
Viral Metagenomic Analysis of the Fecal Samples in Domestic Dogs (Canis lupus familiaris)

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Sample Collection
2.3. Nucleic Acid Extraction, Library Preparation, and Sequencing
2.4. Bioinformatics Analysis
2.5. Phylogenetic Analysis
3. Results
3.1. Overview of the Viral Metagenomics
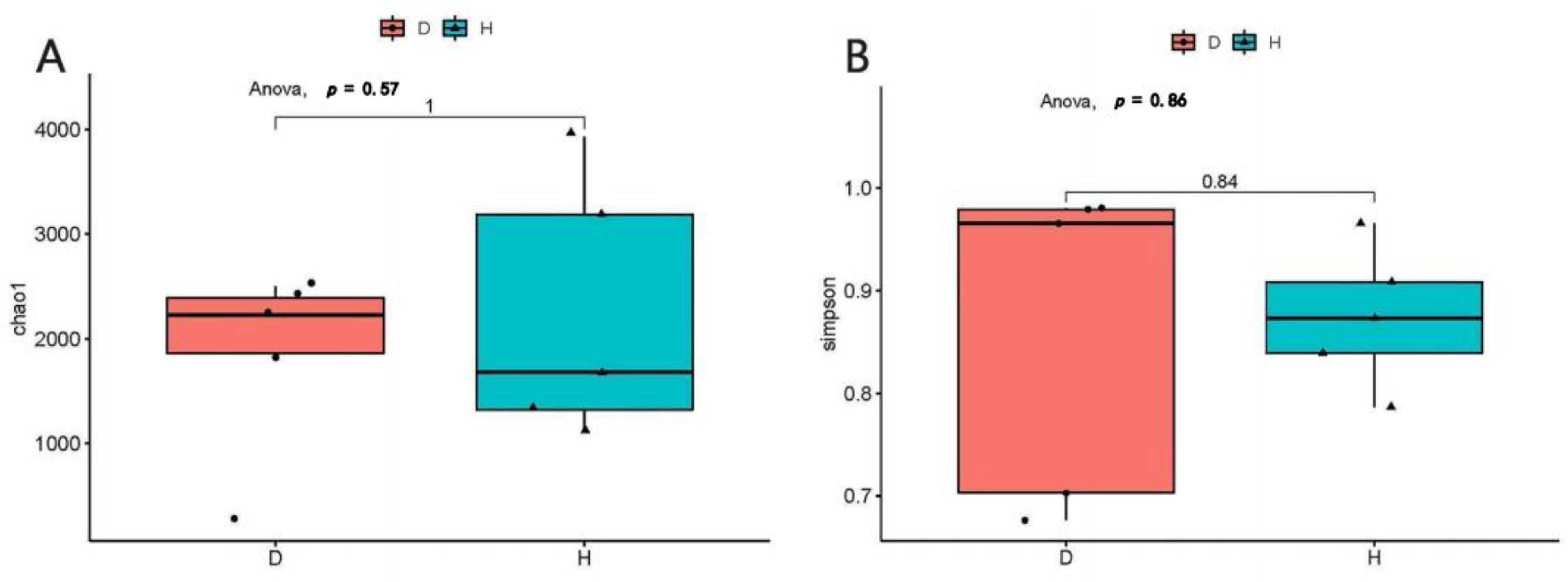
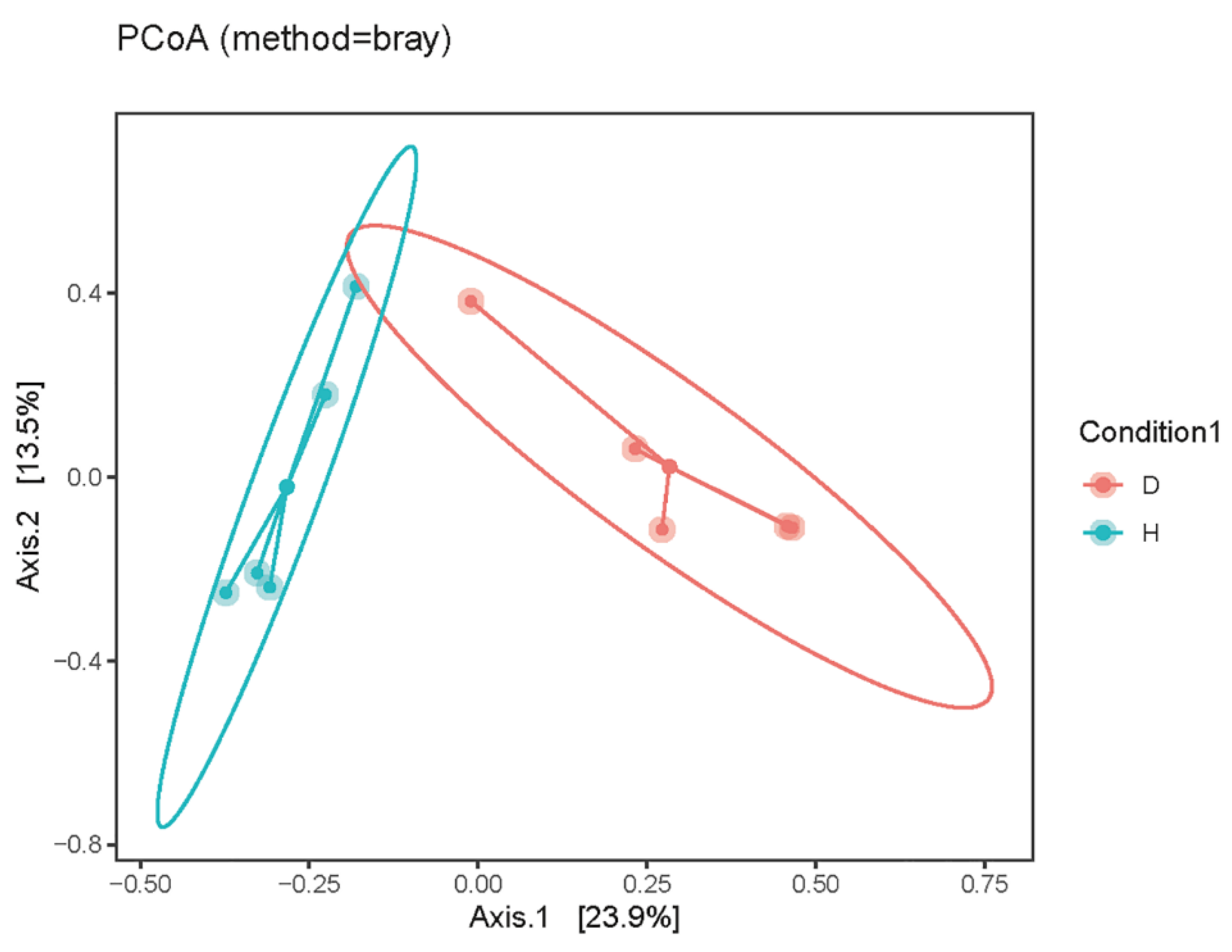
3.2. The Alpha and Beta Diversity of Domestic Dogs’ Gut Virome
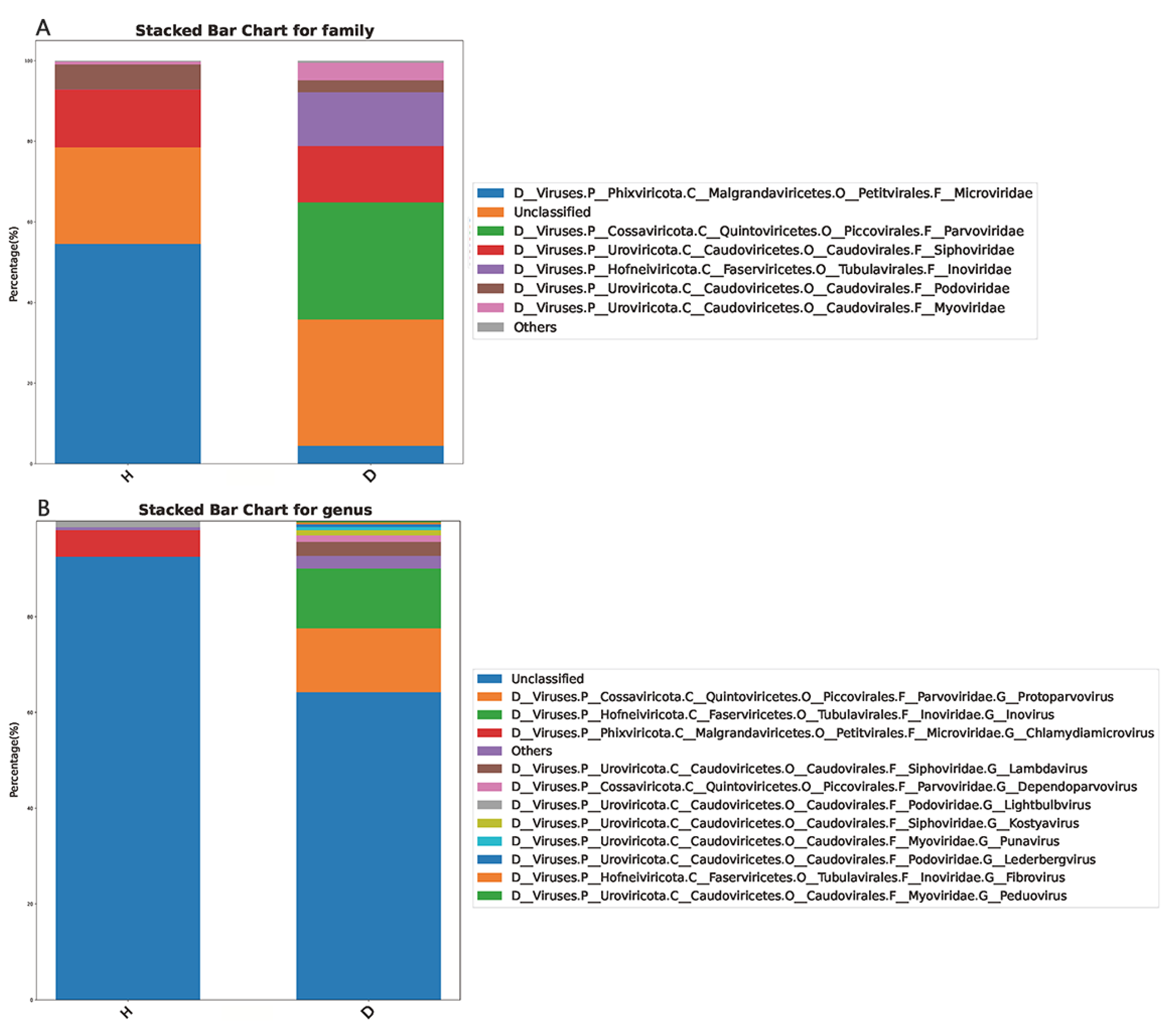
3.3. Taxa Compositions of the Domestic Dogs’s Gut Virome
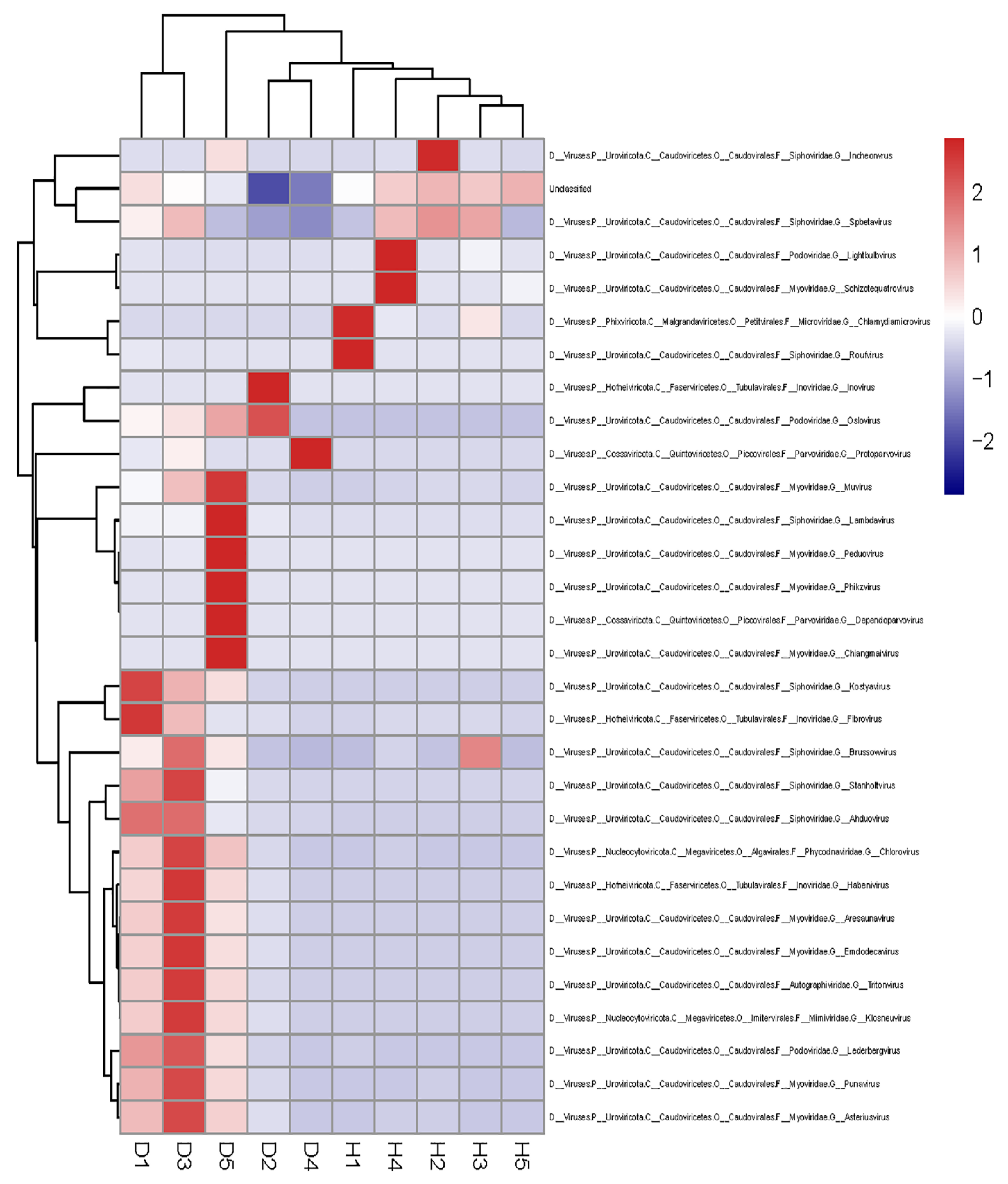
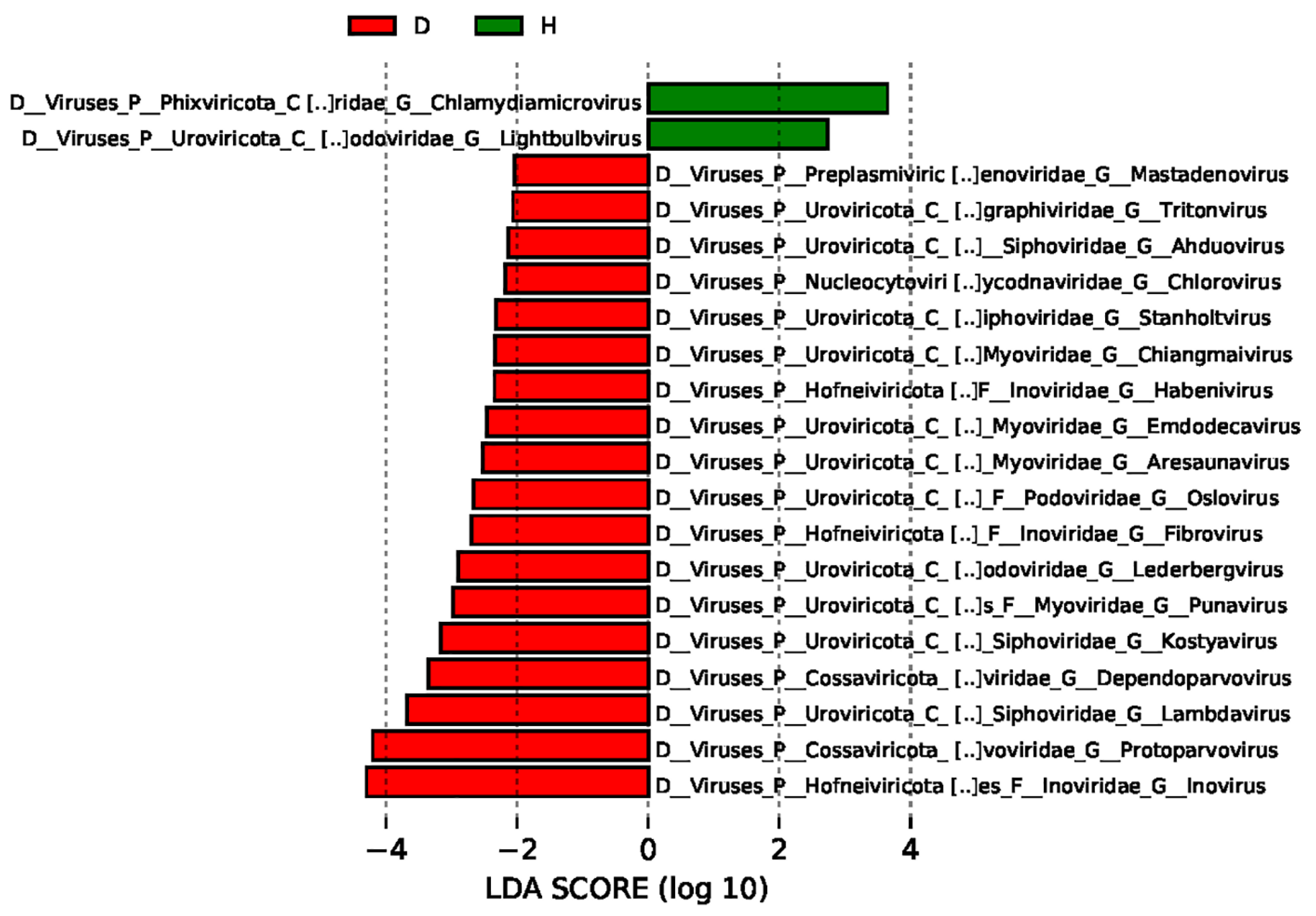
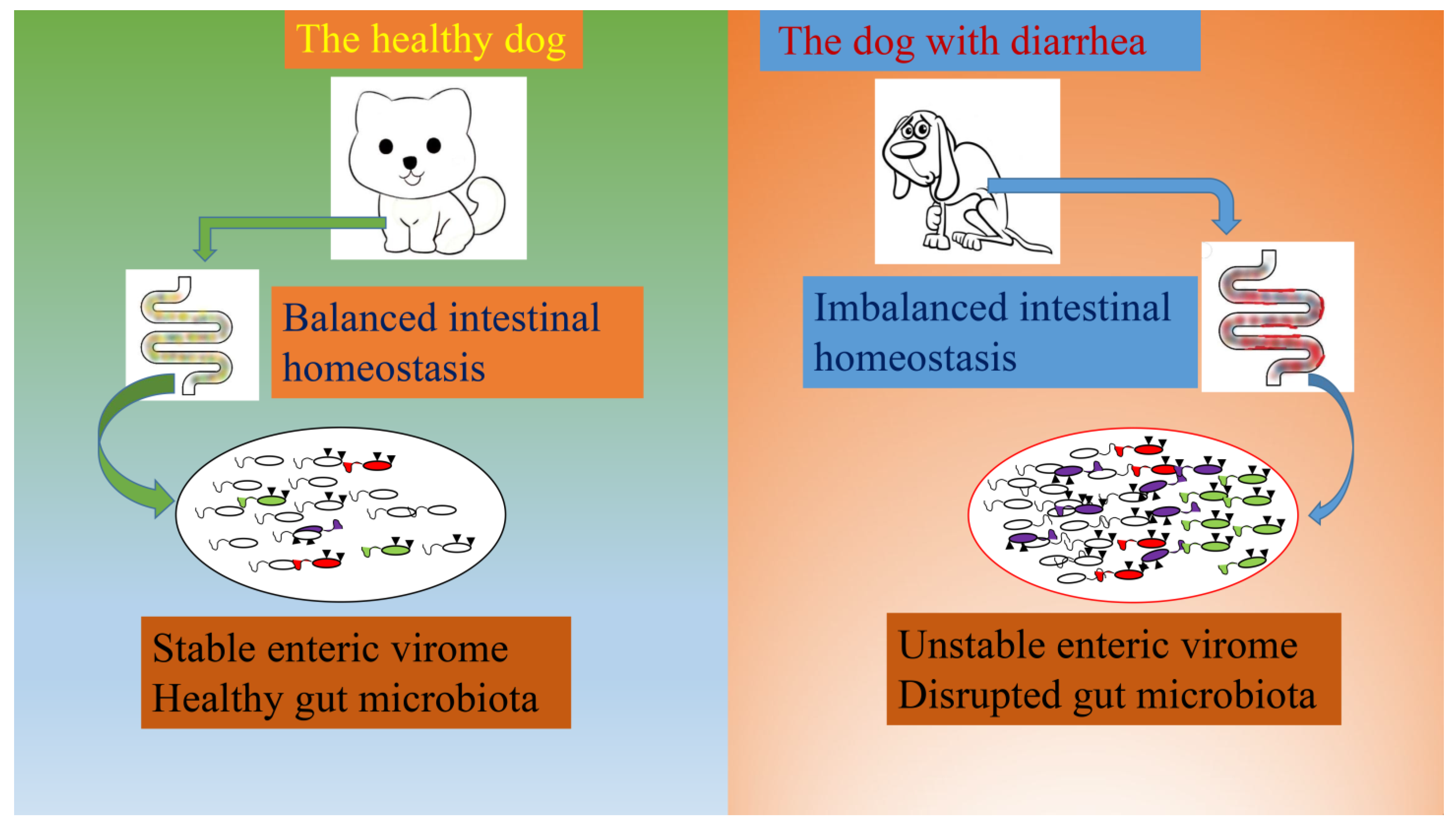
3.4. Comparisons of the Gut Viral Communities in Healthy Dogs and Dogs with Diarrhea
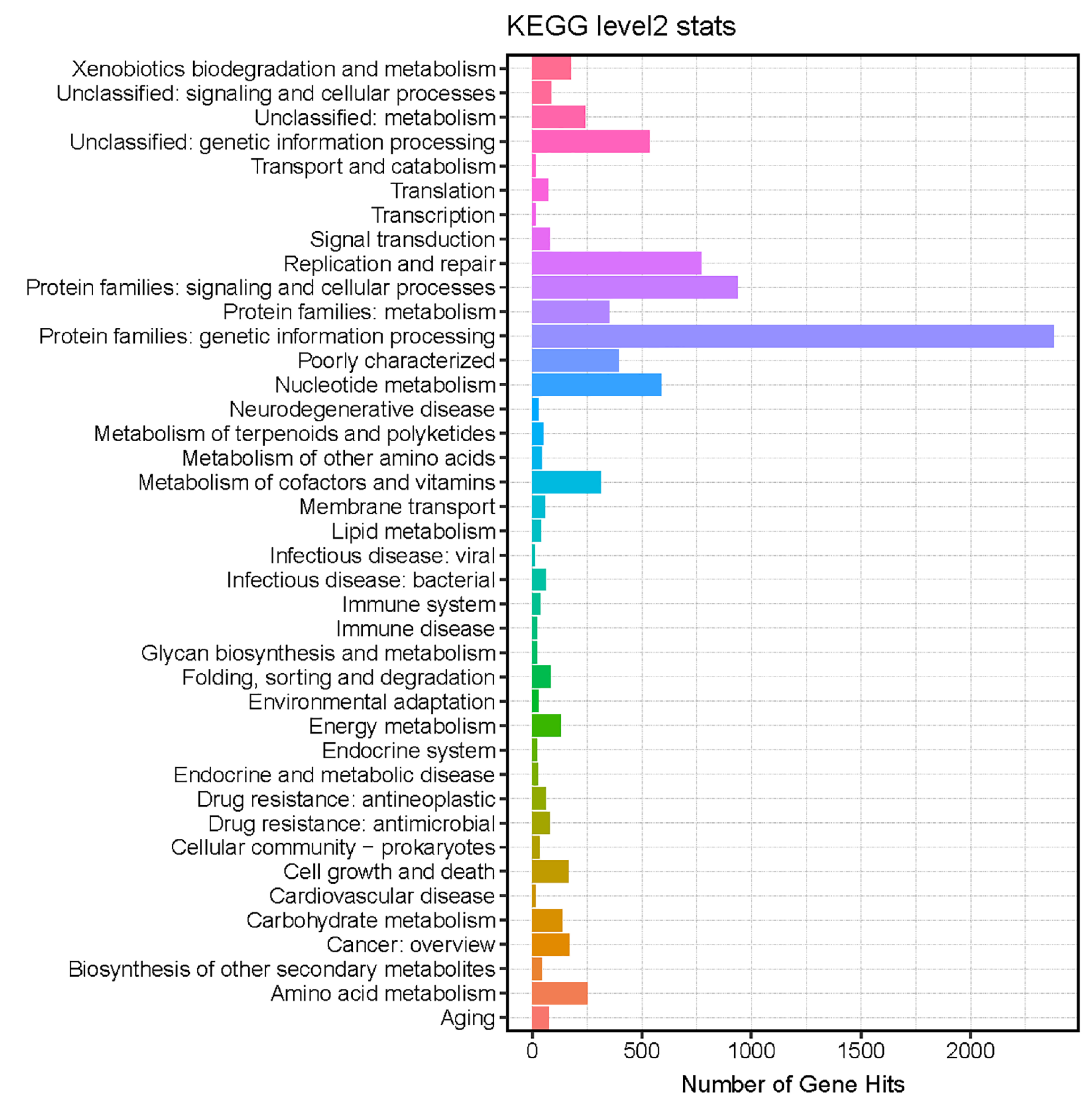
3.5. Functional Annotations of the Identified Viral Genome
3.6. Predictions of the Phages’ Bacterial Hosts
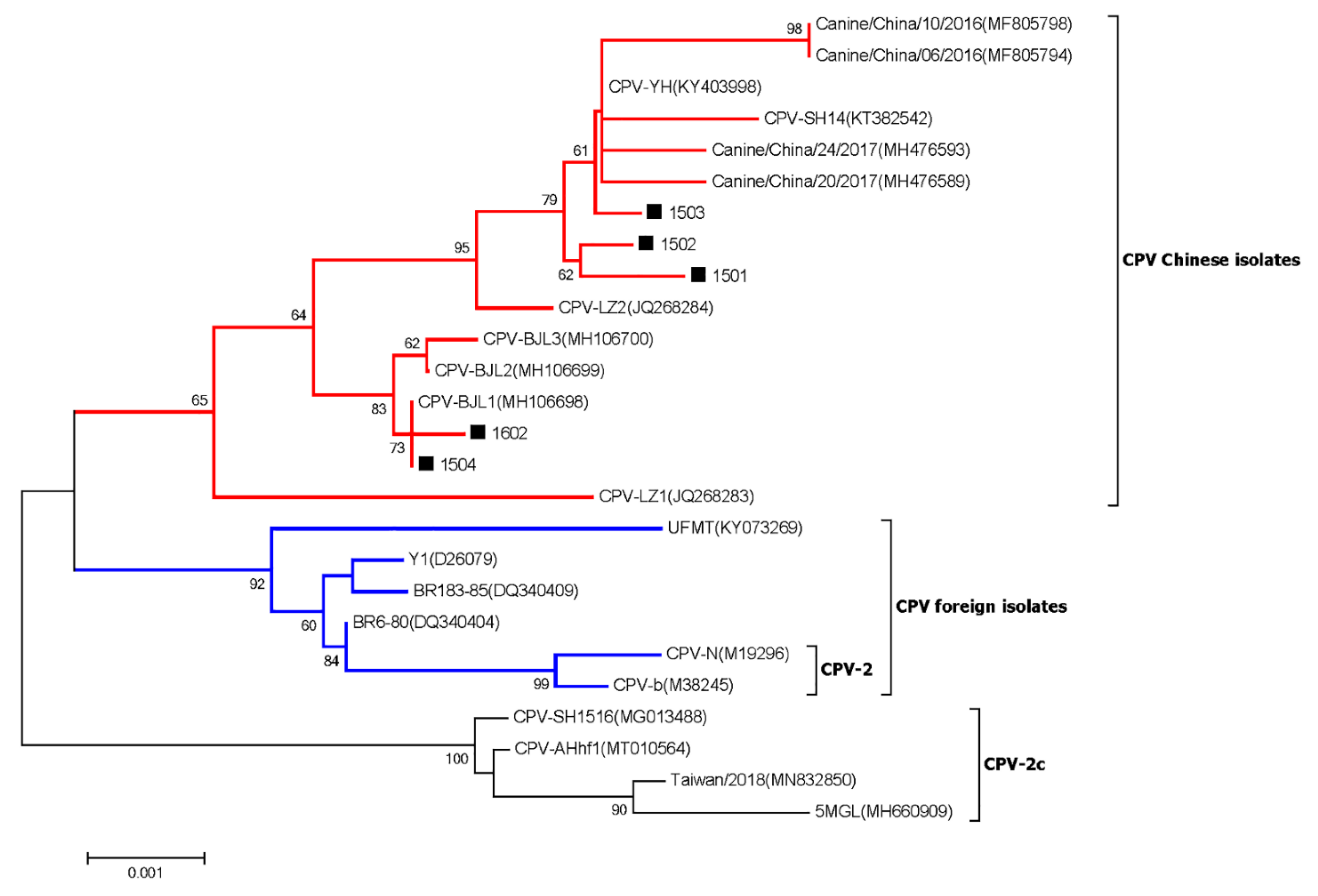
3.7. Canine Parvovirus
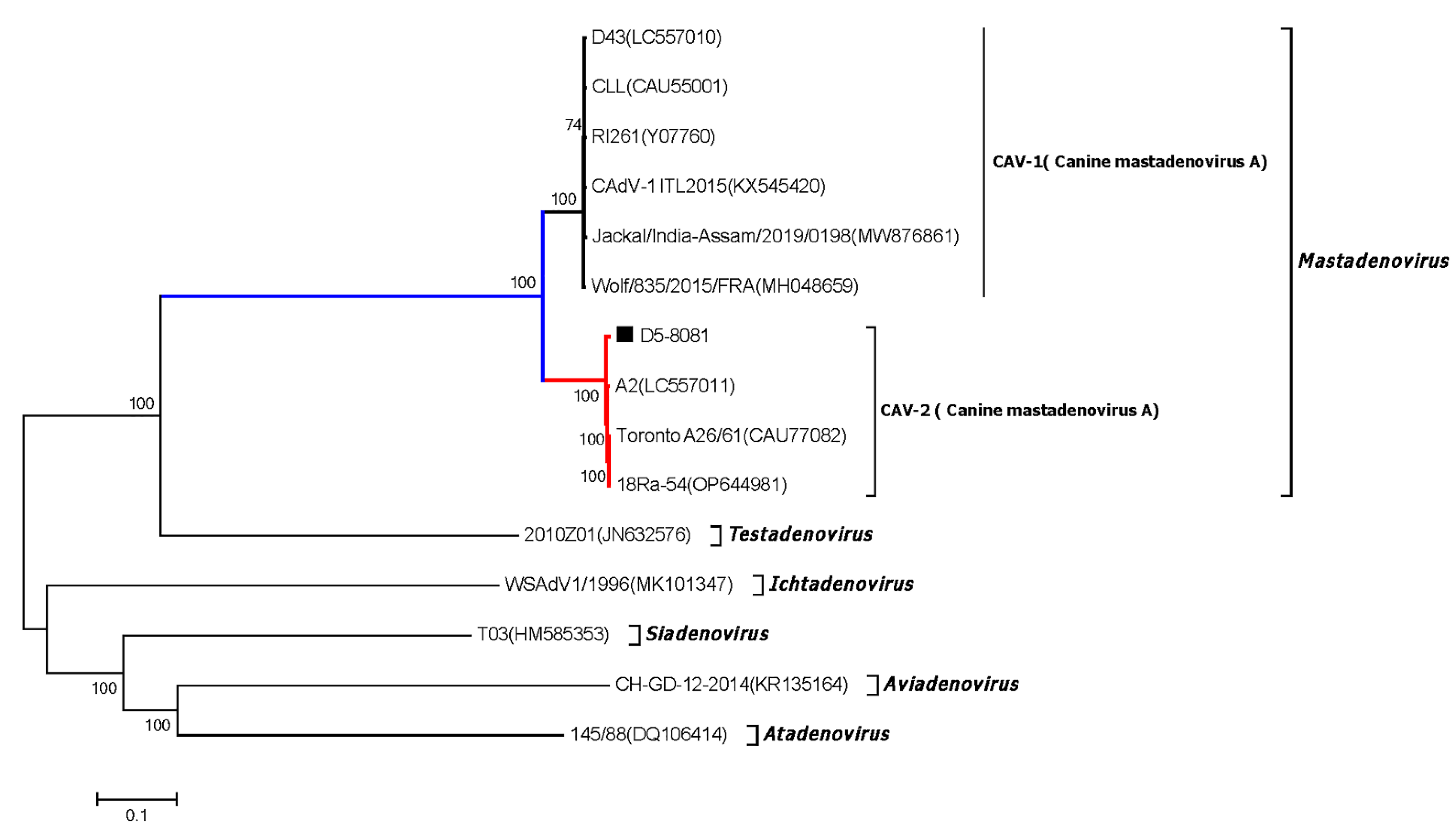
3.8. Canine Adenovirus
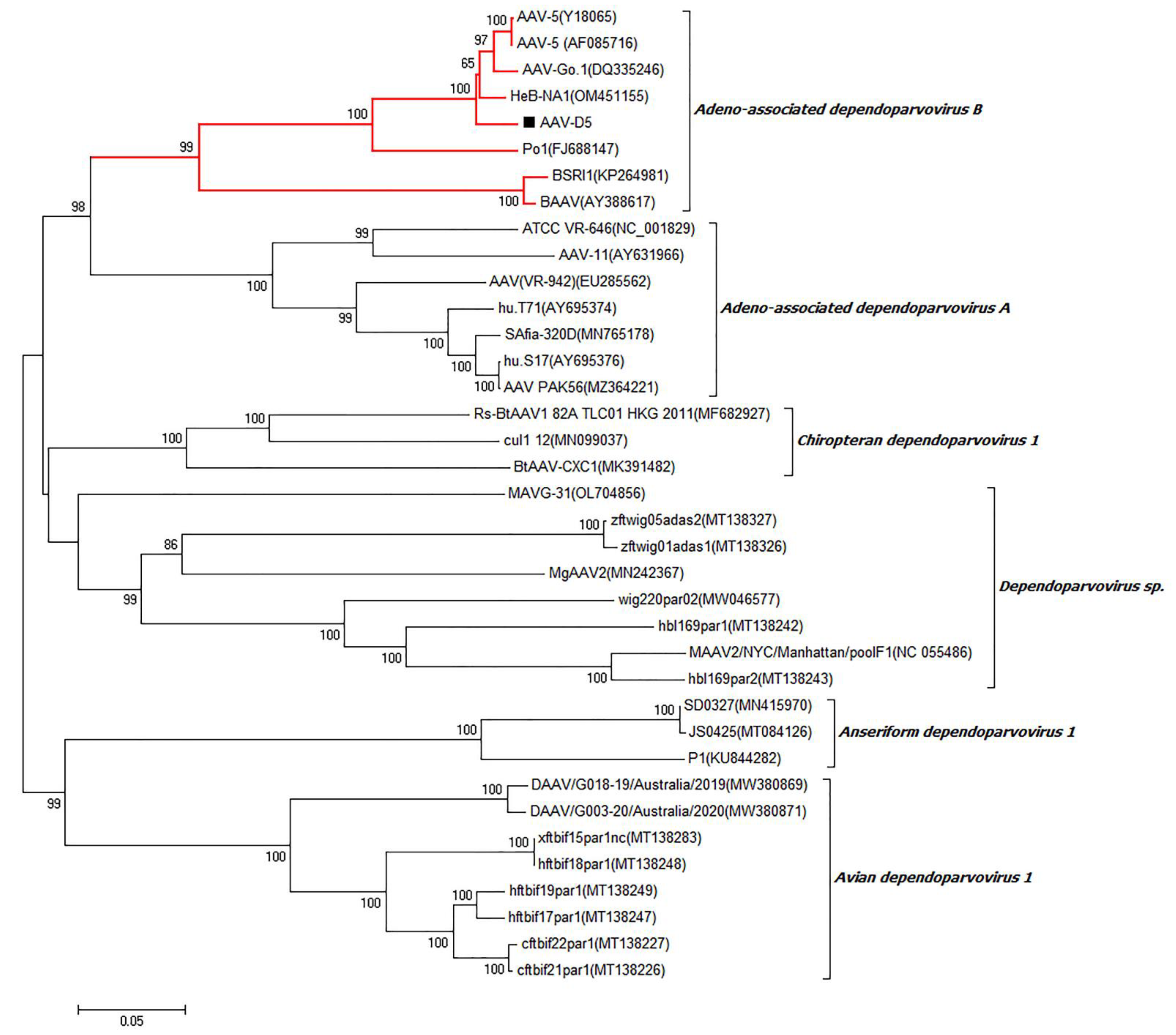
3.9. Adeno-Associated Virus
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ma, Z.S.; Mei, J. Stochastic neutral drifts seem prevalent in driving human virome assembly: Neutral, near-neutral and non-neutral theoretic analyses. Comput. Struct. Biotechnol. J. 2022, 20, 2029–2041. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Clooney, A.G.; Sutton, T.D.S.; Ryan, F.J.; Daly, K.M.; Nolan, J.A.; McDonnell, S.A.; Khokhlova, E.V.; Draper, L.A.; Forde, A.; et al. The Human Gut Virome Is Highly Diverse, Stable, and Individual Specific. Cell Host Microbe. 