
Whole Genomic Sequence Analysis of Human Adenovirus Species C Shows Frequent Recombination in Tianjin, China

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. Virus Isolation
2.3. DNA Extraction and Next-Generation Sequencing
2.4. Phylogenetic Analysis
2.5. Recombination Analysis
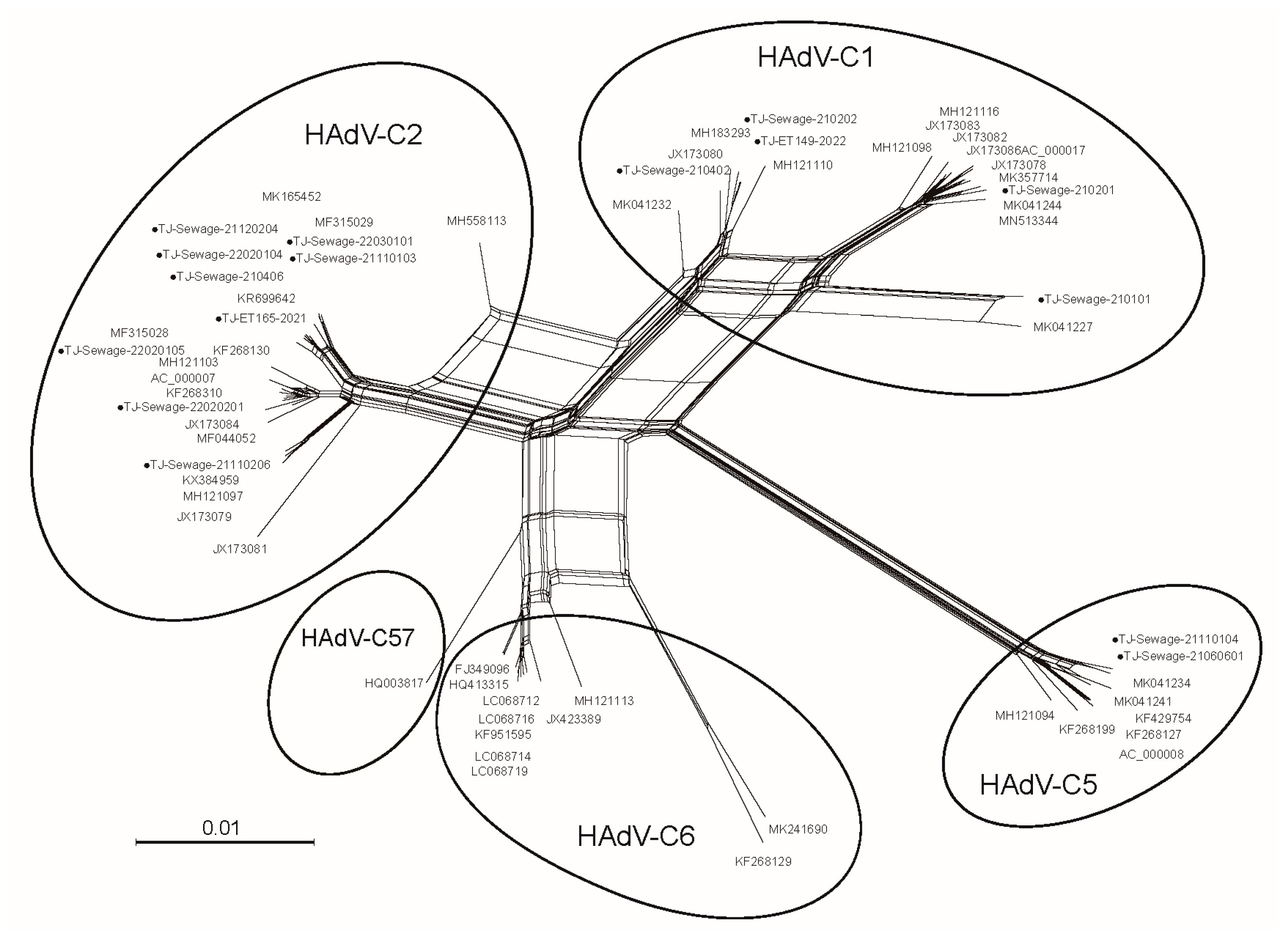
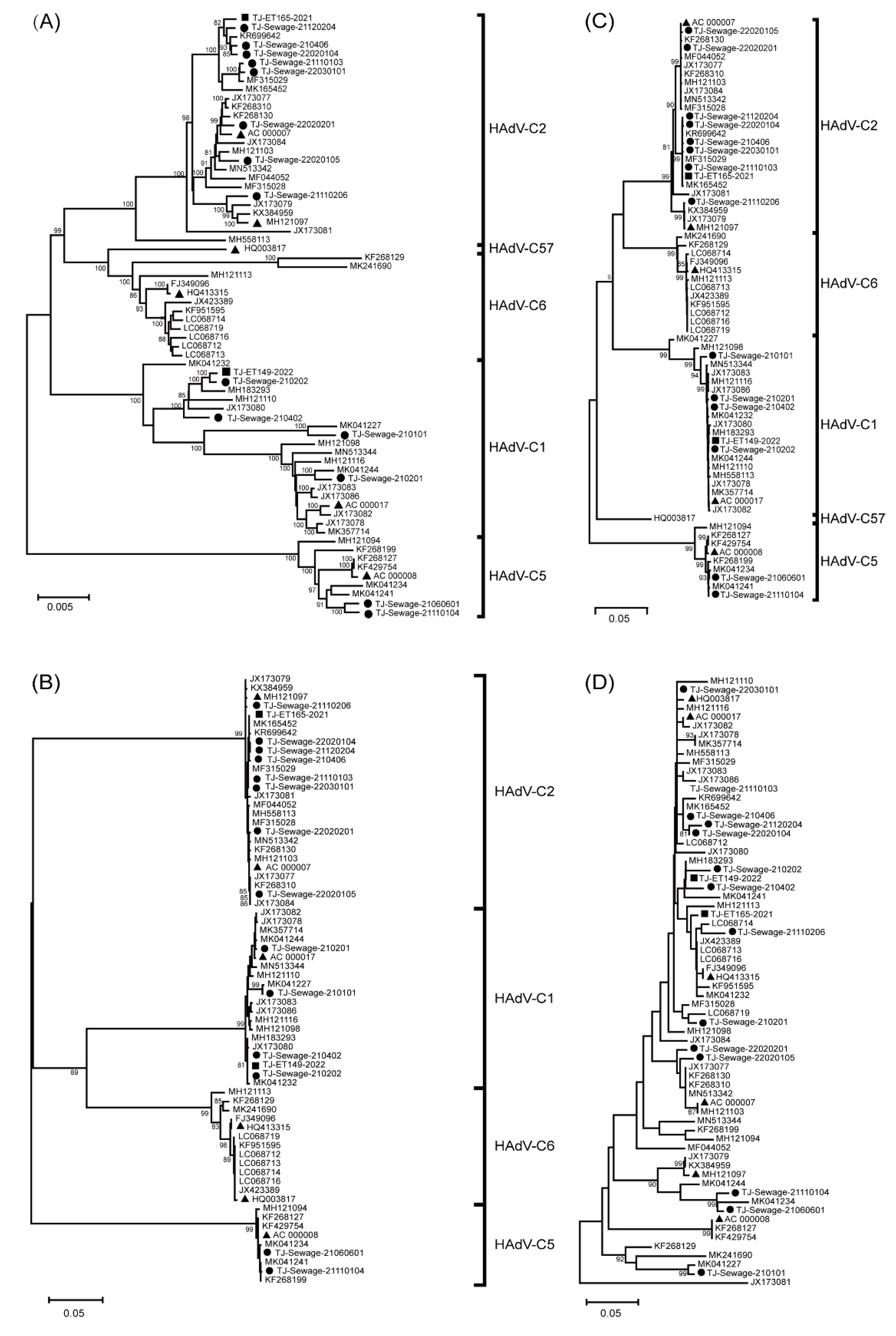
3. Results
3.1. Full-Length Genomic Characterisation
3.2. Phylogenetic Analysis
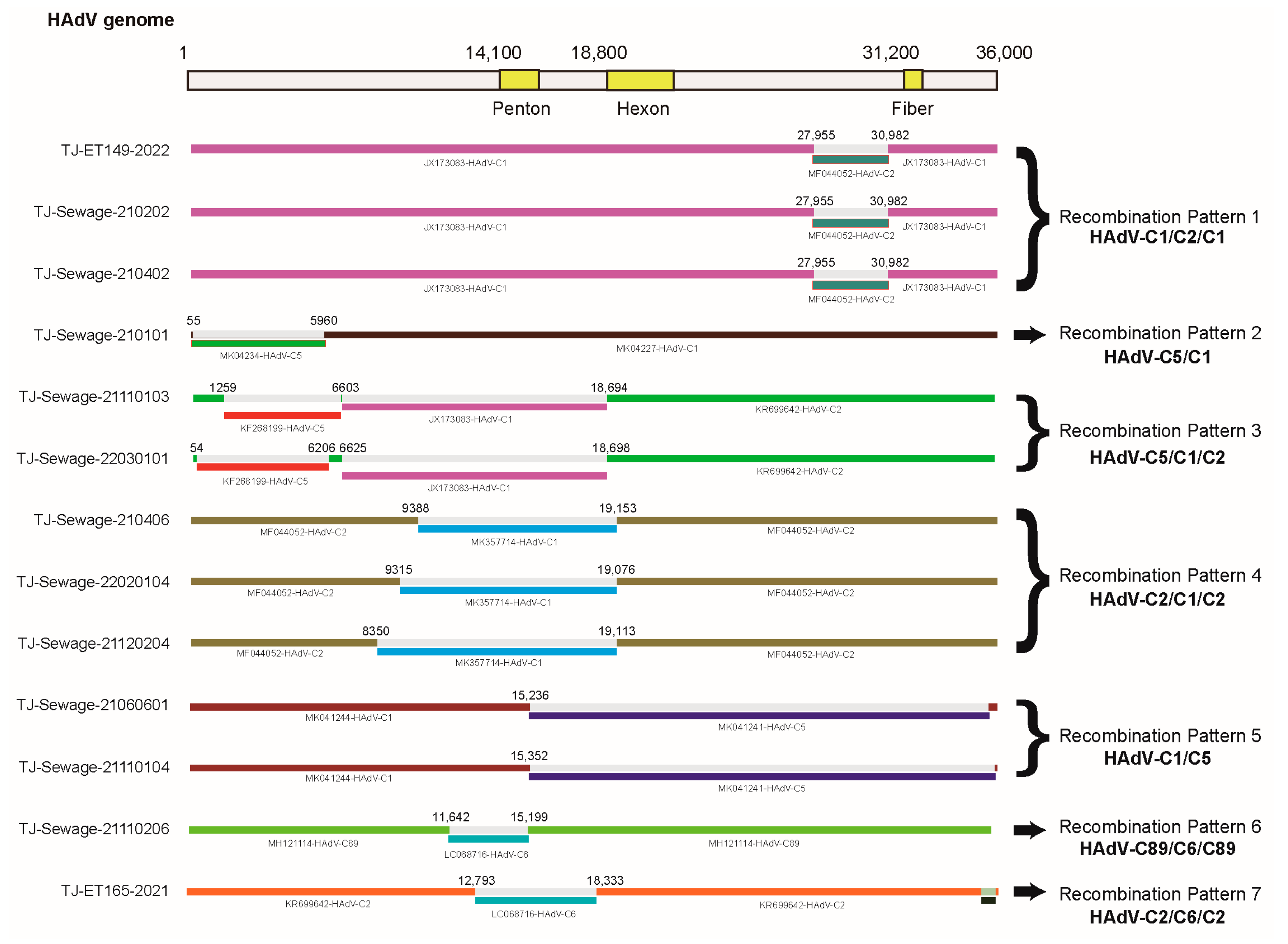
3.3. Recombination Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Benkő, M.; Aoki, K.; Arnberg, N.; Davison, A.; Echavarria, M.; Hess, M.; Jones, M.; Kaján, G.; Kajon, A.; Mittal, S.; et al. ICTV Virus Taxonomy Profile: Adenoviridae 2022. J. Gen. Virol. 2022, 103, 001721. [Google Scholar] [CrossRef]
- Harrach, B.; Benkő, M.; Both, G.W.; Brown, M.; Davison, A.J.; Echavarría, M.; Hess, M.; Jones, M.; Kajon, A.; Lehmkuhl, H.D.; et al. Family Adenoviridae. In Virus Taxonomy: 9th Report of the International Committee on Taxonomy of Viruses; Elsevier: Amsterdam, The Netherlands, 2011; pp. 125–141. [Google Scholar]
- Mao, N.Y.; Zhu, Z.; Zhang, Y.; Xu, W.B. Current status of human adenovirus infection in China. World J. Pediatr. WJP 2022, 18, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.P.; Seto, J.; Liu, E.B.; Dehghan, S.; Hudson, N.R.; Lukashev, A.N.; Ivanova, O.; Chodosh, J.; Dyer, D.W.; Jones, M.S.; et al. Computational analysis of two species C human adenoviruses provides evidence of a novel virus. J. Clin. Microbiol. 2011, 49, 3482–3490. [Google Scholar] [CrossRef]
- Dhingra, A.; Hage, E.; Ganzenmueller, T.; Bottcher, S.; Hofmann, J.; Hamprecht, K.; Obermeier, P.; Rath, B.; Hausmann, F.; Dobner, T.; et al. Molecular Evolution of Human Adenovirus (HAdV) Species C. Sci. Rep. 2019, 9, 1039. [Google Scholar] [CrossRef] [PubMed]
