
Crystal Structures of Inhibitor-Bound Main Protease from Delta- and Gamma-Coronaviruses
 , , and
, , and 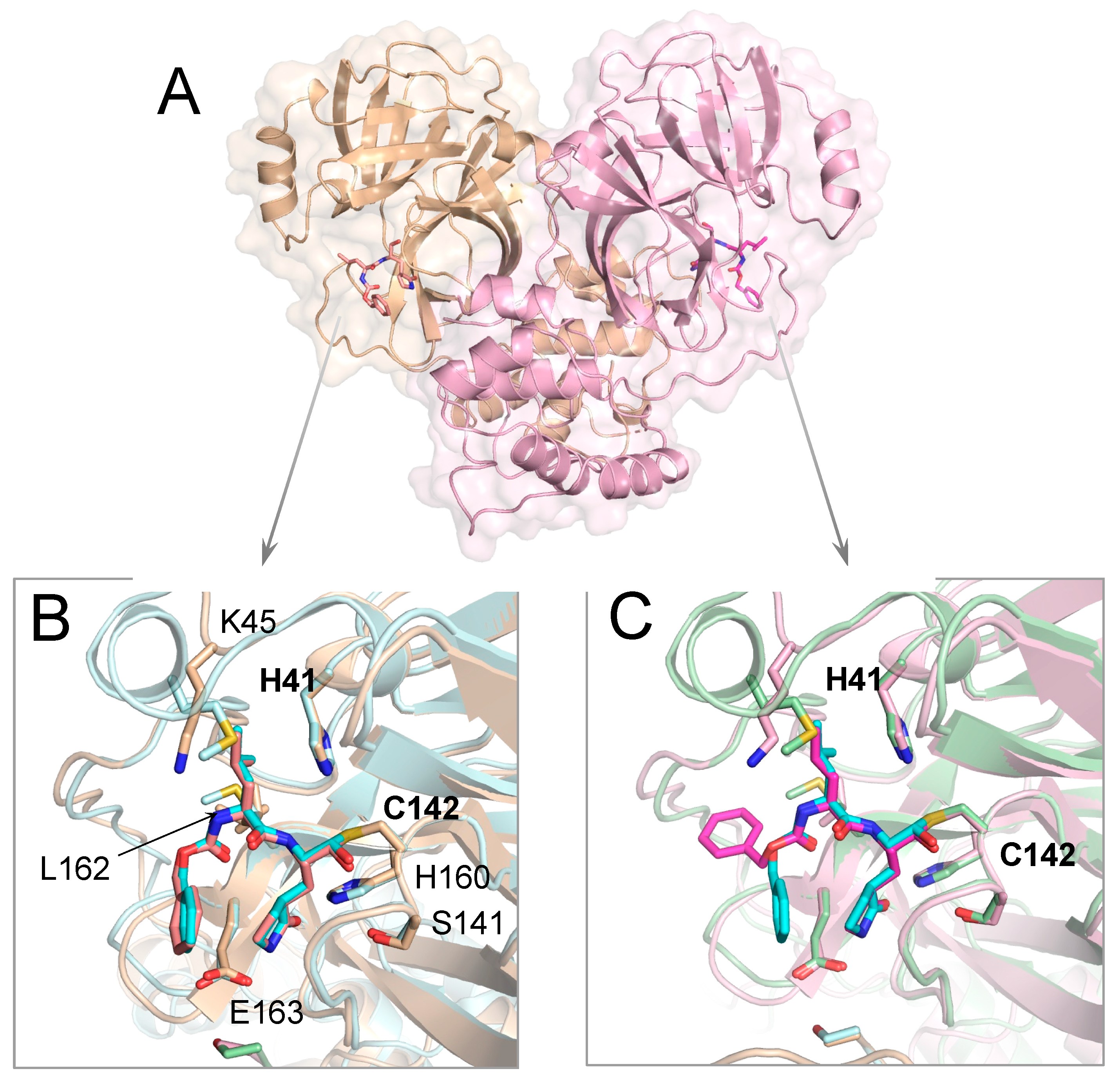{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification of Mpro
2.2. Protein Crystallization
2.3. Data Collection and Structure Determination
3. Results
3.1. Crystal Structure of Inhibitor-Bound Gamma-Coronavirus SW1 Mpro
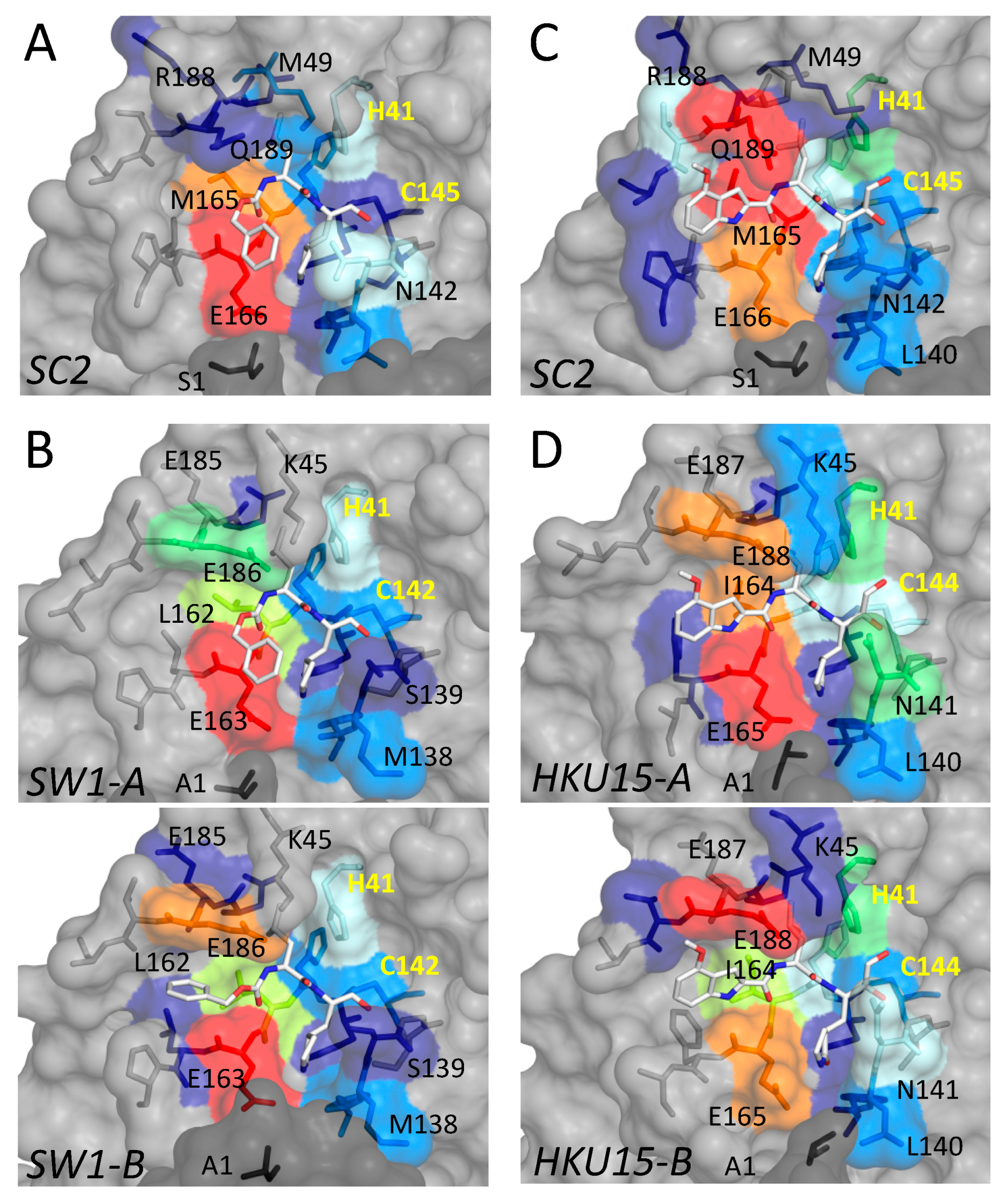
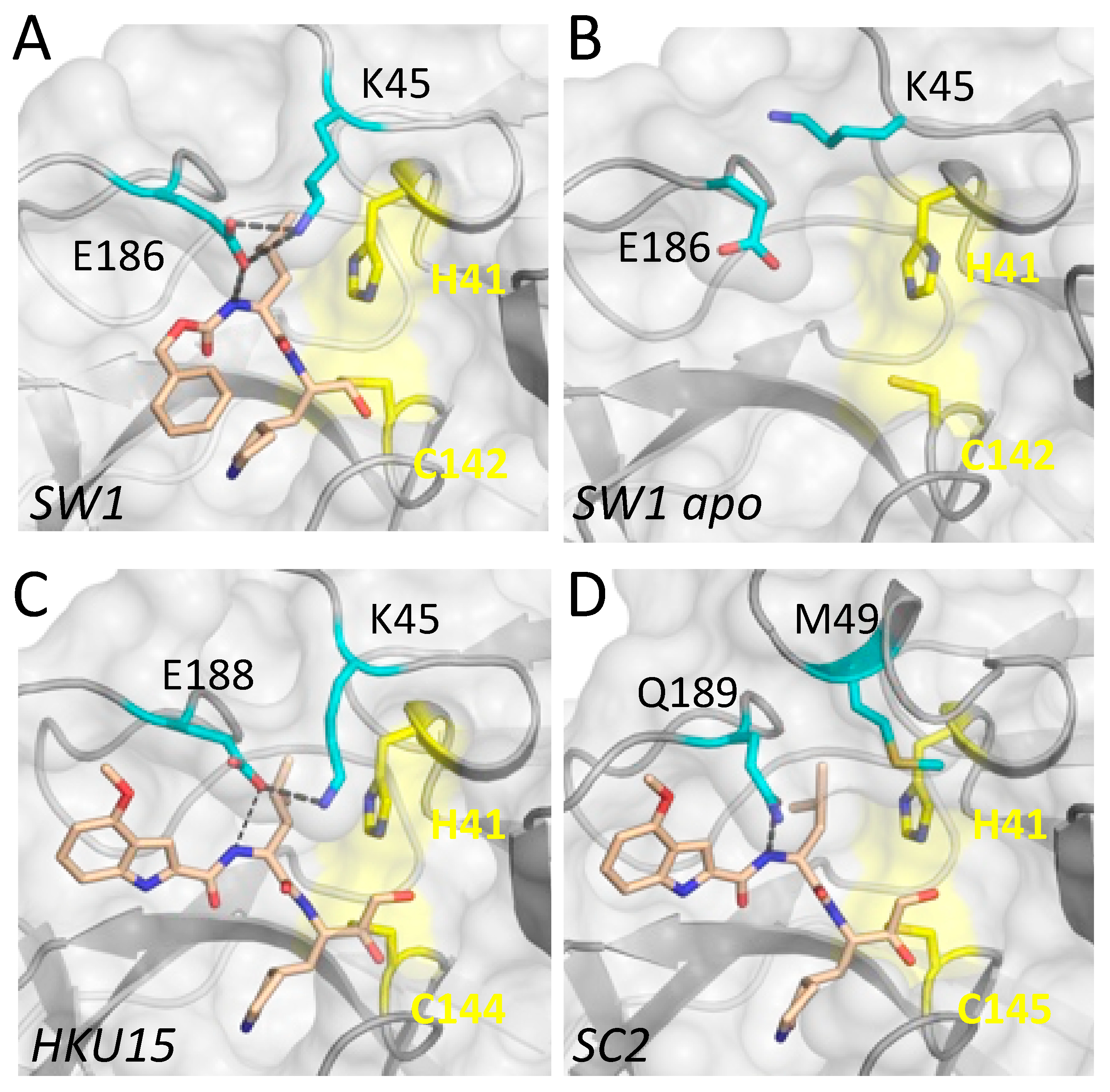
3.2. Inhibitor Binding to Delta and Gamma-Coronavirus Mpro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruiz-Aravena, M.; McKee, C.; Gamble, A.; Lunn, T.; Morris, A.; Snedden, C.E.; Yinda, C.K.; Port, J.R.; Buchholz, D.W.; Yeo, Y.Y.; et al. Ecology, evolution and spillover of coronaviruses from bats. Nat. Rev. Microbiol. 2022, 20, 299–314. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Drosten, C.; Kellam, P.; Memish, Z.A. Evidence for camel-to-human transmission of MERS coronavirus. N. Engl. J. Med. 2014, 371, 1359–1360. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Guan, Y.J.; et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lovell, S.; Tiew, K.C.; Mandadapu, S.R.; Alliston, K.R.; Battaile, K.P.; Groutas, W.C.; Chang, K.O. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol. 2012, 86, 11754–11762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Chen, S.A.; Khan, M.B.; Brassard, R.; Arutyunova, E.; Lamer, T.; Vuong, W.; Fischer, C.; Young, H.S.; Vederas, J.C.; et al. Crystallization of Feline Coronavirus M(pro) With GC376 Reveals Mechanism of Inhibition. Front. Chem. 2022, 10, 852210. [Google Scholar] [CrossRef] [PubMed]
- Iketani, S.; Forouhar, F.; Liu, H.; Hong, S.J.; Lin, F.Y.; Nair, M.S.; Zask, A.; Huang, Y.; Xing, L.; Stockwell, B.R.; et al. Lead compounds for the development of SARS-CoV-2 3CL protease inhibitors. Nat. Commun. 2021, 12, 2016. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Vuong, W.; Khan, M.B.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; McKay, R.T.; van Belkum, M.J.; Joyce, M.A.; et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020, 11, 4282. [Google Scholar] [CrossRef] [PubMed]
- Arutyunova, E.; Khan, M.B.; Fischer, C.; Lu, J.; Lamer, T.; Vuong, W.; van Belkum, M.J.; McKay, R.T.; Tyrrell, D.L.; Vederas, J.C.; et al. N-Terminal Finger Stabilizes the S1 Pocket for the Reversible Feline Drug GC376 in the SARS-CoV-2 M(pro) Dimer. J. Mol. Biol. 2021, 433, 167003. [Google Scholar] [CrossRef] [PubMed]
