
Human Genome Polymorphisms and Computational Intelligence Approach Revealed a Complex Genomic Signature for COVID-19 Severity in Brazilian Patients
 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Material and Methods
2.1. Data Acquisition
2.1.1. Patient Group
2.1.2. Genomic DNA Extraction
2.1.3. DNA Library Preparation and Genotyping
2.2. Data Preprocessing
2.3. Data Analysis and Prognosis
2.4. Feature Selection
2.5. Patient Prognosis
3. Results
3.1. COVID-19 Patient Group
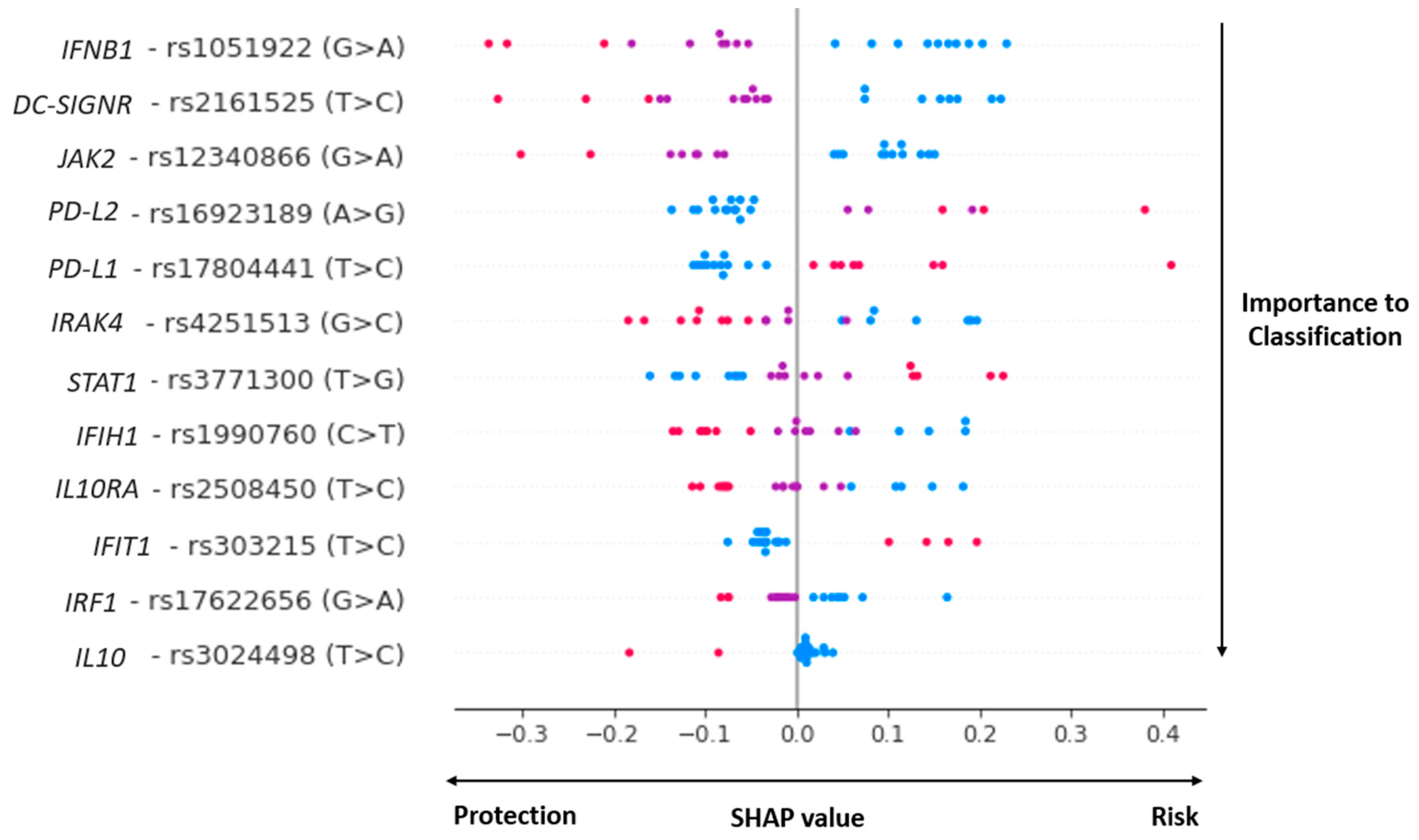