- Ji, T.; Li, L.; Li, W.; Zheng, X.; Ye, X.; Chen, H.; Zhou, Q.; Jia, H.; Chen, B.; Lin, Z.; et al. Emergence and characterization of a putative novel human adenovirus recombinant HAdV-C104 causing pneumonia in Southern China. Virus Evol. 2021, 7, veab018. [Google Scholar] [CrossRef] [PubMed]
- Garnett, C.T.; Talekar, G.; Mahr, J.A.; Huang, W.; Zhang, Y.; Ornelles, D.A.; Gooding, L.R. Latent species C adenoviruses in human tonsil tissues. J. Virol. 2009, 83, 2417–2428. [Google Scholar] [CrossRef]
- Dahling, D.R.; Wright, B.A.; Williams, F.P., Jr. Detection of viruses in environmental samples: Suitability of commercial rotavirus and adenovirus test kits. J. Virol. Methods 1993, 45, 137–147. [Google Scholar] [CrossRef]
- Rigotto, C.; Hanley, K.; Rochelle, P.A.; De Leon, R.; Barardi, C.R.; Yates, M.V. Survival of adenovirus types 2 and 41 in surface and ground waters measured by a plaque assay. Environ. Sci. Technol. 2011, 45, 4145–4150. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.M.; Cui, T.; Dommaraju, K.; Singh, G.; Dehghan, S.; Seto, J.; Shrivastava, S.; Fedorova, N.B.; Gupta, N.; Stockwell, T.B.; et al. Genomic analysis of a large set of currently-and historically-important human adenovirus pathogens. Emerg. Microbes Infect. 2018, 7, 10. [Google Scholar] [CrossRef]
- Zheng, H.; Lu, J.; Zhang, Y.; Yoshida, H.; Guo, X.; Liu, L.; Li, H.; Zeng, H.; Fang, L.; Mo, Y.; et al. Prevalence of nonpolio enteroviruses in the sewage of Guangzhou city, China, from 2009 to 2012. Appl. Environ. Microbiol. 2013, 79, 7679–7683. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mao, N.; Zhang, C.; Ren, B.; Li, H.; Li, N.; Chen, J.; Zhang, R.; Li, H.; Zhu, Z.; et al. Human adenovirus species C recombinant virus continuously circulated in China. Sci. Rep. 2019, 9, 9781. [Google Scholar] [CrossRef]