2019, 26, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, S.G.; Maurice, C.F. Not Just a Passing Phage. Cell Host Microbe. 2019, 26, 448–449. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.R.; Davis, N.; Hoyles, L. Review article: The human intestinal virome in health and disease. Aliment Pharmacol. Ther. 2017, 46, 800–815. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Bushman, F.D. The human virome: Assembly, composition and host interactions. Nat. Rev. Microbiol. 2021, 19, 514–527. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Stockdale, S.R.; Lavelle, A.; Kondova, I.; Heuston, C.; Upadrasta, A.; Khokhlova, E.V.; Ouwerling, B.; Draper, L.A.; Langermans, J.A.M.; et al. Viral biogeography of the mammalian gut and parenchymal organs. Nat. Microbiol. 2022, 7, 1301–1311. [Google Scholar] [CrossRef]
- Tan, J.; Fang, Z.; Wu, S.; Guo, Q.; Jiang, X.; Zhu, H. HoPhage: An ab initio tool for identifying hosts of phage fragments from metaviromes. Bioinformatics 2021, 38, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Johansen, J.; Plichta, D.R.; Nissen, J.N.; Jespersen, M.L.; Shah, S.A.; Deng, L.; Stokholm, J.; Bisgaard, H.; Nielsen, D.S.; Sørensen, S.J.; et al. Genome binning of viral entities from bulk metagenomics data. Nat. Commun. 2022, 13, 965–996. [Google Scholar] [CrossRef]
- Deng, X.; Achari, A.; Federman, S.; Yu, G.; Somasekar, S.; Bártolo, I.; Yagi, S.; Mbala-Kingebeni, P.; Kapetshi, J.; Ahuka-Mundeke, S.; et al. Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance. Nat. Microbiol. 2020, 5, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Li, S.; Yan, Q.; Guo, R.; Zhang, Y.; Chen, F.; Tian, X.; Lv, Q.; Jin, H.; Ma, X.; et al. Optimization and evaluation of viral metagenomic amplification and sequencing procedures toward a genome-level resolution of the human fecal DNA virome. J. Adv. Res. 2022, 16, 1232–1314. [Google Scholar] [CrossRef]
