
SARS-CoV-2 Spike-Mediated Entry and Its Regulation by Host Innate Immunity
 ,
,
Abstract
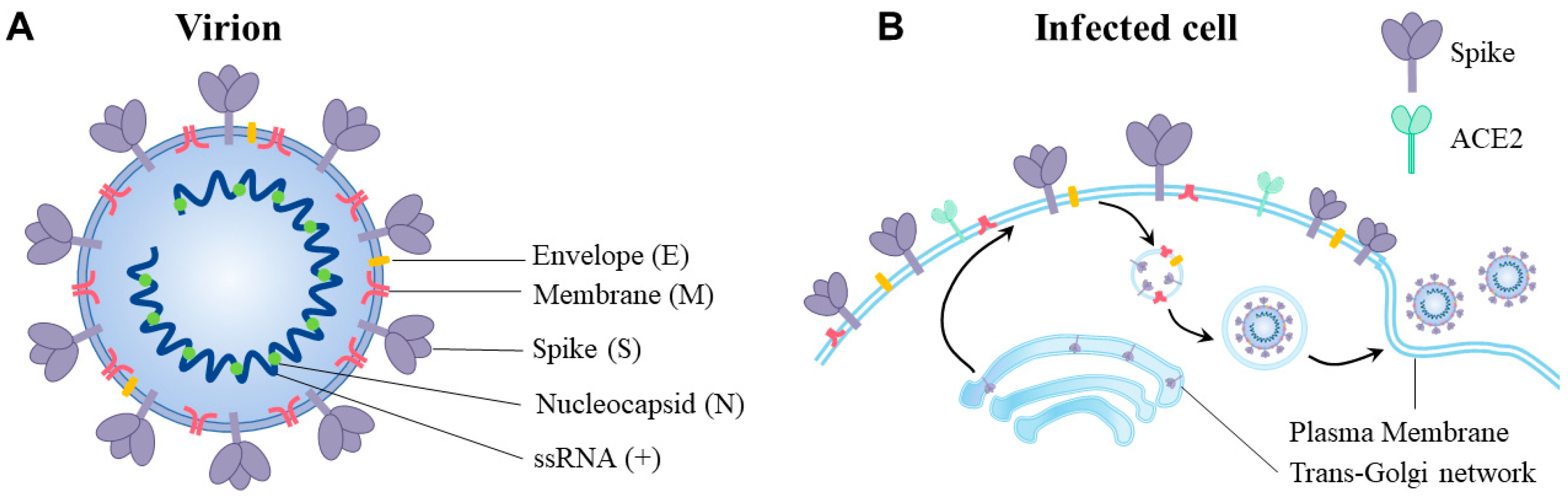
:1. Overview of the SARS-CoV-2 Virus
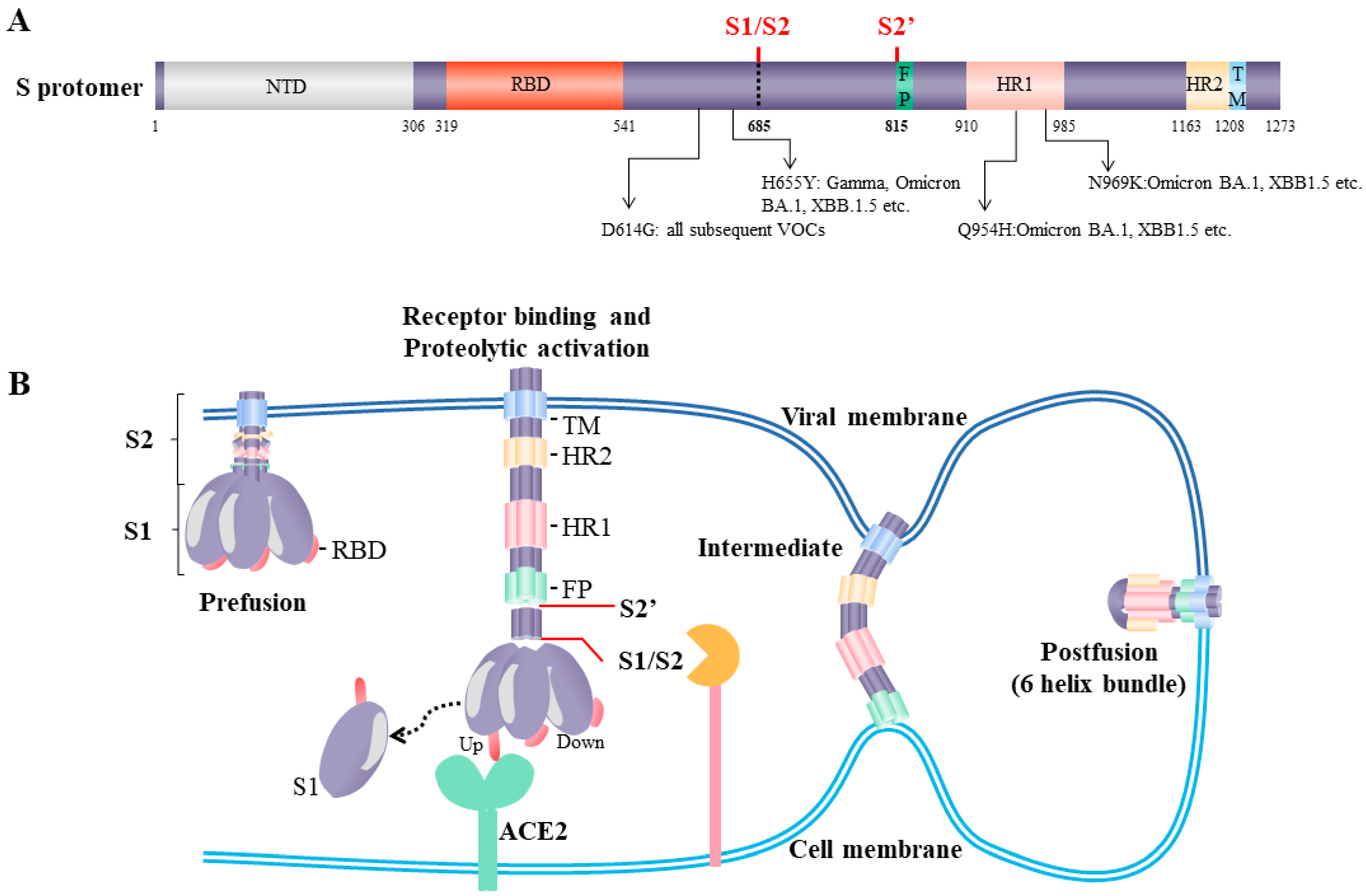
2. The SARS-CoV-2 Spike
3. Proteolytic Priming of the SARS-CoV-2 Spike S1 and S2 Subunits
4. Spike Proteolytic Activation and Utilization of Host Proteases
5. Cellular Innate Immune Responses to Spike-Mediated Entry
6. Inflammatory Processes beyond ISGs
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Worobey, M.; Levy, J.I.; Malpica Serrano, L.; Crits-Christoph, A.; Pekar, J.E.; Goldstein, S.A.; Rasmussen, A.L.; Kraemer, M.U.G.; Newman, C.; Koopmans, M.P.G.; et al. The Huanan Seafood Wholesale Market in Wuhan was the early epicenter of the COVID-19 pandemic. Science 2022, 377, 951–959. [Google Scholar] [CrossRef]
- Pekar, J.E.; Magee, A.; Parker, E.; Moshiri, N.; Izhikevich, K.; Havens, J.L.; Gangavarapu, K.; Malpica Serrano, L.M.; Crits-Christoph, A.; Matteson, N.L.; et al. The molecular epidemiology of multiple zoonotic origins of SARS-CoV-2. Science 2022, 377, 960–966. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Singanayagam, A.; Hakki, S.; Dunning, J.; Madon, K.J.; Crone, M.A.; Koycheva, A.; Derqui-Fernandez, N.; Barnett, J.L.; Whitfield, M.G.; Varro, R.; et al. Community transmission and viral load kinetics of the SARS-CoV-2 delta (B.1.617.2) variant in vaccinated and unvaccinated individuals in the UK: A prospective, longitudinal, cohort study. Lancet Infect. Dis. 2022, 22, 183–195. [Google Scholar] [CrossRef]
- Bergwerk, M.; Gonen, T.; Lustig, Y.; Amit, S.; Lipsitch, M.; Cohen, C.; Mandelboim, M.; Levin, E.G.; Rubin, C.; Indenbaum, V.; et al. COVID-19 Breakthrough Infections in Vaccinated Health Care Workers. N. Engl. J. Med. 2021, 385, 1474–1484. [Google Scholar] [CrossRef]
- Mostaghimi, D.; Valdez, C.N.; Larson, H.T.; Kalinich, C.C.; Iwasaki, A. Prevention of host-to-host transmission by SARS-CoV-2 vaccines. Lancet Infect. Dis. 2022, 22, e52–e58. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; Consortium, C.-G.U.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef]
- Stadnytskyi, V.; Bax, C.E.; Bax, A.; Anfinrud, P. The airborne lifetime of small speech droplets and their potential importance in SARS-CoV-2 transmission. Proc. Natl. Acad. Sci. USA 2020, 117, 11875–11877. [Google Scholar] [CrossRef]
- van Doremalen, N.; Bushmaker, T.; Morris, D.H.; Holbrook, M.G.; Gamble, A.; Williamson, B.N.; Tamin, A.; Harcourt, J.L.; Thornburg, N.J.; Gerber, S.I.; et al. Aerosol and Surface Stability of SARS-CoV-2 as Compared with SARS-CoV-1. N. Engl. J. Med. 2020, 382, 1564–1567. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunyavanich, S.; Do, A.; Vicencio, A. Nasal Gene Expression of Angiotensin-Converting Enzyme 2 in Children and Adults. JAMA 2020, 323, 2427–2429. [Google Scholar] [CrossRef] [PubMed]
- Hikmet, F.; Mear, L.; Edvinsson, A.; Micke, P.; Uhlen, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rao, Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat. Rev. Microbiol. 2021, 19, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Bartenschlager, R. A snapshot of protein trafficking in SARS-CoV-2 infection. Biol. Cell 2022, 115, 2200073. [Google Scholar] [CrossRef] [PubMed]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and functions of coronavirus replication-transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Snijder, E.J.; Decroly, E.; Ziebuhr, J. The Nonstructural Proteins Directing Coronavirus RNA Synthesis and Processing. Adv. Virus Res. 2016, 96, 59–126. [Google Scholar] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Moustaqil, M.; Ollivier, E.; Chiu, H.P.; Van Tol, S.; Rudolffi-Soto, P.; Stevens, C.; Bhumkar, A.; Hunter, D.J.B.; Freiberg, A.N.; Jacques, D.; et al. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): Implications for disease presentation across species. Emerg. Microbes. Infect. 2021, 10, 178–195. [Google Scholar] [CrossRef]
- Thorne, L.G.; Bouhaddou, M.; Reuschl, A.K.; Zuliani-Alvarez, L.; Polacco, B.; Pelin, A.; Batra, J.; Whelan, M.V.X.; Hosmillo, M.; Fossati, A.; et al. Evolution of enhanced innate immune evasion by SARS-CoV-2. Nature 2022, 602, 487–495. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, K.; Qin, B.; Olieric, V.; Wang, M.; Cui, S. Crystal structure of SARS-CoV-2 Orf9b in complex with human TOM70 suggests unusual virus-host interactions. Nat. Commun. 2021, 12, 2843. [Google Scholar] [CrossRef]
- Wu, J.; Shi, Y.; Pan, X.; Wu, S.; Hou, R.; Zhang, Y.; Zhong, T.; Tang, H.; Du, W.; Wang, L.; et al. SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep. 2021, 34, 108761. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- White, J.M.; Whittaker, G.R. Fusion of Enveloped Viruses in Endosomes. Traffic 2016, 17, 593–614. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B.; et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 588, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Turonova, B.; Sikora, M.; Schurmann, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; von Bulow, S.; Gecht, M.; Bagola, K.; Horner, C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Song, Y.; Chen, Y.; Wu, N.; Xu, J.; Sun, C.; Zhang, J.; Weng, T.; Zhang, Z.; Wu, Z.; et al. Molecular Architecture of the SARS-CoV-2 Virus. Cell 2020, 183, 730–738.e13. [Google Scholar] [CrossRef]
