda, M. Disentangling primer interactions improves SARS-CoV-2 genome sequencing by multiplex tiling PCR. PLoS ONE 2020, 15, e0239403. [Google Scholar] [CrossRef]
- Thorburn, F.; Bennett, S.; Modha, S.; Murdoch, D.; Gunson, R.; Murcia, P.R. The use of next generation sequencing in the diagnosis and typing of respiratory infections. J. Clin. Virol. 2015, 69, 96–100. [Google Scholar] [CrossRef]
- Lu, X.; Wang, L.; Sakthivel, S.K.; Whitaker, B.; Murray, J.; Kamili, S.; Lynch, B.; Malapati, L.; Burke, S.A.; Harcourt, J.; et al. US CDC Real-Time Reverse Transcription PCR Panel for Detection of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. 2020, 26, 1654–1665. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Piedra, P.A.; Avadhanula, V.; Durigon, E.L.; Machablishvili, A.; López, M.-R.; Thornburg, N.J.; Peret, T.C.T. Duplex real-time RT-PCR assay for detection and subgroup-specific identification of human respiratory syncytial virus. J. Virol. Methods 2019, 271, 113676. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Kopylova, E.; Noe, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Segata, N.; Waldron, L.; Ballarini, A.; Narasimhan, V.; Jousson, O.; Huttenhower, C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 2012, 9, 811–814. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner. Available online: https://www.osti.gov/servlets/purl/1241166 (accessed on 12 April 2022).
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.; Gamini, R.; Zhang, B.; von Schack, D. Evaluation of two main RNA-seq approaches for gene quantification in clinical RNA sequencing: polyA+ selection versus rRNA depletion. Sci. Rep. 2018, 8, 4781. [Google Scholar] [CrossRef]
- Albert, E.; Torres, I.; Bueno, F.; Huntley, D.; Molla, E.; Fernández-Fuentes, M.Á.; Martínez, M.; Poujois, S.; Forqué, L.; Valdivia, A.; et al. Field evaluation of a rapid antigen test (Panbio™ COVID-19 Ag Rapid Test Device) for COVID-19 diagnosis in primary healthcare centres. Clin. Microbiol. Infect. 2021, 27, 472.e7–472.e10. [Google Scholar] [CrossRef] [PubMed]
- Bal, A.; Pichon, M.; Picard, C.; Casalegno, J.S.; Valette, M.; Schuffenecker, I.; Billard, L.; Vallet, S.; Vilchez, G.; Cheynet, V.; et al. Quality control implementation for universal characterization of DNA and RNA viruses in clinical respiratory samples using single metagenomic next-generation sequencing workflow. BMC Infect. Dis. 2018, 18, 537. [Google Scholar] [CrossRef] [PubMed]
- Dudas, G.; Bedford, T. The ability of single genes vs full genomes to resolve time and space in outbreak analysis. BMC Evol. Biol. 2019, 19, 232. [Google Scholar] [CrossRef]
- Tabor, S.; Richardson, C.C. Selective inactivation of the exonuclease activity of bacteriophage T7 DNA polymerase by in vitro mutagenesis. J. Biol. Chem. 1989, 264, 6447–6458. [Google Scholar] [CrossRef]
- Zhu, B. Bacteriophage T7 DNA polymerase—Sequenase. Front. Microbiol. 2014, 5, 181. [Google Scholar] [CrossRef]