- Chen, F.; Li, S.; Guo, R.; Song, F.; Zhang, Y.; Wang, X.; Huo, X.; Lv, Q.; Ma, X. Meta-analysis of fecal viromes demonstrates high diagnostic potential of the gut viral signatures for colorectal cancer and adenoma risk assessment. J. Adv. Res. 2022, in press. [Google Scholar] [CrossRef]
- Benler, S.; Yutin, N.; Antipov, D.; Rayko, M.; Shmakov, S.; Gussow, A.B.; Pevzner, P.; Koonin, E.V. Thousands of previously unknown phages discovered in whole-community human gut metagenomes. Microbiome 2021, 9, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Yahara, K.; Suzuki, M.; Hirabayashi, A.; Suda, W.; Hattori, M.; Suzuki, Y.; Okazaki, Y. Long-read metagenomics using PromethION uncovers oral bacteriophages and their interaction with host bacteria. Nat. Commun. 2021, 12, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, E.A.; Rodriguez, C.; Hall-Moore, C.; Hoffmann, J.A.; Linneman, L.A.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Holtz, L.R.; Lim, E.S. Longitudinal gut virome analysis identifies specific viral signatures that precede necrotizing enterocolitis onset in preterm infants. Nat. Microbiol. 2022, 7, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Zou, H.; Li, J.; Wei, H. Landscapes of Enteric Virome Signatures in Early-Weaned Piglets. Microbiol. Spectr. 2022, 10, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Li, J.; Zhou, Q.; Xu, Z.; Chen, L.; Zhang, Y.; Xue, C.; Wen, Z.; Cao, Y. A New Bat-HKU2-like Coronavirus in Swine, China, 2017. Emerg. Infect. Dis. 2017, 23, 1607–1619. [Google Scholar] [CrossRef] [Green Version]