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat. Commun. 2020, 11, 5885. [Google Scholar] [CrossRef]
- Beniac, D.R.; Andonov, A.; Grudeski, E.; Booth, T.F. Architecture of the SARS coronavirus prefusion spike. Nat. Struct. Mol. Biol. 2006, 13, 751–752. [Google Scholar] [CrossRef] [Green Version]
- Neuman, B.W.; Adair, B.D.; Yoshioka, C.; Quispe, J.D.; Orca, G.; Kuhn, P.; Milligan, R.A.; Yeager, M.; Buchmeier, M.J. Supramolecular architecture of severe acute respiratory syndrome coronavirus revealed by electron cryomicroscopy. J. Virol. 2006, 80, 7918–7928. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Sui, J.; Huang, I.C.; Kuhn, J.H.; Radoshitzky, S.R.; Marasco, W.A.; Choe, H.; Farzan, M. The S proteins of human coronavirus NL63 and severe acute respiratory syndrome coronavirus bind overlapping regions of ACE2. Virology 2007, 367, 367–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, M.; Gao, J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 13967–13974. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature 2022, 602, 294–299. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, J.; Xiao, T.; Lavine, C.L.; Rawson, S.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; et al. Structural basis for enhanced infectivity and immune evasion of SARS-CoV-2 variants. Science 2021, 373, 642–648. [Google Scholar] [CrossRef]
- Baum, A.; Ajithdoss, D.; Copin, R.; Zhou, A.; Lanza, K.; Negron, N.; Ni, M.; Wei, Y.; Mohammadi, K.; Musser, B.; et al. REGN-COV2 antibodies prevent and treat SARS-CoV-2 infection in rhesus macaques and hamsters. Science 2020, 370, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef]
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 2020, 584, 120–124. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.M.C.; Kopp, M.; Li, D.; Parks, R.; Hsu, A.L.; et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. [Google Scholar] [CrossRef]
- Pang, Y.T.; Acharya, A.; Lynch, D.L.; Pavlova, A.; Gumbart, J.C. SARS-CoV-2 spike opening dynamics and energetics reveal the individual roles of glycans and their collective impact. Commun. Biol. 2022, 5, 1170. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.J.; Mansouri, K.; Stalls, V.; Manne, K.; Watts, B.; Parks, R.; Janowska, K.; Gobeil, S.M.C.; Kopp, M.; Li, D.; et al. Cold sensitivity of the SARS-CoV-2 spike ectodomain. Nat. Struct. Mol. Biol. 2021, 28, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhou, J.; Tian, M.; Huang, M.; Liu, S.; Xie, Y.; Han, P.; Bai, C.; Han, P.; Zheng, A.; et al. Omicron SARS-CoV-2 mutations stabilize spike up-RBD conformation and lead to a non-RBM-binding monoclonal antibody escape. Nat. Commun. 2022, 13, 4958. [Google Scholar] [CrossRef]
- Lai, A.L.; Millet, J.K.; Daniel, S.; Freed, J.H.; Whittaker, G.R. The SARS-CoV Fusion Peptide Forms an Extended Bipartite Fusion Platform that Perturbs Membrane Order in a Calcium-Dependent Manner. J. Mol. Biol. 2017, 429, 3875–3892. [Google Scholar] [CrossRef]
- Madu, I.G.; Roth, S.L.; Belouzard, S.; Whittaker, G.R. Characterization of a highly conserved domain within the severe acute respiratory syndrome coronavirus spike protein S2 domain with characteristics of a viral fusion peptide. J. Virol. 2009, 83, 7411–7421. [Google Scholar] [CrossRef] [Green Version]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Yu, S.; Zheng, X.; Zhou, B.; Li, J.; Chen, M.; Deng, R.; Wong, G.; Lavillette, D.; Meng, G. SARS-CoV-2 spike engagement of ACE2 primes S2′ site cleavage and fusion initiation. Proc. Natl. Acad. Sci. USA 2022, 119, e2111199119. [Google Scholar] [CrossRef]
- Schaefer, S.L.; Jung, H.; Hummer, G. Binding of SARS-CoV-2 Fusion Peptide to Host Endosome and Plasma Membrane. J. Phys. Chem. B 2021, 125, 7732–7741. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, A.; Batchu, K.C.; Matsarskaia, O.; Prevost, S.F.; Russo, D.; Natali, F.; Seydel, T.; Hoffmann, I.; Laux, V.; Haertlein, M.; et al. Strikingly Different Roles of SARS-CoV-2 Fusion Peptides Uncovered by Neutron Scattering. J. Am. Chem. Soc. 2022, 144, 2968–2979. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.J.; Rey, F.A.; Veesler, D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162. [Google Scholar] [CrossRef] [Green Version]
- Wicht, O.; Burkard, C.; de Haan, C.A.; van Kuppeveld, F.J.; Rottier, P.J.; Bosch, B.J. Identification and characterization of a proteolytically primed form of the murine coronavirus spike proteins after fusion with the target cell. J. Virol. 2014, 88, 4943–4952. [Google Scholar] [CrossRef] [Green Version]