- Rivailler, P.; Mao, N.; Zhu, Z.; Xu, W. Recombination analysis of Human mastadenovirus C whole genomes. Sci. Rep. 2019, 9, 2182. [Google Scholar] [CrossRef]
- Mao, N.; Zhu, Z.; Rivailler, P.; Yang, J.; Li, Q.; Han, G.; Yin, J.; Yu, D.; Sun, L.; Jiang, H.; et al. Multiple divergent Human mastadenovirus C co-circulating in mainland of China. Infect. Genet. Evol. 2019, 76, 104035. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, L. Genome Analysis of A Novel Recombinant Human Adenovirus Type 1 in China. Sci. Rep. 2019, 9, 4298. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Lu, R.; Zhao, Y.; Xie, Z.; Shen, J.; Tan, W. Phylogenetic evidence for intratypic recombinant events in a novel human adenovirus C that causes severe acute respiratory infection in children. Sci. Rep. 2016, 6, 23014. [Google Scholar] [CrossRef]
- Mao, N.; Zhu, Z.; Rivailler, P.; Chen, M.; Fan, Q.; Huang, F.; Xu, W. Whole genomic analysis of two potential recombinant strains within Human mastadenovirus species C previously found in Beijing, China. Sci. Rep. 2017, 7, 15380. [Google Scholar] [CrossRef]
- Kosulin, K.; Dworzak, S.; Lawitschka, A.; Matthes-Leodolter, S.; Lion, T. Comparison of different approaches to quantitative adenovirus detection in stool specimens of hematopoietic stem cell transplant recipients. J. Clin. Virol. 2016, 85, 31–36. [Google Scholar] [CrossRef]