- Bzhalava, Z.; Tampuu, A.; Bała, P.; Vicente, R.; Dillner, J. Machine Learning for detection of viral sequences in human metagenomic datasets. BMC Bioinform. 2018, 19, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Casey, E.; Lugli, G.A.; Moore, R.; Kaczorowska, J.; Feehily, C.; Mangifesta, M.; Mancabelli, L.; Ventura, M. Tracing mother-infant transmission of bacteriophages by means of a novel analytical tool for shotgun metagenomic datasets: METAnnotatorX. Microbiome 2018, 6, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Nessler, J.N.; Jo, W.K.; Osterhaus, A.; Ludlow, M.; Tipold, A. Canine Meningoencephalitis of Unknown Origin-The Search for Infectious Agents in the Cerebrospinal Fluid via Deep Sequencing. Front. Vet. Sci. 2021, 8, 45–51. [Google Scholar] [CrossRef]
- Altan, E.; Seguin, M.A.; Leutenegger, C.M.; Phan, T.G.; Deng, X.; Delwart, E. Nasal virome of dogs with respiratory infection signs include novel taupapillomaviruses. Virus Genes. 2019, 55, 191–197. [Google Scholar] [CrossRef]
- Shi, Y.; Tao, J.; Li, B.; Shen, X.; Cheng, J.; Liu, H. The Gut Viral Metagenome Analysis of Domestic Dogs Captures Snapshot of Viral Diversity and Potential Risk of Coronavirus. Front. Vet. Sci. 2021, 8, 69–88. [Google Scholar] [CrossRef]
- Smith, C.S.; Lenz, M.F.; Caldwell, K.; Oakey, J. Identification of a canine coronavirus in Australian racing Greyhounds. J. Vet. Diagn. Investig. 2022, 34, 77–81. [Google Scholar] [CrossRef]
- Weber, M.N.; Cibulski, S.P.; Olegário, J.C.; Puhl, D.E.; Mósena, A.C.S.; Azevedo, S.S.; Canal, C.W. Characterization of dog serum virome from Northeastern Brazil. Virology 2018, 525, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Boros, Á.; Albert, M.; Urbán, P.; Herczeg, R.; Gáspár, G.; Balázs, B.; Cságola, A.; Pankovics, P.; Gyenesei, A.; Reuter, G. Unusual “Asian-origin” 2c to 2b point mutant canine parvovirus (Parvoviridae) and canine astrovirus (Astroviridae) co-infection detected in vaccinated dogs with an outbreak of severe haemorrhagic gastroenteritis with high mortality rate in Hungary. Vet. Res. Commun. 2022, 46, 1355–1361. [Google Scholar] [CrossRef]
- Carrino, M.; Tassoni, L.; Campalto, M.; Cavicchio, L.; Mion, M.; Corrò, M.; Natale, A.; Beato, M.S. Molecular Investigation of Recent Canine Parvovirus-2 (CPV-2) in Italy Revealed Distinct Clustering. Viruses 2022, 14, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yang, S.; Wang, C.; Wang, H.; Gong, G.; Xi, Y.; Pan, J.; Wang, X.; Zeng, J.; Zhang, J.; et al. Gut Virome of the World’s Highest-Elevation Lizard Species (Phrynocephalus erythrurus and Phrynocephalus theobaldi) Reveals Versatile Commensal Viruses. Microbiol. Spectr. 2022, 10, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014, 17, 111–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Li, Y.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 14, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.V.; Lesin, M.; Nikolenko, S.I.; Pevzner, P.A. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Chen, W.; Chen, J.P. Viral Metagenomics Revealed Sendai Virus and Coronavirus Infection of Malayan Pangolins (Manis javanica). Viruses 2019, 11, 979–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hameed, M.; Wahaab, A.; Shan, T.; Wang, X.; Khan, S.; Di, D.; Xiqian, L.; Zhang, J.J.; Anwar, M.N.; Nawaz, M.; et al. A Metagenomic Analysis of Mosquito Virome Collected from Different Animal Farms at Yunnan-Myanmar Border of China. Front. Microbiol. 