- Brenner, T.; Decker, S.O.; Grumaz, S.; Stevens, P.; Bruckner, T.; Schmoch, T.; Pletz, M.W.; Bracht, H.; Hofer, S.; Marx, G.; et al. Next-generation sequencing diagnostics of bacteremia in sepsis (Next GeneSiS-Trial): Study protocol of a prospective, observational, noninterventional, multicenter, clinical trial. Medicine 2018, 97, e9868. [Google Scholar] [CrossRef]
- Peddu, V.; Shean, R.C.; Xie, H.; Shrestha, L.; Perchetti, G.A.; Minot, S.S.; Roychoudhury, P.; Huang, M.-L.; Nalla, A.; Reddy, S.B.; et al. Metagenomic Analysis Reveals Clinical SARS-CoV-2 Infection and Bacterial or Viral Superinfection and Colonization. Clin. Chem. 2020, 66, 966–972. [Google Scholar] [CrossRef]
- Poulsen, C.S.; Ekstrøm, C.T.; Aarestrup, F.M.; Pamp, S.J. Library Preparation and Sequencing Platform Introduce Bias in Metagenomic-Based Characterizations of Microbiomes. Microbiol. Spectr. 2022, 10, e0009022. [Google Scholar] [CrossRef]
- Wilson, M.R.; Naccache, S.N.; Samayoa, E.; Biagtan, M.; Bashir, H.; Yu, G.; Salamat, S.M.; Somasekar, S.; Federman, S.; Miller, S.; et al. Actionable Diagnosis of Neuroleptospirosis by Next-Generation Sequencing. N. Engl. J. Med. 2014, 370, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Peck, M.A.; Sturk-Andreaggi, K.; Thomas, J.T.; Oliver, R.S.; Barritt-Ross, S.; Marshall, C. Developmental validation of a Nextera XT mitogenome Illumina MiSeq sequencing method for high-quality samples. Forensic Sci. Int. Genet. 2018, 34, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Di Giallonardo, F.; Kok, J.; Fernandez, M.; Carter, I.; Geoghegan, J.L.; Dwyer, D.E.; Holmes, E.C.; Eden, J.S. Evolution of Human Respiratory Syncytial Virus (RSV) over Multiple Seasons in New South Wales, Australia. Viruses 2018, 10, 476. [Google Scholar] [CrossRef] [PubMed]
- Eden, J.S.; Rockett, R.; Carter, I.; Rahman, H.; de Ligt, J.; Hadfield, J.; Storey, M.; Ren, X.; Tulloch, R.; Basile, K.; et al. An emergent clade of SARS-CoV-2 linked to returned travellers from Iran. Virus Evol. 2020, 6, veaa027. [Google Scholar] [CrossRef] [PubMed]
- Tulloch, R.L.; Kok, J.; Carter, I.; Dwyer, D.E.; Eden, J.S. An Amplicon-Based Approach for the Whole-Genome Sequencing of Human Metapneumovirus. Viruses 2021, 13, 499. [Google Scholar] [CrossRef]
- Bansal, V. A computational method for estimating the PCR duplication rate in DNA and RNA-seq experiments. BMC Bioinform. 2017, 18, 43. [Google Scholar] [CrossRef]
- Schoonvaere, K.; De Smet, L.; Smagghe, G.; Vierstraete, A.; Braeckman, B.P.; de Graaf, D.C. Unbiased RNA Shotgun Metagenomics in Social and Solitary Wild Bees Detects Associations with Eukaryote Parasites and New Viruses. PLoS ONE 2016, 11, e0168456. [Google Scholar] [CrossRef]
- Meadow, J.F.; Altrichter, A.E.; Bateman, A.C.; Stenson, J.; Brown, G.Z.; Green, J.L.; Bohannan, B.J.M. Humans differ in their personal microbial cloud. PeerJ 2015, 3, e1258. [Google Scholar] [CrossRef]
- Qamar, W.; Khan, M.R.; Arafah, A. Optimization of conditions to extract high quality DNA for PCR analysis from whole blood using SDS-proteinase K method. Saudi J. Biol. Sci. 2017, 24, 1465–1469. [Google Scholar] [CrossRef]