- Stout, A.E.; Millet, J.K.; Stanhope, M.J.; Whittaker, G.R. Furin cleavage sites in the spike proteins of bat and rodent coronaviruses: Implications for virus evolution and zoonotic transfer from rodent species. One Health 2021, 13, 100282. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA 2014, 111, 15214–15219. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.W.; Chao, T.L.; Li, C.L.; Chiu, M.F.; Kao, H.C.; Wang, S.H.; Pang, Y.H.; Lin, C.H.; Tsai, Y.M.; Lee, W.H.; et al. Furin Inhibitors Block SARS-CoV-2 Spike Protein Cleavage to Suppress Virus Production and Cytopathic Effects. Cell Rep. 2020, 33, 108254. [Google Scholar] [CrossRef]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortola, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef] [PubMed]
- Bertram, S.; Glowacka, I.; Müller, M.A.; Lavender, H.; Gnirss, K.; Nehlmeier, I.; Niemeyer, D.; He, Y.; Simmons, G.; Drosten, C.; et al. Cleavage and activation of the severe acute respiratory syndrome coronavirus spike protein by human airway trypsin-like protease. J. Virol. 2011, 85, 13363–13372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glowacka, I.; Bertram, S.; Muller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchrieser, J.; Dufloo, J.; Hubert, M.; Monel, B.; Planas, D.; Rajah, M.M.; Planchais, C.; Porrot, F.; Guivel-Benhassine, F.; Van der Werf, S.; et al. Syncytia formation by SARS-CoV-2-infected cells. Embo J. 2021, 40, e107405. [Google Scholar] [CrossRef] [PubMed]
- Rajah, M.M.; Bernier, A.; Buchrieser, J.; Schwartz, O. The Mechanism and Consequences of SARS-CoV-2 Spike-Mediated Fusion and Syncytia Formation. J. Mol. Biol. 2022, 434, 167280. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Evans, J.P.; King, T.; Zheng, Y.M.; Oltz, E.M.; Whelan, S.P.J.; Saif, L.J.; Peeples, M.E.; Liu, S.L. SARS-CoV-2 spreads through cell-to-cell transmission. Proc. Natl. Acad. Sci. USA 2022, 119, e2111400119. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Li, Q.; Wang, Y.; Shi, Y. Syncytia formation during SARS-CoV-2 lung infection: A disastrous unity to eliminate lymphocytes. Cell Death Differ. 2021, 28, 2019–2021. [Google Scholar] [CrossRef]
- Bussani, R.; Schneider, E.; Zentilin, L.; Collesi, C.; Ali, H.; Braga, L.; Volpe, M.C.; Colliva, A.; Zanconati, F.; Berlot, G.; et al. Persistence of viral RNA, pneumocyte syncytia and thrombosis are hallmarks of advanced COVID-19 pathology. EBioMedicine 2020, 61, 103104. [Google Scholar] [CrossRef]
- Braga, L.; Ali, H.; Secco, I.; Chiavacci, E.; Neves, G.; Goldhill, D.; Penn, R.; Jimenez-Guardeno, J.M.; Ortega-Prieto, A.M.; Bussani, R.; et al. Drugs that inhibit TMEM16 proteins block SARS-CoV-2 spike-induced syncytia. Nature 2021, 594, 88–93. [Google Scholar] [CrossRef]
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef]
- Jaimes, J.A.; Millet, J.K.; Whittaker, G.R. Proteolytic Cleavage of the SARS-CoV-2 Spike Protein and the Role of the Novel S1/S2 Site. iScience 2020, 23, 101212. [Google Scholar] [CrossRef]
- Rajah, M.M.; Hubert, M.; Bishop, E.; Saunders, N.; Robinot, R.; Grzelak, L.; Planas, D.; Dufloo, J.; Gellenoncourt, S.; Bongers, A.; et al. SARS-CoV-2 Alpha, Beta, and Delta variants display enhanced Spike-mediated syncytia formation. EMBO J. 2021, 40, e108944. [Google Scholar] [CrossRef]
- Ssenyange, G.; Kerfoot, M.; Zhao, M.; Farhadian, S.; Chen, S.; Peng, L.; Ren, P.; Dela Cruz, C.S.; Gupta, S.; Sutton, R.E. Development of an efficient reproducible cell-cell transmission assay for rapid quantification of SARS-CoV-2 spike interaction with hACE2. Cell Rep. Methods 2022, 2, 100252. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Teesalu, T.; Sugahara, K.N.; Kotamraju, V.R.; Ruoslahti, E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. USA 2009, 106, 16157–16162. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Mendonca, L.; Yang, Y.; Gao, Y.; Shen, C.; Liu, J.; Ni, T.; Ju, B.; Liu, C.; Tang, X.; et al. The Architecture of Inactivated SARS-CoV-2 with Postfusion Spikes Revealed by Cryo-EM and Cryo-ET. Structure 2020, 28, 1218–1224.e4. [Google Scholar] [CrossRef]
- Ge, J.; Wang, R.; Ju, B.; Zhang, Q.; Sun, J.; Chen, P.; Zhang, S.; Tian, Y.; Shan, S.; Cheng, L.; et al. Antibody neutralization of SARS-CoV-2 through ACE2 receptor mimicry. Nat. Commun. 2021, 12, 250. [Google Scholar] [CrossRef]
- Park, Y.J.; De Marco, A.; Starr, T.N.; Liu, Z.; Pinto, D.; Walls, A.C.; Zatta, F.; Zepeda, S.K.; Bowen, J.E.; Sprouse, K.R.; et al. Antibody-mediated broad sarbecovirus neutralization through ACE2 molecular mimicry. Science 2022, 375, 449–454. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Izard, T.; Farzan, M.; Choe, H.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2020, 592, 116–121. [Google Scholar] [CrossRef]
- Gobeil, S.M.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Manne, K.; Stalls, V.; Kopp, M.F.; Henderson, R.; Edwards, R.J.; et al. D614G Mutation Alters SARS-CoV-2 Spike Conformation and Enhances Protease Cleavage at the S1/S2 Junction. Cell Rep. 2021, 34, 108630. [Google Scholar] [CrossRef]