- Gonzalez, G.; Koyanagi, K.O.; Aoki, K.; Kitaichi, N.; Ohno, S.; Kaneko, H.; Ishida, S.; Watanabe, H. Intertypic modular exchanges of genomic segments by homologous recombination at universally conserved segments in human adenovirus species D. Gene 2014, 547, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, A.N.; Ivanova, O.E.; Eremeeva, T.P.; Iggo, R.D. Evidence of frequent recombination among human adenoviruses. J. Gen. Virol. 2008, 89, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhao, S.; Rao, H. Whole genomic analysis of a potential recombinant human adenovirus type 1 in Qinghai plateau, China. Virol. J. 2020, 17, 111. [Google Scholar] [CrossRef] [PubMed]
- Chroboczek, J.; Bieber, F.; Jacrot, B. The sequence of the genome of adenovirus type 5 and its comparison with the genome of adenovirus type 2. Virology 1992, 186, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Peng, J.; Fang, L.; Zeng, L.; Lin, H.; Xiong, Q.; Liu, Z.; Jiang, H.; Zhang, C.; Yi, L.; et al. Capturing noroviruses circulating in the population: Sewage surveillance in Guangdong, China (2013–2018). Water Res. 2021, 196, 116990. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Lin, X.; Ji, F.; Xiong, P.; Liu, Y.; Wang, S.; Chen, P.; Xu, Q.; Zhang, L.; Tao, Z.; et al. Prevalence and Bayesian Phylogenetics of Enteroviruses Derived From Environmental Surveillance Around Polio Vaccine Switch Period in Shandong Province, China. Food Environ. Virol. 2020, 12, 321–332. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | Genome Sizes | GC Contents (%) |
|---|---|---|
| TJ-ET165-2021 | 35,390 | 55.33 |
| TJ-Sewage-210101 | 35,900 | 55.45 |
| TJ-Sewage-210201 | 35,835 | 55.31 |
| TJ-Sewage-210202 | 35,774 | 55.30 |
| TJ-Sewage-210402 | 35,795 | 55.33 |
| TJ-Sewage-210406 | 35,644 | 55.22 |
| TJ-Sewage-21060601 | 35,765 | 55.22 |