- Yang, L.; Haidar, G.; Zia, H.; Nettles, R.; Qin, S.; Wang, X.; Shah, F.; Rapport, S.F.; Charalampous, T.; Methé, B.; et al. Metagenomic identification of severe pneumonia pathogens in mechanically-ventilated patients: A feasibility and clinical validity study. Respir. Res. 2019, 20, 265. [Google Scholar] [CrossRef]
- Porter, A.F.; Cobbin, J.; Li, C.X.; Eden, J.S.; Holmes, E.C. Metagenomic Identification of Viral Sequences in Laboratory Reagents. Viruses 2021, 13, 2122. [Google Scholar] [CrossRef] [PubMed]
- Tuddenham, R.; Eden, J.S.; Gilbey, T.; Dwyer, D.E.; Jennings, Z.; Holmes, E.C.; Branley, J.M. Human pegivirus in brain tissue of a patient with encephalitis. Diagn. Microbiol. Infect. Dis. 2020, 96, 114898. [Google Scholar] [CrossRef] [PubMed]
- Sikazwe, C.; Neave, M.J.; Michie, A.; Mileto, P.; Wang, J.; Cooper, N.; Levy, A.; Imrie, A.; Baird, R.W.; Currie, B.J.; et al. Molecular detection and characterisation of the first Japanese encephalitis virus belonging to genotype IV acquired in Australia. PLoS Negl. Trop. Dis. 2022, 16, e0010754. [Google Scholar] [CrossRef] [PubMed]
- Waller, C.; Tiemensma, M.; Currie, B.J.; Williams, D.T.; Baird, R.W.; Krause, V.L. Japanese Encephalitis in Australia—A Sentinel Case. N. Engl. J. Med. 2022, 387, 661–662. [Google Scholar] [CrossRef] [PubMed]
- Maamary, J.; Maddocks, S.; Barnett, Y.; Wong, S.; Rodriguez, M.; Hueston, L.; Jeoffreys, N.; Eden, J.S.; Dwyer, D.E.; Floyd, T.; et al. New Detection of Locally Acquired Japanese Encephalitis Virus Using Clinical Metagenomics, New South Wales, Australia. Emerg. Infect. Dis. 2023, 29, 627–630. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | Catalogue | Supplier |
| QIAseq FastSelect-rRNA HMR | 334385 | Qiagen, Hilden, Germany |
| QIAseq FastSelect–5S/16S/23S | 335921 | |
| Invitrogen SuperScript IV VILO Master Mix | 11756050 | Thermo Fisher, Waltham, MA, USA |
| Sequenase Version 2.0 DNA Polymerase | 70775Y200UN | |
| Invitrogen ezDNase Enzyme | 11766051 | |
| Mag-Bind® TotalPure NGS | M1378-01 | Omega Biotek, Norcross, GA, USA |
| Nextera XT DNA Library Preparation Kit | FC-131-1096 | Illumina, San Diego, CA, USA |
| IDT® for Illumina DNA/RNA UD Indexes | 20027213 | |
| iSeq 100 i1 Reagent v2 (300-cycle) | 20031371 |
| In-Reaction | Pre-cDNA Hybridisation | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rRNA | RESP01 | RESP02 | RESP03 | RESP01 | RESP02 | RESP03 | ||||||
| Archaeal:16S | 3.5% | 2.9% | 7.5% | 7.3% | 6.1% | 4.0% | 0.0% | 0.0% | 0.1% | 0.1% | 0.2% | 0.1% |
| Archaeal:23S | 10.9% | 9.5% | 19.2% | 19.9% | 22.0% | 16.2% | 0.1% | 0.1% | 0.8% | 0.7% | 1.6% | 1.4% |
| Bacterial:5S | 0.7% | 0.8% | 0.2% | 0.2% | 0.4% | 0.6% | 1.0% | 1.3% | 0.9% | 0.8% | 1.4% | 1.3% |
| Bacterial:16S | 0.7% | 0.6% | 2.1% | 2.1% | 3.0% | 2.2% | 0.1% | 0.0% | 0.2% | 0.2% | 0.2% | 0.1% |
| Bacterial:23S | 3.5% | 3.1% | 10.1% | 10.7% | 20.7% | 18.1% | 0.2% | 0.3% | 2.2% | 2.1% | 4.0% | 3.9% |