- Ozono, S.; Zhang, Y.; Ode, H.; Sano, K.; Tan, T.S.; Imai, K.; Miyoshi, K.; Kishigami, S.; Ueno, T.; Iwatani, Y.; et al. SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat. Commun. 2021, 12, 848. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cai, Y.; Xiao, T.; Lu, J.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rits-Volloch, S.; Zhu, H.; Woosley, A.N.; et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science 2021, 372, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Escalera, A.; Gonzalez-Reiche, A.S.; Aslam, S.; Mena, I.; Laporte, M.; Pearl, R.L.; Fossati, A.; Rathnasinghe, R.; Alshammary, H.; van de Guchte, A.; et al. Mutations in SARS-CoV-2 variants of concern link to increased spike cleavage and virus transmission. Cell Host Microbe 2022, 30, 373–387.e7. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Chan, J.F.; Liu, H.; Liu, Y.; Chai, Y.; Shi, J.; Shuai, H.; Hou, Y.; Huang, X.; Yuen, T.T.; et al. Spike mutations contributing to the altered entry preference of SARS-CoV-2 omicron BA.1 and BA.2. Emerg. Microbes Infect. 2022, 11, 2275–2287. [Google Scholar] [CrossRef]
- Braun, K.M.; Moreno, G.K.; Halfmann, P.J.; Hodcroft, E.B.; Baker, D.A.; Boehm, E.C.; Weiler, A.M.; Haj, A.K.; Hatta, M.; Chiba, S.; et al. Transmission of SARS-CoV-2 in domestic cats imposes a narrow bottleneck. PLoS Pathog. 2021, 17, e1009373. [Google Scholar] [CrossRef]
- Obermeyer, F.; Jankowiak, M.; Barkas, N.; Schaffner, S.F.; Pyle, J.D.; Yurkovetskiy, L.; Bosso, M.; Park, D.J.; Babadi, M.; MacInnis, B.L.; et al. Analysis of 6.4 million SARS-CoV-2 genomes identifies mutations associated with fitness. Science 2022, 376, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Willett, B.J.; Grove, J.; MacLean, O.A.; Wilkie, C.; De Lorenzo, G.; Furnon, W.; Cantoni, D.; Scott, S.; Logan, N.; Ashraf, S.; et al. SARS-CoV-2 Omicron is an immune escape variant with an altered cell entry pathway. Nat. Microbiol. 2022, 7, 1161–1179. [Google Scholar] [CrossRef]
- Halfmann, P.J.; Iida, S.; Iwatsuki-Horimoto, K.; Maemura, T.; Kiso, M.; Scheaffer, S.M.; Darling, T.L.; Joshi, A.; Loeber, S.; Singh, G.; et al. SARS-CoV-2 Omicron virus causes attenuated disease in mice and hamsters. Nature 2022, 603, 687–692. [Google Scholar] [CrossRef]
- Meng, B.; Abdullahi, A.; Ferreira, I.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S.; et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef]
- Qu, P.; Evans, J.P.; Kurhade, C.; Zeng, C.; Zheng, Y.M.; Xu, K.; Shi, P.Y.; Xie, X.; Liu, S.L. Determinants and Mechanisms of the Low Fusogenicity and High Dependence on Endosomal Entry of Omicron Subvariants. mBio 2023, 10, e0317622. [Google Scholar] [CrossRef]
- Pastorio, C.; Zech, F.; Noettger, S.; Jung, C.; Jacob, T.; Sanderson, T.; Sparrer, K.M.J.; Kirchhoff, F. Determinants of Spike infectivity, processing, and neutralization in SARS-CoV-2 Omicron subvariants BA.1 and BA.2. Cell Host Microbe 2022, 30, 1255–1268.e5. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wang, C.; White, K.I.; Pfuetzner, R.A.; Esquivies, L.; Brunger, A.T. Structural conservation among variants of the SARS-CoV-2 spike postfusion bundle. Proc. Natl. Acad. Sci. USA 2022, 119, e2119467119. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Chan, J.F.; Wang, L.; Jiao, F.; Chik, K.K.; Chu, H.; Lan, Q.; Xu, W.; Wang, Q.; Wang, C.; et al. Peptide-based pan-CoV fusion inhibitors maintain high potency against SARS-CoV-2 Omicron variant. Cell Res. 2022, 32, 404–406. [Google Scholar] [CrossRef]
- Belouzard, S.; Chu, V.C.; Whittaker, G.R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. USA 2009, 106, 5871–5876. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, E.; Matrosovich, T.; Beyerle, M.; Klenk, H.D.; Garten, W.; Matrosovich, M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 2006, 80, 9896–9898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef]
- Limburg, H.; Harbig, A.; Bestle, D.; Stein, D.A.; Moulton, H.M.; Jaeger, J.; Janga, H.; Hardes, K.; Koepke, J.; Schulte, L.; et al. TMPRSS2 Is the Major Activating Protease of Influenza A Virus in Primary Human Airway Cells and Influenza B Virus in Human Type II Pneumocytes. J. Virol. 2019, 93, e00649-19. [Google Scholar] [CrossRef] [Green Version]
- Koch, J.; Uckeley, Z.M.; Doldan, P.; Stanifer, M.; Boulant, S.; Lozach, P.Y. TMPRSS2 expression dictates the entry route used by SARS-CoV-2 to infect host cells. EMBO J. 2021, 40, e107821. [Google Scholar] [CrossRef]
- Matsuyama, S.; Ujike, M.; Morikawa, S.; Tashiro, M.; Taguchi, F. Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc. Natl. Acad. Sci. USA 2005, 102, 12543–12547. [Google Scholar] [CrossRef] [Green Version]
- Essalmani, R.; Jain, J.; Susan-Resiga, D.; Andreo, U.; Evagelidis, A.; Derbali, R.M.; Huynh, D.N.; Dallaire, F.; Laporte, M.; Delpal, A.; et al. Distinctive Roles of Furin and TMPRSS2 in SARS-CoV-2 Infectivity. J. Virol. 2022, 96, e0012822. [Google Scholar] [CrossRef] [PubMed]