2021, 11, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Tang, J.; Chen, Z.; Li, Q.; Huang, Z.; Wang, Q.; Meng, C.; Wang, Y.; Liu, G. Genetic characterization of the complete genome of a mutant canine parvovirus isolated in China. Arch. Virol. 2018, 163, 521–525. [Google Scholar] [CrossRef]
- Beller, L.; Matthijnssens, J. What is (not) known about the dynamics of the human gut virome in health and disease. Curr. Opin. Virol. 2019, 37, 52–57. [Google Scholar] [CrossRef]
- Fahsbender, E.; Altan, E.; Seguin, M.A.; Young, P.; Estrada, M.; Leutenegger, C.; Delwart, E. Chapparvovirus DNA Found in 4% of Dogs with Diarrhea. Viruses 2019, 11, 398–405. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; He, Y.; Chen, X.; Kalim, U.; Wang, Y.; Yang, S.; Qi, H.; Cheng, H.; Lu, X.; Wang, X.; et al. Viral Metagenomics Reveals Diverse Viruses in the Feces Samples of Raccoon Dogs. Front. Vet. Sci. 2021, 8, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cui, L.; Deng, X.; Yu, X.; Zhang, Z.; Yang, Z.; Delwart, E.; Zhang, W.; Hua, X. Canine bufavirus in faeces and plasma of dogs with diarrhoea, China. Emerg. Microbes. Infect. 2019, 8, 245–247. [Google Scholar] [CrossRef] [Green Version]
- Moreno, P.S.; Wagner, J.; Kirkwood, C.D.; Gilkerson, J.R.; Mansfield, C.S. Characterization of the fecal virome in dogs with chronic enteropathy. Vet. Microbiol. 2018, 221, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Hoon-Hanks, L.L.; McGrath, S.; Tyler, K.L.; Owen, C.; Stenglein, M.D. Metagenomic Investigation of Idiopathic Meningoencephalomyelitis in Dogs. J. Vet. Intern. Med. 2018, 32, 324–330. [Google Scholar] [CrossRef] [Green Version]