| Eukaryotic:5.8S | 0.5% | 0.5% | 1.1% | 1.1% | 0.9% | 0.8% | 0.0% | 0.0% | 0.1% | 0.1% | 0.1% | 0.1% |
| Eukaryotic:18S | 14.0% | 11.9% | 22.0% | 22.1% | 11.2% | 7.7% | 0.3% | 0.4% | 0.9% | 0.8% | 0.6% | 0.5% |
| Eukaryotic:28S | 8.5% | 6.7% | 12.6% | 13.0% | 6.6% | 4.4% | 0.2% | 0.2% | 0.7% | 0.7% | 0.4% | 0.4% |
| rRNA levels | 0.0% | 5.0% | 10.0% | 15.0% | 20.0% | 25.0% | ||||||
| Library. | Group | Virus | Type | Extraction Method | Library Yield (nM) | Data Output (Reads) |
|---|---|---|---|---|---|---|
| RAPID01 | COVID-19 | SARS-CoV-2 | Nasopharyngeal swab | Zymo Quick-RNA Viral | 8 | 16,810,302 |
| RAPID02 | SARS-CoV-2 | Nasopharyngeal swab | Zymo Quick-RNA Viral | 34.7 | 11,620,222 | |
| RAPID03 | SARS-CoV-2 | Nasopharyngeal swab | Zymo Quick-RNA Viral | 2.8 | 18,322,864 | |
| RAPID04 | SARS-CoV-2 | Nasopharyngeal swab | Zymo Quick-RNA Viral | 2.4 | 12,707,642 | |
| RAPID05 | SARS-CoV-2 | Nasopharyngeal swab | Zymo Quick-RNA Viral | 2.2 | 15,327,662 | |
| RAPID06 | SARS-CoV-2 | Nasopharyngeal swab | Zymo Quick-RNA Viral | 13.2 | 15,271,010 | |
| RAPID07 | SARS-CoV-2 | Nasopharyngeal swab | Zymo Quick-RNA Viral | 3.1 | 11,147,058 | |
| RAPID08 | SARS-CoV-2 | Nasopharyngeal swab | Zymo Quick-RNA Viral | 6.1 | 9,453,054 | |
| RAPID09 | SARS-CoV-2 | Nasopharyngeal swab | Zymo Quick-RNA Viral | 15.2 | 17,326,098 | |
| RAPID10 | SARS-CoV-2 | Nasopharyngeal swab | Zymo Quick-RNA Viral | 9.1 | 15,531,486 | |
| RAPID11 | SARS-CoV-2 | Nasopharyngeal swab | Zymo Quick-RNA Viral | 1.6 | 10,903,012 | |
| RAPID12 | SARS-CoV-2 | Nasopharyngeal swab | Zymo Quick-RNA Viral | 16.2 | 12,670,186 | |
| RAPID13 | Influenza A | pdmH1N1 | Viral culture | Zymo Quick-RNA Viral | 29.8 | 15,200,408 |
| RAPID14 | pdmH1N1 | Viral culture | Zymo Quick-RNA Viral | 3.5 | 12,405,816 | |
| RAPID15 | Mock community | None | Mixed culture | ZymoBIOMICS DNA/RNA Miniprep | 44.5 | 13,541,676 |
| RAPID16 | None | Mixed culture | ZymoBIOMICS DNA/RNA Miniprep | 86 | 12,427,300 | |
| RAPID17 | Kids SARI | Unknown | Nasopharyngeal aspirate | ZymoBIOMICS DNA/RNA Miniprep | 6.6 | 16,321,598 |
| RAPID18 | Unknown | Nasopharyngeal aspirate | ZymoBIOMICS DNA/RNA Miniprep | 1.5 | 17,340,092 | |
| RAPID19 | Unknown | Nasopharyngeal aspirate | ZymoBIOMICS DNA/RNA Miniprep | 1 | 15,464,422 | |
| RAPID20 | Unknown | Nasopharyngeal aspirate | ZymoBIOMICS DNA/RNA Miniprep | 8.3 | 16,563,150 | |
| RAPID21 | Unknown | Nasopharyngeal aspirate | ZymoBIOMICS DNA/RNA Miniprep | 1.6 | 24,661,800 | |
| RAPID22 | Unknown | Nasopharyngeal aspirate | ZymoBIOMICS DNA/RNA Miniprep | 8.8 | 14,490,708 | |
| RAPID23 | Unknown | Nasopharyngeal aspirate | ZymoBIOMICS DNA/RNA Miniprep | 3.3 | 28,187,178 | |
| RAPID24 | Unknown | Nasopharyngeal aspirate | ZymoBIOMICS DNA/RNA Miniprep | 3.8 | 19,042,138 | |
| RAPID25 | RSV | RSV | Nasopharyngeal swab | Roche MagNA Pure 96 Viral NA | 84.2 | 15,287,586 |
| RAPID26 | RSV | Nasopharyngeal swab | Roche MagNA Pure 96 Viral NA | 94.1 | 19,473,790 | |
| RAPID27 | RSV | Nasopharyngeal swab | Roche MagNA Pure 96 Viral NA | 97.5 | 16,302,456 | |