- Ou, T.; Mou, H.; Zhang, L.; Ojha, A.; Choe, H.; Farzan, M. Hydroxychloroquine-mediated inhibition of SARS-CoV-2 entry is attenuated by TMPRSS2. PLoS Pathog. 2021, 17, e1009212. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Mosbauer, K.; Hofmann-Winkler, H.; Kaul, A.; Kleine-Weber, H.; Kruger, N.; Gassen, N.C.; Muller, M.A.; Drosten, C.; Pohlmann, S. Chloroquine does not inhibit infection of human lung cells with SARS-CoV-2. Nature 2020, 585, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Wettstein, L.; Knaff, P.M.; Kersten, C.; Muller, P.; Weil, T.; Conzelmann, C.; Muller, J.A.; Bruckner, M.; Hoffmann, M.; Pohlmann, S.; et al. Peptidomimetic inhibitors of TMPRSS2 block SARS-CoV-2 infection in cell culture. Commun. Biol. 2022, 5, 681. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, M.; Damalanka, V.C.; Tartell, M.A.; Chung, D.H.; Lourenco, A.L.; Pwee, D.; Mayer Bridwell, A.E.; Hoffmann, M.; Voss, J.; Karmakar, P.; et al. A novel class of TMPRSS2 inhibitors potently block SARS-CoV-2 and MERS-CoV viral entry and protect human epithelial lung cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2108728118. [Google Scholar] [CrossRef] [PubMed]
- Shapira, T.; Monreal, I.A.; Dion, S.P.; Buchholz, D.W.; Imbiakha, B.; Olmstead, A.D.; Jager, M.; Desilets, A.; Gao, G.; Martins, M.; et al. A TMPRSS2 inhibitor acts as a pan-SARS-CoV-2 prophylactic and therapeutic. Nature 2022, 605, 340–348. [Google Scholar] [CrossRef]
- Zhao, H.; Lu, L.; Peng, Z.; Chen, L.L.; Meng, X.; Zhang, C.; Ip, J.D.; Chan, W.M.; Chu, A.W.; Chan, K.H.; et al. SARS-CoV-2 Omicron variant shows less efficient replication and fusion activity when compared with Delta variant in TMPRSS2-expressed cells. Emerg. Microbes Infect. 2022, 11, 277–283. [Google Scholar] [CrossRef]
- Suzuki, R.; Yamasoba, D.; Kimura, I.; Wang, L.; Kishimoto, M.; Ito, J.; Morioka, Y.; Nao, N.; Nasser, H.; Uriu, K.; et al. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature 2022, 603, 700–705. [Google Scholar] [CrossRef]
- Chen, D.Y.; Chin, C.V.; Kenney, D.; Tavares, A.H.; Khan, N.; Conway, H.L.; Liu, G.; Choudhary, M.C.; Gertje, H.P.; O’Connell, A.K.; et al. Spike and nsp6 are key determinants of SARS-CoV-2 Omicron BA.1 attenuation. Nature 2023. [Google Scholar] [CrossRef]
- Bottcher-Friebertshauser, E.; Freuer, C.; Sielaff, F.; Schmidt, S.; Eickmann, M.; Uhlendorff, J.; Steinmetzer, T.; Klenk, H.D.; Garten, W. Cleavage of influenza virus hemagglutinin by airway proteases TMPRSS2 and HAT differs in subcellular localization and susceptibility to protease inhibitors. J. Virol. 2010, 84, 5605–5614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreutzberger, A.J.B.; Sanyal, A.; Saminathan, A.; Bloyet, L.M.; Stumpf, S.; Liu, Z.; Ojha, R.; Patjas, M.T.; Geneid, A.; Scanavachi, G.; et al. SARS-CoV-2 requires acidic pH to infect cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2209514119. [Google Scholar] [CrossRef] [PubMed]
- Fraser, B.J.; Beldar, S.; Seitova, A.; Hutchinson, A.; Mannar, D.; Li, Y.; Kwon, D.; Tan, R.; Wilson, R.P.; Leopold, K.; et al. Structure and activity of human TMPRSS2 protease implicated in SARS-CoV-2 activation. Nat. Chem. Biol. 2022, 18, 963–971. [Google Scholar] [CrossRef]
- Fuentes-Prior, P. Priming of SARS-CoV-2 S protein by several membrane-bound serine proteinases could explain enhanced viral infectivity and systemic COVID-19 infection. J. Biol. Chem. 2021, 296, 100135. [Google Scholar] [CrossRef]
- Kishimoto, M.; Uemura, K.; Sanaki, T.; Sato, A.; Hall, W.W.; Kariwa, H.; Orba, Y.; Sawa, H.; Sasaki, M. TMPRSS11D and TMPRSS13 Activate the SARS-CoV-2 Spike Protein. Viruses 2021, 13, 384. [Google Scholar] [CrossRef]
- Zang, R.; Gomez Castro, M.F.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef] [Green Version]
- Bosch, B.J.; Bartelink, W.; Rottier, P.J. Cathepsin L functionally cleaves the severe acute respiratory syndrome coronavirus class I fusion protein upstream of rather than adjacent to the fusion peptide. J. Virol. 2008, 82, 8887–8890. [Google Scholar] [CrossRef] [Green Version]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 infects cells after viral entry via clathrin-mediated endocytosis. J. Biol. Chem. 2021, 296, 100306. [Google Scholar] [CrossRef]
- Daniloski, Z.; Jordan, T.X.; Wessels, H.H.; Hoagland, D.A.; Kasela, S.; Legut, M.; Maniatis, S.; Mimitou, E.P.; Lu, L.; Geller, E.; et al. Identification of Required Host Factors for SARS-CoV-2 Infection in Human Cells. Cell 2021, 184, 92–105.e16. [Google Scholar] [CrossRef]