3.2. Genomic Aspects
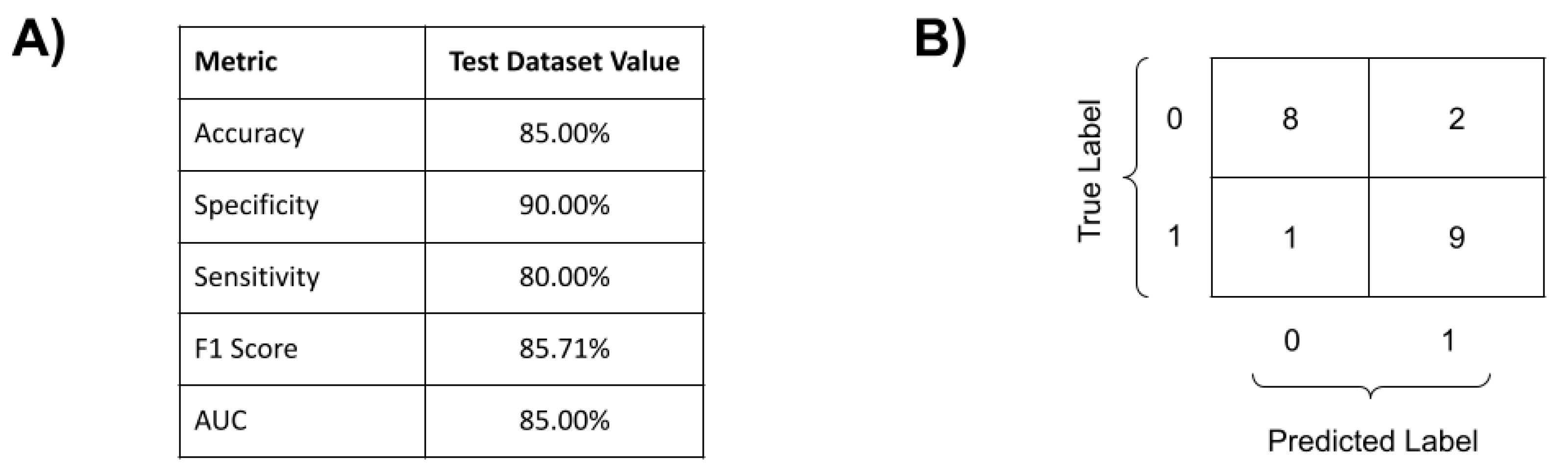
3.3. COVID-19 Genomic Classifier
4. Discussion
4.1. COVID-19 Genomic Prognosis Classifier
4.2. Molecular Biology of the Complex COVID-19 Prognosis Classifier
4.2.1. Viral Recognition
4.2.2. IL10, IFNB and JAK-STAT Pathways
4.2.3. Virus Replication
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johns Hopkins University COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). Available online: https://coronavirus.jhu.edu/map.html (accessed on 22 January 2023).