- Polovitzer, J.; Guija-De-Arespacochaga, A.; Auer, A.; Künzel, F. Successful treatment of coagulation disorders and hypoalbuminaemia in a puppy with Infectious Canine Hepatitis. Tierärztliche Prax. Ausg. K Kleintiere/Heimtiere 2022, 50, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.N.; Everett, J.K.; Kafle, S.; Roche, A.M.; Raymond, H.E.; Leiby, J.; Wood, C.; Assenmacher, C.A.; Merricks, E.P.; Long, C.T.; et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021, 39, 47–55. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raw Reads | Clean Reads | Canis lupus Familiaris Reads | Surplus Reads | Total Contigs |
|---|---|---|---|---|
| 384,381,025 | 263,337,595 | 14,466,745 | 248,870,850 | 20,158 |
| The Predicted Host Taxa of the Phages | Count |
|---|---|
| P__Proteobacteria.C__Epsilonproteobacteria.O__Campylobacterales.F__Campylobacteraceae.G__Campylobacter | 1523 |
| P__Proteobacteria.C__Gammaproteobacteria.O__Enterobacterales.F__Enterobacteriaceae.G__Escherichia | 655 |
| P__Proteobacteria.C__Gammaproteobacteria.O__Enterobacterales.F__Enterobacteriaceae.G__Salmonella | 115 |
| P__Proteobacteria.C__Gammaproteobacteria.O__Pseudomonadales.F__Pseudomonadaceae.G__Pseudomonas | 48 |
| P__Proteobacteria.C__Gammaproteobacteria.O__Pseudomonadales.F__Moraxellaceae.G__Acinetobacter | 23 |
| P__Proteobacteria.C__Gammaproteobacteria.O__Pseudomonadales.F__Moraxellaceae.G__Moraxella | 21 |
| P__Firmicutes.C__Clostridia.O__Eubacteriales.F__Lachnospiraceae.G__Mediterraneibacter | 20 |
| P__Firmicutes.C__Bacilli.O__Bacillales.F__Listeriaceae.G__Listeria | 14 |
| P__Firmicutes.C__Clostridia.O__Eubacteriales.F__Clostridiaceae.G__Clostridium | 14 |
| P__Proteobacteria.C__Gammaproteobacteria.O__Enterobacterales.F__Enterobacteriaceae.G__Klebsiella | 14 |