- Williams, C.G.; Jureka, A.S.; Silvas, J.A.; Nicolini, A.M.; Chvatal, S.A.; Carlson-Stevermer, J.; Oki, J.; Holden, K.; Basler, C.F. Inhibitors of VPS34 and fatty-acid metabolism suppress SARS-CoV-2 replication. Cell Rep. 2021, 36, 109479. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.M.; Zhu, Y.; Zhang, L.; Zhong, G.; Tai, L.; Liu, S.; Yin, G.; Lu, J.; He, Q.; Li, M.J.; et al. Novel cleavage sites identified in SARS-CoV-2 spike protein reveal mechanism for cathepsin L-facilitated viral infection and treatment strategies. Cell Discov. 2022, 8, 53. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.M.; Yang, W.L.; Yang, F.Y.; Zhang, L.; Huang, W.J.; Hou, W.; Fan, C.F.; Jin, R.H.; Feng, Y.M.; Wang, Y.C.; et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target Ther. 2021, 6, 134. [Google Scholar] [CrossRef]
- Richards, C.M.; Jabs, S.; Qiao, W.; Varanese, L.D.; Schweizer, M.; Mosen, P.R.; Riley, N.M.; Klussendorf, M.; Zengel, J.R.; Flynn, R.A.; et al. The human disease gene LYSET is essential for lysosomal enzyme transport and viral infection. Science 2022, 378, eabn5648. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. beta-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535.e14. [Google Scholar] [CrossRef]
- Chen, D.; Zheng, Q.; Sun, L.; Ji, M.; Li, Y.; Deng, H.; Zhang, H. ORF3a of SARS-CoV-2 promotes lysosomal exocytosis-mediated viral egress. Dev. Cell 2021, 56, 3250–3263.e5. [Google Scholar] [CrossRef]
- Jocher, G.; Grass, V.; Tschirner, S.K.; Riepler, L.; Breimann, S.; Kaya, T.; Oelsner, M.; Hamad, M.S.; Hofmann, L.I.; Blobel, C.P.; et al. ADAM10 and ADAM17 promote SARS-CoV-2 cell entry and spike protein-mediated lung cell fusion. EMBO Rep. 2022, 23, e54305. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pohlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [Green Version]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef] [Green Version]
- Harte, J.V.; Wakerlin, S.L.; Lindsay, A.J.; McCarthy, J.V.; Coleman-Vaughan, C. Metalloprotease-Dependent S2′-Activation Promotes Cell-Cell Fusion and Syncytiation of SARS-CoV-2. Viruses 2022, 14, 2094. [Google Scholar] [CrossRef]
- Yamamoto, M.; Gohda, J.; Kobayashi, A.; Tomita, K.; Hirayama, Y.; Koshikawa, N.; Seiki, M.; Semba, K.; Akiyama, T.; Kawaguchi, Y.; et al. Metalloproteinase-Dependent and TMPRSS2-Independent Cell Surface Entry Pathway of SARS-CoV-2 Requires the Furin Cleavage Site and the S2 Domain of Spike Protein. mBio 2022, 13, e0051922. [Google Scholar] [PubMed]
- Benlarbi, M.; Laroche, G.; Fink, C.; Fu, K.; Mulloy, R.P.; Phan, A.; Ariana, A.; Stewart, C.M.; Prevost, J.; Beaudoin-Bussieres, G.; et al. Identification and differential usage of a host metalloproteinase entry pathway by SARS-CoV-2 Delta and Omicron. iScience 2022, 25, 105316. [Google Scholar] [CrossRef] [PubMed]
- Belouzard, S.; Madu, I.; Whittaker, G.R. Elastase-mediated activation of the severe acute respiratory syndrome coronavirus spike protein at discrete sites within the S2 domain. J. Biol. Chem. 2010, 285, 22758–22763. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Attia, Z.; Woodfint, R.M.; Zeng, C.; Kim, S.H.; Steiner, H.E.; Shukla, R.K.; Liyanage, N.P.M.; Ghimire, S.; Li, J.; et al. Inhibition of elastase enhances the adjuvanticity of alum and promotes anti-SARS-CoV-2 systemic and mucosal immunity. Proc. Natl. Acad. Sci. USA 2021, 118, e2102435118. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Mercadante, M.; Nilsson-Payant, B.; Johnson, J.L.; Jaimes, J.A.; Muecksch, F.; Weisblum, Y.; Bram, Y.; Chandar, V.; Whittaker, G.R.; et al. Coagulation factors directly cleave SARS-CoV-2 spike and enhance viral entry. Elife 2022, 11, e77444. [Google Scholar] [CrossRef] [PubMed]
- Wettstein, L.; Immenschuh, P.; Weil, T.; Conzelmann, C.; Almeida-Hernandez, Y.; Hoffmann, M.; Kempf, A.; Nehlmeier, I.; Lotke, R.; Petersen, M.; et al. Native and activated antithrombin inhibits TMPRSS2 activity and SARS-CoV-2 infection. J. Med. Virol. 2023, 95, e28124. [Google Scholar] [CrossRef] [PubMed]
- Schultze, J.L.; Aschenbrenner, A.C. COVID-19 and the human innate immune system. Cell 2021, 184, 1671–1692. [Google Scholar] [CrossRef]
- Das, T.; Yang, X.; Lee, H.; Garst, E.; Valencia, E.; Chandran, K.; Im, W.; Hang, H. S-palmitoylation and sterol interactions mediate antiviral specificity of IFITM. ACS Chem. Biol. 2022, 17, 2109–2120. [Google Scholar] [CrossRef]
- Nchioua, R.; Schundner, A.; Kmiec, D.; Prelli Bozzo, C.; Zech, F.; Koepke, L.; Graf, A.; Krebs, S.; Blum, H.; Frick, M.; et al. SARS-CoV-2 Variants of Concern Hijack IFITM2 for Efficient Replication in Human Lung Cells. J. Virol. 2022, 96, e0059422. [Google Scholar] [CrossRef]
- Shi, G.; Kenney, A.D.; Kudryashova, E.; Zani, A.; Zhang, L.; Lai, K.K.; Hall-Stoodley, L.; Robinson, R.T.; Kudryashov, D.S.; Compton, A.A.; et al. Opposing activities of IFITM proteins in SARS-CoV-2 infection. EMBO J. 2021, 40, e106501. [Google Scholar] [CrossRef]
- Prelli Bozzo, C.; Nchioua, R.; Volcic, M.; Koepke, L.; Kruger, J.; Schutz, D.; Heller, S.; Sturzel, C.M.; Kmiec, D.; Conzelmann, C.; et al. IFITM proteins promote SARS-CoV-2 infection and are targets for virus inhibition in vitro. Nat. Commun. 2021, 12, 4584. [Google Scholar] [CrossRef]