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Buitrago-Garcia, D.; Egli-Gany, D.; Counotte, M.J.; Hossmann, S.; Imeri, H.; Ipekci, A.M.; Salanti, G.; Low, N. Occurrence and Transmission Potential of Asymptomatic and Presymptomatic SARSCoV-2 Infections: A Living Systematic Review and Meta-Analysis. PLoS Med. 2020, 17, e1003346. [Google Scholar] [CrossRef]
- Zsichla, L.; Müller, V. Risk Factors of Severe COVID-19: A Review of Host, Viral and Environmental Factors. Viruses 2023, 15, 175. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.E.K.; Karjalainen, J.; Liao, R.G.; Neale, B.M.; Daly, M.; Ganna, A.; Pathak, G.A.; Andrews, S.J.; Kanai, M.; Veerapen, K.; et al. Mapping the Human Genetic Architecture of COVID-19. Nature 2021, 600, 472–477. [Google Scholar] [CrossRef]
- Li, X.; Xu, S.; Yu, M.; Wang, K.; Tao, Y.; Zhou, Y.; Shi, J.; Zhou, M.; Wu, B.; Yang, Z.; et al. Risk Factors for Severity and Mortality in Adult COVID-19 Inpatients in Wuhan. J. Allergy Clin. Immunol. 2020, 146, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients with COVID-19 in Wuhan, China: A Retrospective Cohort Study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Asteris, P.G.; Gavriilaki, E.; Touloumenidou, T.; Koravou, E.E.; Koutra, M.; Papayanni, P.G.; Pouleres, A.; Karali, V.; Lemonis, M.E.; Mamou, A.; et al. Genetic Prediction of ICU Hospitalization and Mortality in COVID-19 Patients Using Artificial Neural Networks. J. Cell. Mol. Med. 2022, 26, 1445–1455. [Google Scholar] [CrossRef]
- Benetti, E.; Tita, R.; Spiga, O.; Ciolfi, A.; Birolo, G.; Bruselles, A.; Doddato, G.; Giliberti, A.; Marconi, C.; Musacchia, F.; et al. ACE2 Gene Variants May Underlie Interindividual Variability and Susceptibility to COVID-19 in the Italian Population. Eur. J. Hum. Genet. 2020, 28, 1602–1614. [Google Scholar] [CrossRef]
- Hou, Y.; Zhao, J.; Martin, W.; Kallianpur, A.; Chung, M.K.; Jehi, L.; Sharifi, N.; Erzurum, S.; Eng, C.; Cheng, F. New Insights into Genetic Susceptibility of COVID-19: An ACE2 and TMPRSS2 Polymorphism Analysis. BMC Med. 2020, 18, 216. [Google Scholar] [CrossRef]
- Suryamohan, K.; Diwanji, D.; Stawiski, E.W.; Gupta, R.; Miersch, S.; Liu, J.; Chen, C.; Jiang, Y.P.; Fellouse, F.A.; Sathirapongsasuti, J.F.; et al. Human ACE2 Receptor Polymorphisms and Altered Susceptibility to SARS-CoV-2. Commun. Biol. 2021, 4, 475. [Google Scholar] [CrossRef]
- Franke Genomewide Association Study of Severe COVID-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [CrossRef] [PubMed]
- Wang, F.; Huang, S.; Gao, R.; Zhou, Y.; Lai, C.; Li, Z.; Xian, W.; Qian, X.; Li, Z.; Huang, Y.; et al. Initial Whole-Genome Sequencing and Analysis of the Host Genetic Contribution to COVID-19 Severity and Susceptibility. Cell Discov. 2020, 6, 83. [Google Scholar] [CrossRef] [PubMed]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic Mechanisms of Critical Illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Secolin, R.; de Araujo, T.K.; Gonsales, M.C.; Rocha, C.S.; Naslavsky, M.; de Marco, L.; Bicalho, M.A.C.; Vazquez, V.L.; Zatz, M.; Silva, W.A.; et al. Genetic Variability in COVID-19-Related Genes in the Brazilian Population. Hum. Genome Var. 2021, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Platt, C.D.; Habiballah, S.; Nguyen, A.A.; Elkins, M.; Weeks, S.; Peters, Z.; Day-Lewis, M.; Novak, T.; Armant, M.; et al. Mechanisms Underlying Genetic Susceptibility to Multisystem Inflammatory Syndrome in Children (MIS-C). J. Allergy Clin. Immunol. 2021, 148, 732–738.e1. [Google Scholar] [CrossRef] [PubMed]