| TJ-Sewage-21110103 | 35,786 | 55.27 |
| TJ-Sewage-21110104 | 35,761 | 55.18 |
| TJ-Sewage-21110206 | 35,773 | 55.21 |
| TJ-Sewage-21120204 | 35,815 | 55.27 |
| TJ-ET149-2022 | 35,751 | 55.31 |
| TJ-Sewage-22020104 | 35,772 | 55.24 |
| TJ-Sewage-22020105 | 35,790 | 55.22 |
| TJ-Sewage-22020201 | 35,774 | 55.23 |
| TJ-Sewage-22030101 | 35,790 | 55.27 |
| Sequence | Nucleotide Identity (%) | |||||
|---|---|---|---|---|---|---|
| HAdV-C1 | HAdV-C2 | HAdV-C5 | HAdV-C6 | HAdV-C57 | HAdV-C89 | |
| TJ-ET165-2021 | 98.45 | 99.08 | 98.62 | 98.91 | 97.54 | 98.87 |
| TJ-Sewage-210101 | 97.31 | 97.51 | 98.22 | 98.19 | 96.69 | 97.58 |
| TJ-Sewage-210201 | 99.31 | 98.20 | 98.36 | 96.93 | 97.27 | 98.26 |
| TJ-Sewage-210202 | 99.03 | 98.23 | 98.59 | 97.04 | 97.57 | 98.37 |
| TJ-Sewage-210402 | 99.03 | 98.42 | 98.51 | 97.12 | 97.59 | 98.43 |
| TJ-Sewage-210406 | 98.59 | 99.05 | 98.61 | 98.75 | 97.54 | 98.90 |
| TJ-Sewage-21060601 | 98.44 | 98.53 | 99.00 | 98.58 | 98.63 | 98.70 |
| TJ-Sewage-21110103 | 98.63 | 98.97 | 98.68 | 98.33 | 97.48 | 98.80 |
| TJ-Sewage-21110104 | 98.47 | 98.53 | 98.99 | 98.62 | 98.68 | 98.71 |
| TJ-Sewage-21110206 | 98.20 | 98.94 | 98.62 | 98.77 | 97.46 | 99.52 |
| TJ-Sewage-21120204 | 98.55 | 99.07 | 98.62 | 98.85 | 97.52 | 98.85 |
| TJ-ET149-2022 | 99.03 | 98.28 | 98.66 | 97.07 | 97.56 | 98.41 |
| TJ-Sewage-22020104 | 98.55 | 99.03 | 98.57 | 98.74 | 97.53 | 98.82 |
| TJ-Sewage-22020105 | 98.41 | 99.50 | 98.58 | 98.18 | 97.57 | 98.80 |
| TJ-Sewage-22020201 | 98.19 | 99.68 | 98.86 | 98.60 | 97.44 | 98.94 |
| TJ-Sewage-22030101 | 98.28 | 98.94 | 98.62 | 98.28 | 97.46 | 98.77 |
| Sequence | Most Closely Identical Strain in GenBank | Nucleotide Identity (%) |
|---|---|---|
| TJ-ET165-2021 | ON054624 | 99.82 |