| RAPID28 | RSV | Nasopharyngeal swab | Roche MagNA Pure 96 Viral NA | 80.1 | 14,524,914 | |
| RAPID29 | RSV | Nasopharyngeal swab | Roche MagNA Pure 96 Viral NA | 67.9 | 17,923,728 | |
| RAPID30 | RSV | Nasopharyngeal swab | Roche MagNA Pure 96 Viral NA | 49.3 | 13,466,538 | |
| RAPID31 | RSV | Nasopharyngeal swab | Roche MagNA Pure 96 Viral NA | 56.7 | 12,192,164 | |
| RAPID32 | RSV | Nasopharyngeal swab | Roche MagNA Pure 96 Viral NA | 61.8 | 17,199,224 | |
| RAPID33 | Kids unknown | Unknown | Nasopharyngeal aspirate | ZymoBIOMICS DNA/RNA Miniprep | 22.2 | 14,839,480 |
| RAPID34 | Unknown | Nasopharyngeal swab | Zymo Quick-RNA Viral | 31.4 | 14,806,470 | |
| RAPID35 | Unknown | Nasopharyngeal swab | Zymo Quick-RNA Viral | 70.1 | 12,576,818 | |
| RAPID36 | Unknown | Nasopharyngeal aspirate | Zymo Quick-RNA Viral | 10.9 | 13,418,996 | |
| RAPID37 | Unknown | Vomitus | Zymo Quick-RNA Viral | 11.8 | 9,862,060 | |
| RAPID38 | Unknown | Nasopharyngeal swab | Zymo Quick-RNA Viral | 48.1 | 11,384,408 | |
| RAPID39 | Unknown | Nasopharyngeal swab | Zymo Quick-RNA Viral | 134.5 | 20,986,506 | |
| RAPID40 | NTC | NTC | Water | N/A | 6.4 | 26,137,100 |
| Library. | Virus | Type # | SARS-CoV2 | RSV-A | RSV-B | Flu-A pdmH1N1 | Flu-C | Rhinovirus | GB Virus C | CMV | HHV7 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| RAPID01 | SARS-CoV-2 | NP swab | 5.80 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID02 | SARS-CoV-2 | NP swab | 3.49 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID03 | SARS-CoV-2 | NP swab | 3.50 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID04 | SARS-CoV-2 | NP swab | 5.89 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID05 | SARS-CoV-2 | NP swab | 3.98 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID06 | SARS-CoV-2 | NP swab | 5.92 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID07 | SARS-CoV-2 | NP swab | 5.82 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 1.15 | 0.00 | 0.00 |
| RAPID08 | SARS-CoV-2 | NP swab | 5.93 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID09 | SARS-CoV-2 | NP swab | 5.98 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID10 | SARS-CoV-2 | NP swab | 4.90 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID11 | SARS-CoV-2 | NP swab | 4.81 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID12 | SARS-CoV-2 | NP swab | 5.98 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID13 | pdmH1N1 | Culture | 0.00 | 0.00 | 0.00 | 5.94 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID14 | pdmH1N1 | Culture | 0.00 | 0.00 | 0.00 | 5.83 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID15 | None | Culture | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID16 | None | Culture | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID17 | Unknown | NP aspirate | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.62 | 0.00 |