- John, C.C.; Ponnusamy, V.; Krishnan Chandrasekaran, S.; Ra, N. A Survey on Mathematical, Machine Learning and Deep Learning Models for COVID-19 Transmission and Diagnosis. IEEE Rev. Biomed. Eng. 2022, 15, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Comito, C.; Pizzuti, C. Artificial Intelligence for Forecasting and Diagnosing COVID-19 Pandemic: A Focused Review. Artif. Intell. Med. 2022, 128, 102286. [Google Scholar] [CrossRef]
- Wang, G.; Liu, X.; Shen, J.; Wang, C.; Li, Z.; Ye, L.; Wu, X.; Chen, T.; Wang, K.; Zhang, X.; et al. A Deep-Learning Pipeline for the Diagnosis and Discrimination of Viral, Non-Viral and COVID-19 Pneumonia from Chest X-Ray Images. Nat. Biomed. Eng. 2021, 5, 509–521. [Google Scholar] [CrossRef]
- Fang, C.; Bai, S.; Chen, Q.; Zhou, Y.; Xia, L.; Qin, L.; Gong, S.; Xie, X.; Zhou, C.; Tu, D.; et al. Deep Learning for Predicting COVID-19 Malignant Progression. Med. Image Anal. 2021, 72, 102096. [Google Scholar] [CrossRef]
- Fallerini, C.; Picchiotti, N.; Baldassarri, M.; Zguro, K.; Daga, S.; Fava, F.; Benetti, E.; Amitrano, S.; Bruttini, M.; Palmieri, M.; et al. Common, Low-Frequency, Rare, and Ultra-Rare Coding Variants Contribute to COVID-19 Severity. Hum. Genet. 2022, 141, 147–173. [Google Scholar] [CrossRef]
- Sun, C.; Bai, Y.; Chen, D.; He, L.; Zhu, J.; Ding, X.; Luo, L.; Ren, Y.; Xing, H.; Jin, X.; et al. Accurate Classification of COVID-19 Patients with Different Severity via Machine Learning. Clin. Transl. Med. 2021, 11, e323. [Google Scholar] [CrossRef] [PubMed]
- Davi, C.; Pastor, A.; Oliveira, T.; Neto, F.B.D.L.; Braga-Neto, U.; Bigham, A.W.; Bamshad, M.; Marques, E.T.A.; Acioli-Santos, B. Severe Dengue Prognosis Using Human Genome Data and Machine Learning. IEEE Trans. Biomed. Eng. 2019, 66, 2861–2868. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Petrazzini, B.O.; Naya, H.; Lopez-Bello, F.; Vazquez, G.; Spangenberg, L. Evaluation of Different Approaches for Missing Data Imputation on Features Associated to Genomic Data. BioData Min. 2021, 14, 44. [Google Scholar] [CrossRef]
- Fernández-Martínez, J.L.; Fernández-Muñiz, Z. The Curse of Dimensionality in Inverse Problems. J. Comput. Appl. Math. 2020, 369, 112571. [Google Scholar] [CrossRef]
- Guyon, I.; Weston, J.; Barnhill, S. Gene Selection for Cancer Classification Using Support Vector Machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Pedregosa, F.; Michel, V.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Vanderplas, J.; Cournapeau, D.; Pedregosa, F.; Varoquaux, G.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Ambrish, G.; Ganesh, B.; Ganesh, A.; Srinivas, C.; Dhanraj; Mensinkal, K. Logistic Regression Technique for Prediction of Cardiovascular Disease. Glob. Transit. Proc. 2022, 3, 127–130. [Google Scholar] [CrossRef]
- Cover, T.M.; Hart, P.E. Nearest Neighbor Pattern Classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L.; Friedman, J.H.; Olshen, R.A.; Stone, C.J. Classification and Regression Trees (The Wadsworth Statistics/Probability Series); Routledge: Oxfordshire, UK, 1983; Volume 1. [Google Scholar]