- Mihindukulasuriya, K.A.; Wu, G.; St Leger, J.; Nordhausen, R.W.; Wang, D. Identification of a novel coronavirus from a beluga whale by using a panviral microarray. J. Virol. 2008, 82, 5084–5088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Chen, C.; Wang, Z.; Han, X.; Shi, P.; Zhou, K.; Liu, X.; Xiao, Y.; Cai, Y.; Huang, J.; et al. The Structure of the Porcine Deltacoronavirus Main Protease Reveals a Conserved Target for the Design of Antivirals. Viruses 2022, 14, 486. [Google Scholar] [CrossRef]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of two coronavirus main proteases: Implications for substrate binding and antiviral drug design. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Lockbaum, G.J.; Reyes, A.C.; Lee, J.M.; Tilvawala, R.; Nalivaika, E.A.; Ali, A.; Kurt Yilmaz, N.; Thompson, P.R.; Schiffer, C.A. Crystal Structure of SARS-CoV-2 Main Protease in Complex with the Non-Covalent Inhibitor ML188. Viruses 2021, 13, 174. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, N.W.; Grosse-Kunstleve, R.W.; Adams, P.D. electronic Ligand Builder and Optimization Workbench (eLBOW): A tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 1074–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunger, A.T. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B., 3rd; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Shaqra, A.M.; Zvornicanin, S.N.; Huang, Q.Y.J.; Lockbaum, G.J.; Knapp, M.; Tandeske, L.; Bakan, D.T.; Flynn, J.; Bolon, D.N.A.; Moquin, S.; et al. Defining the substrate envelope of SARS-CoV-2 main protease to predict and avoid drug resistance. Nat. Commun. 2022, 13, 3556. [Google Scholar] [CrossRef]