| RAPID18 | Unknown | NP aspirate | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID19 | Unknown | NP aspirate | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID20 | Unknown | NP aspirate | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID21 | Unknown | NP aspirate | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID22 | Unknown | NP aspirate | 0.00 | 0.00 | 0.00 | 0.00 | 5.77 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID23 | Unknown | NP aspirate | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID24 | Unknown | NP aspirate | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID25 | RSV | NP swab | 0.00 | 0.00 | 2.64 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID26 | RSV | NP swab | 0.00 | 3.96 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID27 | RSV | NP swab | 0.00 | 0.00 | 3.25 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID28 | RSV | NP swab | 0.00 | 2.89 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID29 | RSV | NP swab | 0.00 | 0.00 | 4.09 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID30 | RSV | NP swab | 0.00 | 5.25 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID31 | RSV | NP swab | 0.00 | 4.67 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID32 | RSV | NP swab | 0.00 | 0.00 | 3.14 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID33 | Unknown | NP aspirate | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 5.61 | 0.00 | 0.00 | 0.00 |
| RAPID34 | Unknown | NP swab | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 5.94 | 0.00 | 0.00 | 0.00 |
| RAPID35 | Unknown | NP swab | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 6.00 | 0.00 | 0.00 | 0.00 |
| RAPID36 | Unknown | NP aspirate | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 2.35 |
| RAPID37 | Unknown | Vomitus | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| RAPID38 | Unknown | NP swab | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 4.79 | 0.00 | 0.00 | 0.00 |
| RAPID39 | Unknown | NP swab | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 5.98 | 0.00 | 0.00 | 0.00 |
| RAPID40 | NTC | Water | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Log-RPM | 6.00 | 5.00 | 4.00 | 3.00 | 2.00 | 1.00 | 0.00 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tulloch, R.L.; Kim, K.; Sikazwe, C.; Michie, A.; Burrell, R.; Holmes, E.C.; Dwyer, D.E.; Britton, P.N.; Kok, J.; Eden, J.-S. RAPIDprep: A Simple, Fast Protocol for RNA Metagenomic Sequencing of Clinical Samples. Viruses 2023, 15, 1006. https://doi.org/10.3390/v15041006
Tulloch RL, Kim K, Sikazwe C, Michie A, Burrell R, Holmes EC, Dwyer DE, Britton PN, Kok J, Eden J-S. RAPIDprep: A Simple, Fast Protocol for RNA Metagenomic Sequencing of Clinical Samples. Viruses. 2023; 15(4):1006. https://doi.org/10.3390/v15041006
Chicago/Turabian StyleTulloch, Rachel L., Karan Kim, Chisha Sikazwe, Alice Michie, Rebecca Burrell, Edward C. Holmes, Dominic E. Dwyer, Philip N. Britton, Jen Kok, and John-Sebastian Eden. 2023. "RAPIDprep: A Simple, Fast Protocol for RNA Metagenomic Sequencing of Clinical Samples" Viruses 15, no. 4: 1006. https://doi.org/10.3390/v15041006