- Boser, B.E.; Guyon, I.M.; Vapnik, V.N. A Training Algorithm for Optimal Margin Classifiers. ACM 1992, 144–152. [Google Scholar] [CrossRef]
- Lundberg, S.M.; Allen, P.G.; Lee, S.-I. A Unified Approach to Interpreting Model Predictions. In Proceedings of the 31st Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017; pp. 1–10. [Google Scholar]
- Zietz, M.; Zucker, J.; Tatonetti, N.P. Associations between Blood Type and COVID-19 Infection, Intubation, and Death. Nat. Commun. 2020, 11, 5761. [Google Scholar] [CrossRef] [PubMed]
- Adamidi, E.S.; Mitsis, K.; Nikita, K.S. Artificial Intelligence in Clinical Care amidst COVID-19 Pandemic: A Systematic Review. Comput. Struct. Biotechnol. J. 2021, 19, 2833–2850. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Matuozzo, D.; le Pen, J.; Lee, D.; Moens, L.; Asano, T.; Bohlen, J.; Liu, Z.; Moncada-Velez, M.; Kendir-Demirkol, Y.; et al. Recessive Inborn Errors of Type I IFN Immunity in Children with COVID-19 Pneumonia. J. Exp. Med. 2022, 219, e20220131. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.L.; Pilling, L.C.; Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Kuchel, G.A.; Melzer, D. APOE E4 Genotype Predicts Severe COVID-19 in the UK Biobank Community Cohort. J. Gerontol.-Ser. A Biol. Sci. Med. Sci. 2020, 75, 2231–2232. [Google Scholar] [CrossRef]
- Pena, S.D.J.; di Pietro, G.; Fuchshuber-Moraes, M.; Genro, J.P.; Hutz, M.H.; Kehdy, F.D.S.G.; Kohlrausch, F.; Magno, L.A.V.; Montenegro, R.C.; Moraes, M.O.; et al. The Genomic Ancestry of Individuals from Different Geographical Regions of Brazil Is More Uniform than Expected. PLoS ONE 2011, 6, e17063. [Google Scholar] [CrossRef] [Green Version]
- Domsgen, E.; Lind, K.; Kong, L.; Hühn, M.H.; Rasool, O.; van Kuppeveld, F.; Korsgren, O.; Lahesmaa, R.; Flodström-Tullberg, M. An IFIH1 Gene Polymorphism Associated with Risk for Autoimmunity Regulates Canonical Antiviral Defence Pathways in Coxsackievirus Infected Human Pancreatic Islets. Sci. Rep. 2016, 6, 39378. [Google Scholar] [CrossRef] [Green Version]
- Soilleux, E.J.; Barten, R.; Trowsdale, J. Cutting Edge: DC-SIGN; a Related Gene, DC-SIGNR; and CD23 Form a Cluster on 19p13. J. Immunol. 2000, 165, 2937–2942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashirova, A.A.; Geijtenbeek, T.B.H.; van Duijnhoven, G.C.F.; van Vliet, S.J.; Eilering, J.B.G.; Martin, M.P.; Wu, L.; Martin, T.D.; Viebig, N.; Knolle, P.A.; et al. A Dendritic Cell-Specific Intercellular Adhesion Molecule 3-Grabbing Nonintegrin (DC-SIGN)-Related Protein Is Highly Expressed on Human Liver Sinusoidal Endothelial Cells and Promotes HIV-1 Infection. J. Exp. Med. 2001, 193, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Saito, T. IRAK-4-a Shared NF-ΚB Activator in Innate and Acquired Immunity. Trends Immunol. 2006, 27, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, M.; O’Garra, A. The Regulation of IL-10 Production by Immune Cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Carey, A.J.; Tan, C.K.; Ulett, G.C. Infection-Induced IL-10 and JAK-STAT. JAKSTAT 2012, 1, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Solinas, C.; Aiello, M.; Rozali, E.; Lambertini, M.; Willard-Gallo, K.; Migliori, E. Programmed Cell Death-Ligand 2: A Neglected But Important Target in the Immune Response to Cancer? Transl. Oncol. 2020, 13, 100811. [Google Scholar] [CrossRef]