- Ho, C.Y.; Yu, J.X.; Wang, Y.C.; Lin, Y.C.; Chiu, Y.F.; Gao, J.Y.; Lai, S.J.; Chen, M.J.; Huang, W.C.; Tien, N.; et al. A Structural Comparison of SARS-CoV-2 Main Protease and Animal Coronaviral Main Protease Reveals Species-Specific Ligand Binding and Dimerization Mechanism. Int. J. Mol. Sci. 2022, 23, 5669. [Google Scholar] [CrossRef]
- Flynn, J.M.; Samant, N.; Schneider-Nachum, G.; Barkan, D.T.; Yilmaz, N.K.; Schiffer, C.A.; Moquin, S.A.; Dovala, D.; Bolon, D.N.A. Comprehensive fitness landscape of SARS-CoV-2 M(pro) reveals insights into viral resistance mechanisms. Elife 2022, 11, e77433. [Google Scholar] [CrossRef]
- Wang, L.; Maddox, C.; Terio, K.; Lanka, S.; Fredrickson, R.; Novick, B.; Parry, C.; McClain, A.; Ross, K. Detection and Characterization of New Coronavirus in Bottlenose Dolphin, United States, 2019. Emerg. Infect. Dis. 2020, 26, 1610–1612. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Tsang, A.K.; Hui, S.W.; Fan, R.Y.; Martelli, P.; Yuen, K.Y. Discovery of a novel bottlenose dolphin coronavirus reveals a distinct species of marine mammal coronavirus in Gammacoronavirus. J. Virol. 2014, 88, 1318–1331. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wang, M.; et al. Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhama, K.; Patel, S.K.; Sharun, K.; Pathak, M.; Tiwari, R.; Yatoo, M.I.; Malik, Y.S.; Sah, R.; Rabaan, A.A.; Panwar, P.K.; et al. SARS-CoV-2 jumping the species barrier: Zoonotic lessons from SARS, MERS and recent advances to combat this pandemic virus. Travel Med. Infect. Dis. 2020, 37, 101830. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zvornicanin, S.N.; Shaqra, A.M.; Huang, Q.J.; Ornelas, E.; Moghe, M.; Knapp, M.; Moquin, S.; Dovala, D.; Schiffer, C.A.; Kurt Yilmaz, N. Crystal Structures of Inhibitor-Bound Main Protease from Delta- and Gamma-Coronaviruses. Viruses 2023, 15, 781. https://doi.org/10.3390/v15030781
Zvornicanin SN, Shaqra AM, Huang QJ, Ornelas E, Moghe M, Knapp M, Moquin S, Dovala D, Schiffer CA, Kurt Yilmaz N. Crystal Structures of Inhibitor-Bound Main Protease from Delta- and Gamma-Coronaviruses. Viruses. 2023; 15(3):781. https://doi.org/10.3390/v15030781
Chicago/Turabian StyleZvornicanin, Sarah N., Ala M. Shaqra, Qiuyu J. Huang, Elizabeth Ornelas, Mallika Moghe, Mark Knapp, Stephanie Moquin, Dustin Dovala, Celia A. Schiffer, and Nese Kurt Yilmaz. 2023. "Crystal Structures of Inhibitor-Bound Main Protease from Delta- and Gamma-Coronaviruses" Viruses 15, no. 3: 781. https://doi.org/10.3390/v15030781