| TJ-Sewage-210101 | MK041227 | 99.41 |
| TJ-Sewage-210201 | MK041244 | 99.59 |
| TJ-Sewage-210202 | MT263140 | 99.77 |
| TJ-Sewage-210402 | MH183293 | 99.37 |
| TJ-Sewage-210406 | ON054624 | 99.90 |
| TJ-Sewage-21060601 | MK041234 | 99.36 |
| TJ-Sewage-21110103 | MF315029 | 99.89 |
| TJ-Sewage-21110104 | MK041242 | 99.29 |
| TJ-Sewage-21110206 | MH121114 | 99.63 |
| TJ-Sewage-21120204 | ON054624 | 99.82 |
| TJ-ET149-2022 | MT263140 | 99.78 |
| TJ-Sewage-22020104 | ON054624 | 99.88 |
| TJ-Sewage-22020105 | MH121084 | 99.85 |
| TJ-Sewage-22020201 | MZ151863 | 99.78 |
| TJ-Sewage-22030101 | MF315029 | 99.88 |
| Recombinant Strain | Parent Major/Minor | Recombinant Region in Alignment | Model (Average p Value) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| RDP | GENECONV | BootScan | MaxChi | Chimaera | SiScan | 3Seq | |||
| TJ-Sewage-210202 | JX173083/ MF044052 | 28256–31639 | 3.604 × 10−183 | 7.272 × 10−186 | 1.532 × 10−179 | 5.392 × 10−49 | 2.865 × 10−44 | 1.688 × 10−52 | 4.419 × 10−19 |
| TJ-Sewage-210402 | |||||||||
| TJ-ET149-2022 | |||||||||
| TJ-Sewage-210101 | MK041227/ MK041234 | 100–6023 | 3.914 × 10−66 | 5.280 × 10−61 | 4.680 × 10−53 | 4.268 × 10−19 | 5.374 × 10−17 | 1.318 × 10−18 | 2.220 × 10−15 |
| TJ-Sewage-21110103 | KR699642/ JX173083 | 6740–18910 | 3.421 × 10−46 | 3.657 × 10−18 | 1.226 × 10−15 | 3.064 × 10−12 | 9.972 × 10−14 | 3.117 × 10−12 | 1.914 × 10−5 |
| KR699642/ KF268199 | 1361–6739 | 1.391 × 10−43 | 1.610 × 10−5 | 3.268 × 10−29 | 3.555 × 10−10 | 6.242 × 10−4 | 1.415 × 10−7 | 2.220 × 10−15 | |
| TJ-Sewage-22030101 | KR699642/ JX173083 | 6762–18910 | 5.498 × 10−46 | 4.178 × 10−19 | 6.486 × 10−15 | 2.483 × 10−12 | 3.145 × 10−04 | 2.221 × 10−12 | 3.111 × 10−05 |