- Alghamdi, J.; Alaamery, M.; Barhoumi, T.; Rashid, M.; Alajmi, H.; Aljasser, N.; Alhendi, Y.; Alkhalaf, H.; Alqahtani, H.; Algablan, O.; et al. Interferon-induced transmembrane protein-3 genetic variant rs12252 is associated with COVID-19 mortality. Genomics 2021, 113, 1733–1741. [Google Scholar] [CrossRef]
- Spence, J.S.; He, R.; Hoffmann, H.H.; Das, T.; Thinon, E.; Rice, C.M.; Peng, T.; Chandran, K.; Hang, H.C. IFITM3 directly engages and shuttles incoming virus particles to lysosomes. Nat. Chem. Biol. 2019, 15, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Zu, S.; Deng, Y.Q.; Zhou, C.; Li, J.; Li, L.; Chen, Q.; Li, X.F.; Zhao, H.; Gold, S.; He, J.; et al. 25-Hydroxycholesterol is a potent SARS-CoV-2 inhibitor. Cell Res. 2020, 30, 1043–1045. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, W.; Hui, H.; Tiwari, S.K.; Zhang, Q.; Croker, B.A.; Rawlings, S.; Smith, D.; Carlin, A.F.; Rana, T.M. Cholesterol 25-Hydroxylase inhibits SARS-CoV-2 and other coronaviruses by depleting membrane cholesterol. EMBO J. 2020, 39, e106057. [Google Scholar] [CrossRef]
- Zang, R.; Case, J.B.; Yutuc, E.; Ma, X.; Shen, S.; Gomez Castro, M.F.; Liu, Z.; Zeng, Q.; Zhao, H.; Son, J.; et al. Cholesterol 25-hydroxylase suppresses SARS-CoV-2 replication by blocking membrane fusion. Proc. Natl. Acad. Sci. USA 2020, 117, 32105–32113. [Google Scholar]
- Zhao, X.; Zheng, S.; Chen, D.; Zheng, M.; Li, X.; Li, G.; Lin, H.; Chang, J.; Zeng, H.; Guo, J.T. LY6E Restricts Entry of Human Coronaviruses, Including Currently Pandemic SARS-CoV-2. J. Virol. 2020, 94, e00562-20. [Google Scholar]
- Yu, J.; Liang, C.; Liu, S.L. CD4-Dependent Modulation of HIV-1 Entry by LY6E. J. Virol. 2019, 93, e01866-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Liang, C.; Liu, S.L. Interferon-inducible LY6E Protein Promotes HIV-1 Infection. J. Biol. Chem. 2017, 292, 4674–4685. [Google Scholar] [CrossRef] [Green Version]
- Mar, K.B.; Rinkenberger, N.R.; Boys, I.N.; Eitson, J.L.; McDougal, M.B.; Richardson, R.B.; Schoggins, J.W. LY6E mediates an evolutionarily conserved enhancement of virus infection by targeting a late entry step. Nat. Commun. 2018, 9, 3603. [Google Scholar] [CrossRef]
- Pfaender, S.; Mar, K.B.; Michailidis, E.; Kratzel, A.; Boys, I.N.; V’Kovski, P.; Fan, W.; Kelly, J.N.; Hirt, D.; Ebert, N.; et al. LY6E impairs coronavirus fusion and confers immune control of viral disease. Nat. Microbiol. 2020, 5, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Hackett, B.A.; Cherry, S. Flavivirus internalization is regulated by a size-dependent endocytic pathway. Proc. Natl. Acad. Sci. USA 2018, 115, 4246–4251. [Google Scholar] [CrossRef] [Green Version]
- Mihelic, M.; Dobersek, A.; Guncar, G.; Turk, D. Inhibitory fragment from the p41 form of invariant chain can regulate activity of cysteine cathepsins in antigen presentation. J. Biol. Chem. 2008, 283, 14453–14460. [Google Scholar] [CrossRef] [Green Version]
- Barrowman, J.; Michaelis, S. ZMPSTE24, an integral membrane zinc metalloprotease with a connection to progeroid disorders. Biol. Chem. 2009, 390, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Wang, L.; Li, S.; Dorf, M.E. ZMPSTE24 defends against influenza and other pathogenic viruses. J. Exp. Med. 2017, 214, 919–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shilagardi, K.; Spear, E.D.; Abraham, R.; Griffin, D.E.; Michaelis, S. The Integral Membrane Protein ZMPSTE24 Protects Cells from SARS-CoV-2 Spike-Mediated Pseudovirus Infection and Syncytia Formation. mBio 2022, 13, e0254322. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Kanneganti, T.D. Innate immunity: The first line of defense against SARS-CoV-2. Nat. Immunol. 2022, 23, 165–176. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-kappaB pathway. Elife 2021, 10, e68563. [Google Scholar] [CrossRef]
- Yamada, T.; Sato, S.; Sotoyama, Y.; Orba, Y.; Sawa, H.; Yamauchi, H.; Sasaki, M.; Takaoka, A. RIG-I triggers a signaling-abortive anti-SARS-CoV-2 defense in human lung cells. Nat. Immunol. 2021, 22, 820–828. [Google Scholar] [CrossRef]
- Bauernfried, S.; Scherr, M.J.; Pichlmair, A.; Duderstadt, K.E.; Hornung, V. Human NLRP1 is a sensor for double-stranded RNA. Science 2021, 371, eabd0811. [Google Scholar] [CrossRef]
- Planes, R.; Pinilla, M.; Santoni, K.; Hessel, A.; Passemar, C.; Lay, K.; Paillette, P.; Valadao, A.C.; Robinson, K.S.; Bastard, P.; et al. Human NLRP1 is a sensor of pathogenic coronavirus 3CL proteases in lung epithelial cells. Mol. Cell 2022, 82, 2385–2400.e9. [Google Scholar] [CrossRef]
- Junqueira, C.; Crespo, A.; Ranjbar, S.; de Lacerda, L.B.; Lewandrowski, M.; Ingber, J.; Parry, B.; Ravid, S.; Clark, S.; Schrimpf, M.R.; et al. FcgammaR-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature 2022, 606, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Lempp, F.A.; Soriaga, L.B.; Montiel-Ruiz, M.; Benigni, F.; Noack, J.; Park, Y.J.; Bianchi, S.; Walls, A.C.; Bowen, J.E.; Zhou, J.; et al. Lectins enhance SARS-CoV-2 infection and influence neutralizing antibodies. Nature 2021, 598, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.; Shen, M.; Yu, Z.; Ge, W.; Chen, K.; Tian, M.; Xiao, F.; Wang, Z.; Wang, J.; Jia, Y.; et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat. Commun. 2021, 12, 4664. [Google Scholar] [CrossRef] [PubMed]