- Kali, S.K.; Dröge, P.; Murugan, P. Interferon β, an Enhancer of the Innate Immune Response against SARS-CoV-2 Infection. Microb. Pathog. 2021, 158, 105105. [Google Scholar] [CrossRef]
- García-Sastre, A. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe 2017, 22, 176–184. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT Signaling Pathway: From Bench to Clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Kimura, T.; Katoh, H.; Kayama, H.; Saiga, H.; Okuyama, M.; Okamoto, T.; Umemoto, E.; Matsuura, Y.; Yamamoto, M.; Takeda, K. Ifit1 Inhibits Japanese Encephalitis Virus Replication through Binding to 5′ Capped 2′-O Unmethylated RNA. J. Virol. 2013, 87, 9997–10003. [Google Scholar] [CrossRef] [Green Version]
- Fensterl, V.; Sen, G.C. Interferon-Induced Ifit Proteins: Their Role in Viral Pathogenesis. J. Virol. 2015, 89, 2462–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohoff, M.; Mak, T.W. Roles of Interferon-Regulatory Factors in T-Helper-Cell Differentiation. Nat. Rev. Immunol. 2005, 5, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, D.; Tamura, T.; Yanai, H.; Taniguchi, T. Regulation of Immunity and Oncogenesis by the IRF Transcription Factor Family. Cancer Immunol. Immunother. 2010, 59, 489–510. [Google Scholar] [CrossRef] [PubMed]
- Dias Junior, A.G.; Sampaio, N.G.; Rehwinkel, J. A Balancing Act: MDA5 in Antiviral Immunity and Autoinflammation. Trends Microbiol. 2019, 27, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.P.; Look, D.C.; Shi, L.; Hickey, M.; Pewe, L.; Netland, J.; Farzan, M.; Wohlford-Lenane, C.; Perlman, S.; McCray, P.B. ACE2 Receptor Expression and Severe Acute Respiratory Syndrome Coronavirus Infection Depend on Differentiation of Human Airway Epithelia. J. Virol. 2005, 79, 14614–14621. [Google Scholar] [CrossRef] [Green Version]
- Chan, V.S.F.; Chan, K.Y.K.; Chen, Y.; Poon, L.L.M.; Cheung, A.N.Y.; Zheng, B.; Chan, K.H.; Mak, W.; Ngan, H.Y.S.; Xu, X.; et al. Homozygous L-SIGN (CLEC4M) Plays a Protective Role in SARS Coronavirus Infection. Nat. Genet. 2006, 38, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.A.; Dorfman, D.M.; Ma, F.-R.; Sullivan, E.L.; Munoz, O.; Wood, C.R.; Greenfield, E.A.; Freeman, G.J. Blockade of Programmed Death-1 Ligands on Dendritic Cells Enhances T Cell Activation and Cytokine Production. J. Immunol. 2003, 170, 1257–1266. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Zhang, H.; Dauphars, D.J.; He, Y.W. A Potential Role of Interleukin 10 in COVID-19 Pathogenesis. Trends Immunol. 2021, 42, 3–5. [Google Scholar] [CrossRef]
- Ishige, T.; Igarashi, Y.; Hatori, R.; Tatsuki, M.; Sasahara, Y.; Takizawa, T.; Arakawa, H. IL-10RA Mutation as a Risk Factor of Severe Influenza-Associated Encephalopathy: A Case Report. Pediatrics 2018, 141, e20173548. [Google Scholar] [CrossRef]
- Zhou, H.; Tang, Y.D.; Zheng, C. Revisiting IRF1-Mediated Antiviral Innate Immunity. Cytokine Growth Factor Rev. 2022, 64, 1–6. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Age (Years) | Male | Female | Mild COVID-19 Patients | Severe COVID-19 Patients |
|---|---|---|---|---|
| 11–21 | 1 (1.6%) | 1 (2.8%) | 2 (4.2%) | - |
| 22–30 | 4 (6.6%) | 3 (8.6%) | 5 (10.4%) | 2 (4.2%) |
| 31–40 | 11 (18.1%) | 4 (11.4%) | 3 (6.2%) | 12 (25%) |