| KR699642/ KF268199 | 144–6206 | 9.919 × 10−43 | 1.592 × 10−05 | 1.700 × 10−28 | 4.405 × 10−3 | 1.203 × 10−3 | 2.814E × 10−7 | 2.220 × 10−15 | |
| TJ-Sewage-21110104 | MK041244/MK041241 | 15439–36508 | 1.795 × 10−52 | 4.478 × 10−18 | 9.904 × 10−25 | 6.752 × 10−13 | 9.492 × 10−18 | 2.382 × 10−121 | 1.193 × 10−67 |
| TJ-Sewage-21060601 | 15439–36227 | ||||||||
| TJ-Sewage-210406 | MF044052/ MK357714 | 10299–19306 | 1.125 × 10−44 | 2.795 × 10−52 | 3.560 × 10−20 | 1.174 × 10−12 | 1.465 × 10−10 | 2.858 × 10−13 | 8.715 × 10−11 |
| TJ-Sewage-22020104 | 9473–19306 | ||||||||
| TJ-Sewage-21120204 | 8450–19306 | ||||||||
| TJ-Sewage-21110206 | MH121114/ LC068716 | 11758–15320 | 1.321 × 10−35 | 2.689 × 10−27 | 8.306 × 10−33 | 1.169 × 10−10 | 8.263 × 10−11 | 2.595 × 10−09 | 3.330 × 10−15 |
| TJ-ET165-2021 | KR699642/ LC068716 | 12982–18546 | 1.754 × 10−33 | 7.011 × 10−22 | 1.141 × 10−23 | 3.703 × 10−11 | 5.590 × 10−11 | —— | 3.330 × 10−15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, Y.; Zhuang, Z.; Liu, Y.; Tan, Z.; Gao, X.; Li, X.; Yang, D. Whole Genomic Sequence Analysis of Human Adenovirus Species C Shows Frequent Recombination in Tianjin, China. Viruses 2023, 15, 1004. https://doi.org/10.3390/v15041004
Lei Y, Zhuang Z, Liu Y, Tan Z, Gao X, Li X, Yang D. Whole Genomic Sequence Analysis of Human Adenovirus Species C Shows Frequent Recombination in Tianjin, China. Viruses. 2023; 15(4):1004. https://doi.org/10.3390/v15041004
Chicago/Turabian StyleLei, Yue, Zhichao Zhuang, Yang Liu, Zhaolin Tan, Xin Gao, Xiaoyan Li, and Dongjing Yang. 2023. "Whole Genomic Sequence Analysis of Human Adenovirus Species C Shows Frequent Recombination in Tianjin, China" Viruses 15, no. 4: 1004. https://doi.org/10.3390/v15041004