- Yalcinkaya, M.; Liu, W.; Islam, M.N.; Kotini, A.G.; Gusarova, G.A.; Fidler, T.P.; Papapetrou, E.P.; Bhattacharya, J.; Wang, N.; Tall, A.R. Modulation of the NLRP3 inflammasome by SARS-CoV-2 Envelope protein. Sci. Rep. 2021, 11, 24432. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Zhu, Z.; Lin, H.; Wang, S.; Zhang, P.; Li, Y.; Li, L.; Wang, J.; Zhao, Y.; Han, J. Pyroptosis of syncytia formed by fusion of SARS-CoV-2 spike and ACE2-expressing cells. Cell Discov. 2021, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- Tahtinen, S.; Tong, A.J.; Himmels, P.; Oh, J.; Paler-Martinez, A.; Kim, L.; Wichner, S.; Oei, Y.; McCarron, M.J.; Freund, E.C.; et al. IL-1 and IL-1ra are key regulators of the inflammatory response to RNA vaccines. Nat. Immunol. 2022, 23, 532–542. [Google Scholar] [CrossRef]
- Hilligan, K.L.; Namasivayam, S.; Clancy, C.S.; O’Mard, D.; Oland, S.D.; Robertson, S.J.; Baker, P.J.; Castro, E.; Garza, N.L.; Lafont, B.A.P.; et al. Intravenous administration of BCG protects mice against lethal SARS-CoV-2 challenge. J. Exp. Med. 2022, 219, e20211862. [Google Scholar] [CrossRef]
- Counoupas, C.; Johansen, M.D.; Stella, A.O.; Nguyen, D.H.; Ferguson, A.L.; Aggarwal, A.; Bhattacharyya, N.D.; Grey, A.; Hutchings, O.; Patel, K.; et al. A single dose, BCG-adjuvanted COVID-19 vaccine provides sterilising immunity against SARS-CoV-2 infection. NPJ Vaccines 2021, 6, 143. [Google Scholar] [CrossRef]
- Rivas, M.N.; Ebinger, J.E.; Wu, M.; Sun, N.; Braun, J.; Sobhani, K.; Van Eyk, J.E.; Cheng, S.; Arditi, M. BCG vaccination history associates with decreased SARS-CoV-2 seroprevalence across a diverse cohort of health care workers. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, E.; Khan, N.; Tran, K.A.; Ulndreaj, A.; Pernet, E.; Fontes, G.; Lupien, A.; Desmeules, P.; McIntosh, F.; Abow, A.; et al. BCG vaccination provides protection against IAV but not SARS-CoV-2. Cell Rep. 2022, 38, 110502. [Google Scholar] [PubMed]
- Pepin, J.; Labbe, A.C.; Carignan, A.; Parent, M.E.; Yu, J.; Grenier, C.; Beauchemin, S.; De Wals, P.; Valiquette, L.; Rousseau, M.C. Does BCG provide long-term protection against SARS-CoV-2 infection? A case-control study in Quebec, Canada. Vaccine 2021, 39, 7300–7307. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Phenotypes | Wuhan-Hu-1, Alpha, Beta, Delta | Omicron and Subvariants BA.1, BA.2 … BQ.1.1 and XBB.1.5 |
|---|---|---|
| Conserved mutations | D614G | D614G, H655Y, Q954H and N969K |
| S1/S2 cleavage | Strong | Reduced |
| Plasma membrane entry | Yes | No |
| Endocytic entry | Yes | Yes |
| TMPRSS2 Usage | Yes | No |
| Predominantly targeted tissues | Nasal cavity, lung | Nasal cavity |
| Risk of lung pathogenicity | High | Low |
| ISG Name | Subcellular Localization | Affect Step of Virus Entry | Molecular Mechanisms | Other Coronaviruses |
|---|---|---|---|---|
| IFITMs 1 | Early and late endosome membranes, plasma membrane | Semi-fusion stage at the target cell plasma membrane or endosomes | Reduce the fluidity of the plasma membrane or expanding the curvature of the outer membrane lobule | hCoV-229E, SARS-CoV, MERS-CoV |
| CH25H | Endoplasmic reticulum membrane, plasma membrane, cytosol | Reduces spike-mediated cell-cell fusion | Converts cholesterol to 25HC, leading the depletion of accessible cholesterol from the plasma membrane | SARS-CoV, MERS-CoV, HCoV-229E, HCoV-OC43 |
| LY6E | Glycosylphosphatidyl-inositol (GPI)-anchored cell surface | Directly blocks membrane fusion | Directly blocks membrane fusion, depending on its GPI anchoring, being less dependent on the expression of host proteases, such as TMPRSS2. | SARS-CoV, SARS-CoV-2, MERS-CoV, MHV |
| CD74 2 | Plasma membrane | Inhibits cathepsin-mediated entry | Functional thyroglobulin type-1 domain inhibits cysteine proteinases | hCoV-229E, hCoV-OC43, SARS-CoV, MERS-CoV |
| ZMPSTE24 | Plasma membrane, endomembranes | Bingding, membrane fusion | Mediates cleavage and shedding of the ACE2 ectodomain interacts with IFITMs | hCoV-229E, hCoV-OC43, SARS-CoV, MERS-CoV |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, S.; Hu, H.; Ai, Q.; Bai, R.; Ma, K.; Zhou, M.; Wang, S. SARS-CoV-2 Spike-Mediated Entry and Its Regulation by Host Innate Immunity. Viruses 2023, 15, 639. https://doi.org/10.3390/v15030639
Yu S, Hu H, Ai Q, Bai R, Ma K, Zhou M, Wang S. SARS-CoV-2 Spike-Mediated Entry and Its Regulation by Host Innate Immunity. Viruses. 2023; 15(3):639. https://doi.org/10.3390/v15030639
Chicago/Turabian StyleYu, Shi, Huina Hu, Qiangyun Ai, Rong Bai, Kaixiong Ma, Minmin Zhou, and Shaobo Wang. 2023. "SARS-CoV-2 Spike-Mediated Entry and Its Regulation by Host Innate Immunity" Viruses 15, no. 3: 639. https://doi.org/10.3390/v15030639