| P__Fusobacteria.C__Fusobacteriia.O__Fusobacteriales.F__Fusobacteriaceae.G__Fusobacterium | 11 |
| P__Firmicutes.C__Bacilli.O__Lactobacillales.F__Streptococcaceae.G__Streptococcus | 8 |
| P__Proteobacteria.C__Gammaproteobacteria.O__Xanthomonadales.F__Xanthomonadaceae.G__Xanthomonas | 8 |
| P__Proteobacteria.C__Betaproteobacteria.O__Neisseriales.F__Neisseriaceae.G__Neisseria | 7 |
| P__Actinobacteria.C__Actinomycetia.O__Bifidobacteriales.F__Bifidobacteriaceae.G__Bifidobacterium | 6 |
| P__Actinobacteria.C__Actinomycetia.O__Streptomycetales.F__Streptomycetaceae.G__Streptomyces | 6 |
| P__Firmicutes.C__Bacilli.O__Bacillales.F__Staphylococcaceae.G__Staphylococcus | 6 |
| P__Firmicutes.C__Bacilli.O__Lactobacillales.F__Enterococcaceae.G__Enterococcus | 6 |
| P__Firmicutes.C__Bacilli.O__Lactobacillales.F__Lactobacillaceae.G__Lacticaseibacillus | 6 |
| P__Firmicutes.C__Clostridia.O__Eubacteriales.F__Oscillospiraceae.G__Faecalibacterium | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Li, Z.; Li, C.; Ma, Y.; Sun, Q.; Zhang, H.; Niu, G.; Wei, J.; Yao, H.; Ma, Z. Viral Metagenomic Analysis of the Fecal Samples in Domestic Dogs (Canis lupus familiaris). Viruses 2023, 15, 685. https://doi.org/10.3390/v15030685
Wang H, Li Z, Li C, Ma Y, Sun Q, Zhang H, Niu G, Wei J, Yao H, Ma Z. Viral Metagenomic Analysis of the Fecal Samples in Domestic Dogs (Canis lupus familiaris). Viruses. 2023; 15(3):685. https://doi.org/10.3390/v15030685
Chicago/Turabian StyleWang, Hongyan, Zongjie Li, Chuanfeng Li, Yanfeng Ma, Qing Sun, Hailong Zhang, Guangbin Niu, Jianchao Wei, Huochun Yao, and Zhiyong Ma. 2023. "Viral Metagenomic Analysis of the Fecal Samples in Domestic Dogs (Canis lupus familiaris)" Viruses 15, no. 3: 685. https://doi.org/10.3390/v15030685