| 41–50 | 30 (49.1%) | 13 (37.2%) | 22 (45.8%) | 21 (43.7%) |
| 51–70 | 15 (24.6%) | 12 (34.3%) | 14 (29.2%) | 13 (27.1%) |
| 71–90 | - | 2 (5.7%) | 2 (4.2%) | - |
| Total | 61 | 35 | 48 | 48 |
| SNP (RS Code/Alleles) | Function/Location | Gene | Protein Function | References |
|---|---|---|---|---|
| Viral recognition | ||||
| rs1990760 C>T | missense_variant | IFIH1 | MDA5 is an intracellular sensor of viral RNA. | [43] |
| rs2161525 T>C | intron_variant | DC-SIGNR | A C-type lectin that functions in cell adhesion and pathogen recognition. | [44,45] |
| rs4251513 G>C | intron_variant | IRAK4 | A kinase that activates NF-kappaB in both the Toll-like receptor (TLR) and T-cell receptor (TCR) signaling pathways. | [46] |
| IL-10 and IFN Pathways | ||||
| rs3024498 T>C | non_coding_transcript_variant | IL10 | A cytokine with pleiotropic effects in immune regulation and inflammation. | [47] |
| rs2508450 T>C | intron_variant | IL10RA | Receptor for interleukin 10. | [48] |
| rs16923189 A>G | 5_prime_UTR_variant | PD-L2 | Negative regulation of interleukin-10 production. | [49] |
| rs17804441 T>C | intron_variant | PD-L1 | Inhibitory receptor-ligand expressed by T-cells and B cells, and various types of tumor cells. | [49] |
| rs1051922 G>A | “sotp gained”, coding sequence. | IFNB1 | A cytokine that belongs to the interferon family of signaling proteins. | [50,51] |
| rs12340866 G>A | intron variant | JAK2 | The non-receptor tyrosine kinase of the JAK/STAT pathway. | [52] |
| rs3771300 T>G | genic_downstream_transcript | STAT1 | Member of the STAT protein family. | [52] |
| Viral Replication | ||||
| rs17804441 T>C | intron_variant | PD-L1 | Positive regulator of ISG expression. | [23] |
| rs303215 T>C | intron_variant | IFIT1 | An interferon-induced protein that inhibits viral replication and translational initiation. | [53,54] |
| rs17622656 G>A | intron_variant | IRF1 | A transcriptional regulator that activates the genes involved in both innate and acquired immune responses. | [55,56] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastor, A.F.; Docena, C.; Rezende, A.M.; Oliveira, F.R.d.S.; Sena, M.d.A.; Morais, C.N.L.d.; Bresani-Salvi, C.C.; Vasconcelos, L.R.S.; Valença, K.D.C.; Mariz, C.d.A.; et al. Human Genome Polymorphisms and Computational Intelligence Approach Revealed a Complex Genomic Signature for COVID-19 Severity in Brazilian Patients. Viruses 2023, 15, 645. https://doi.org/10.3390/v15030645
Pastor AF, Docena C, Rezende AM, Oliveira FRdS, Sena MdA, Morais CNLd, Bresani-Salvi CC, Vasconcelos LRS, Valença KDC, Mariz CdA, et al. Human Genome Polymorphisms and Computational Intelligence Approach Revealed a Complex Genomic Signature for COVID-19 Severity in Brazilian Patients. Viruses. 2023; 15(3):645. https://doi.org/10.3390/v15030645
Chicago/Turabian StylePastor, André Filipe, Cássia Docena, Antônio Mauro Rezende, Flávio Rosendo da Silva Oliveira, Marília de Albuquerque Sena, Clarice Neuenschwander Lins de Morais, Cristiane Campello Bresani-Salvi, Luydson Richardson Silva Vasconcelos, Kennya Danielle Campelo Valença, Carolline de Araújo Mariz, and et al. 2023. "Human Genome Polymorphisms and Computational Intelligence Approach Revealed a Complex Genomic Signature for COVID-19 Severity in Brazilian Patients" Viruses 15, no. 3: 645. https://doi.org/10.3390/v15030645