
HIV-1 Gag Binds the Multi-Aminoacyl-tRNA Synthetase Complex via the EPRS Subunit
 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids, Protein Expression and Purification
2.2. RNA In Vitro Transcription and Labeling
2.3. Fluorescence-Quenching and Fluorescence Anisotropy (FA) Assays
2.4. Cell Culture and Stable Cell Line Generation
2.5. Immunoprecipitation (IP) and Western Blotting
2.6. Streptavidin Pull-Down Assay
2.7. Immunofluorescence Microscopy
2.8. Virus Production and Infectivity Assays
2.9. Quantitative Real-Time (qRT)-PCR and Luciferase Assays
3. Results
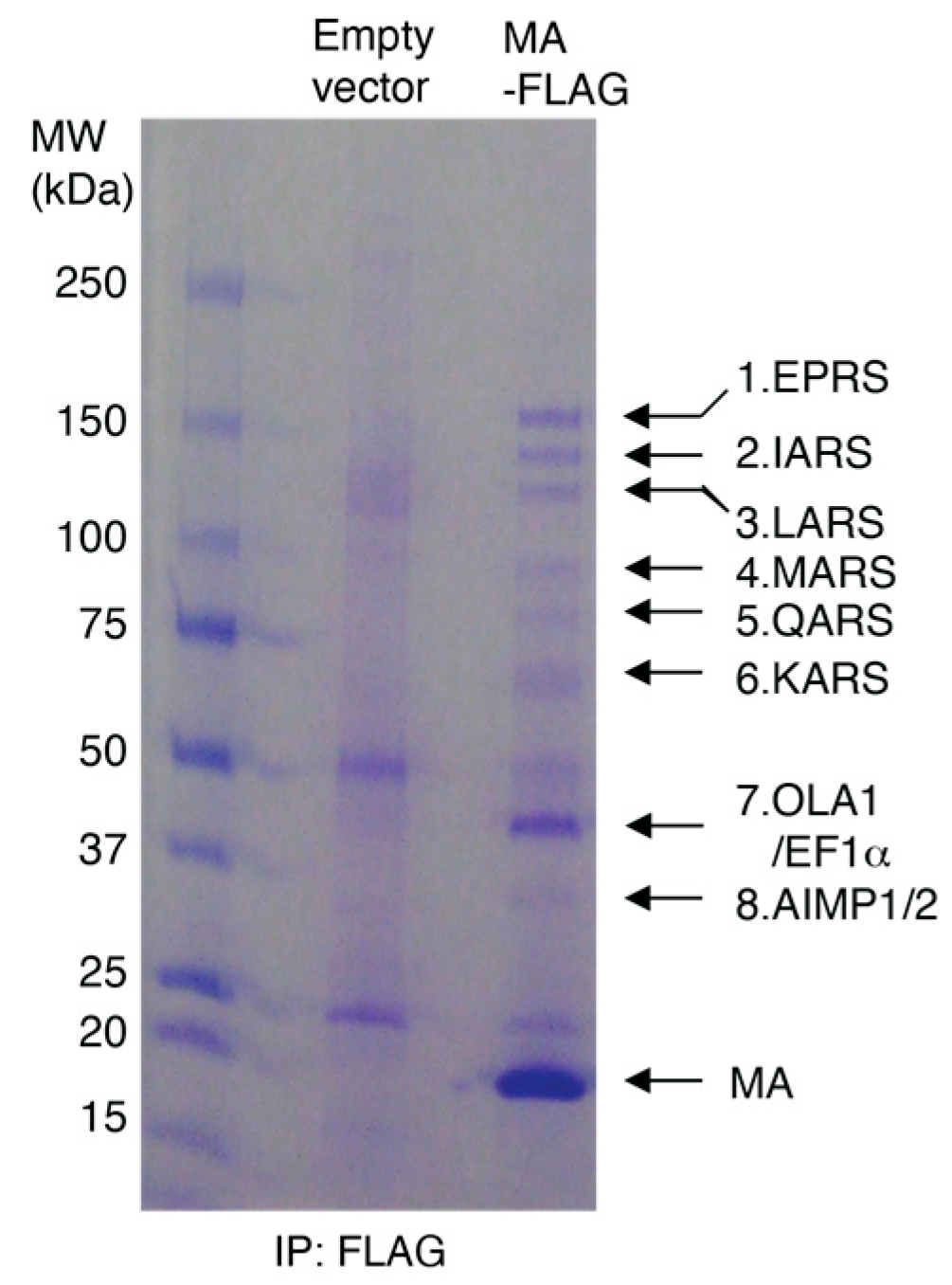
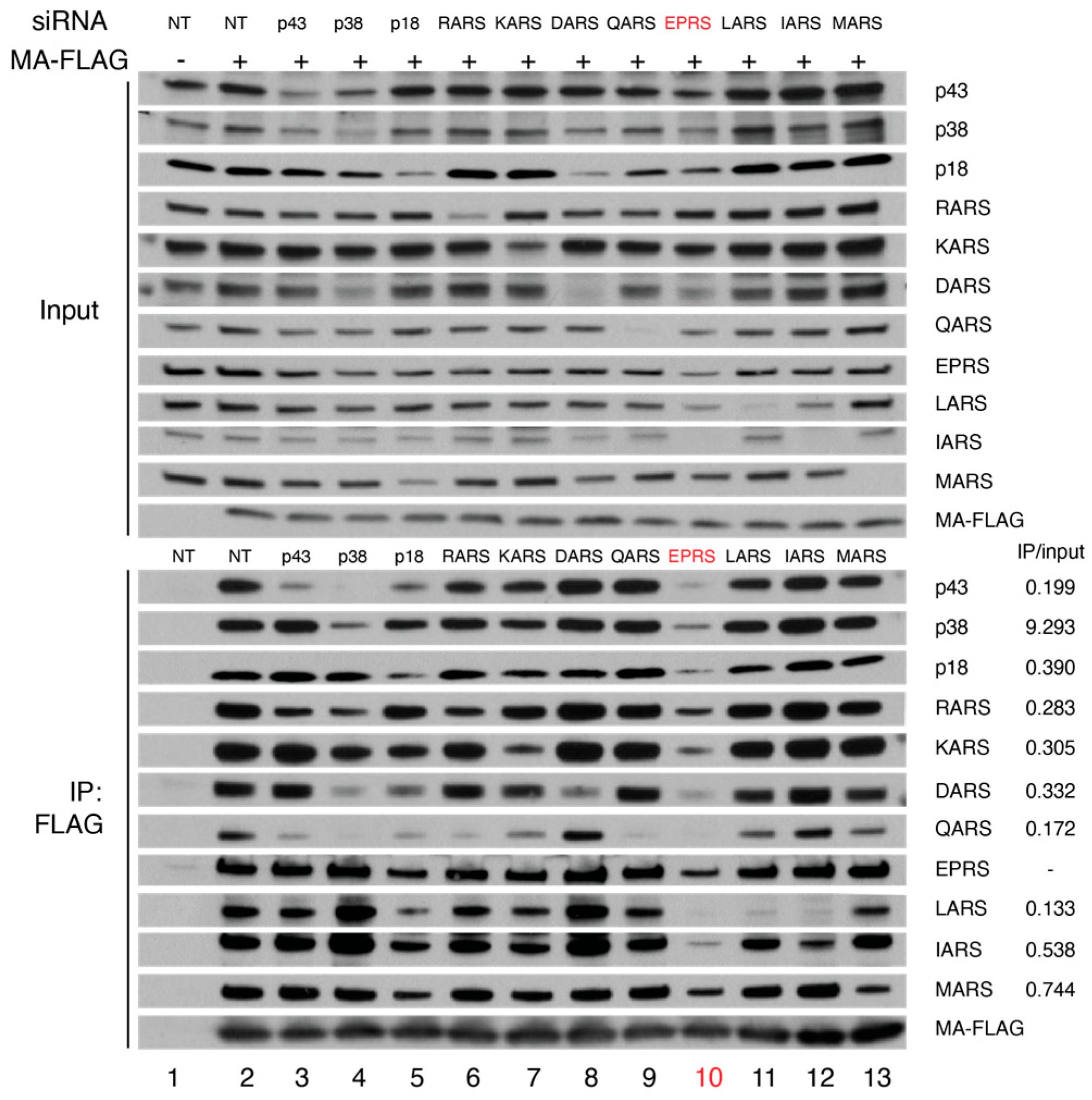
3.1. HIV-1 MA Interacts with the MSC Primarily through EPRS


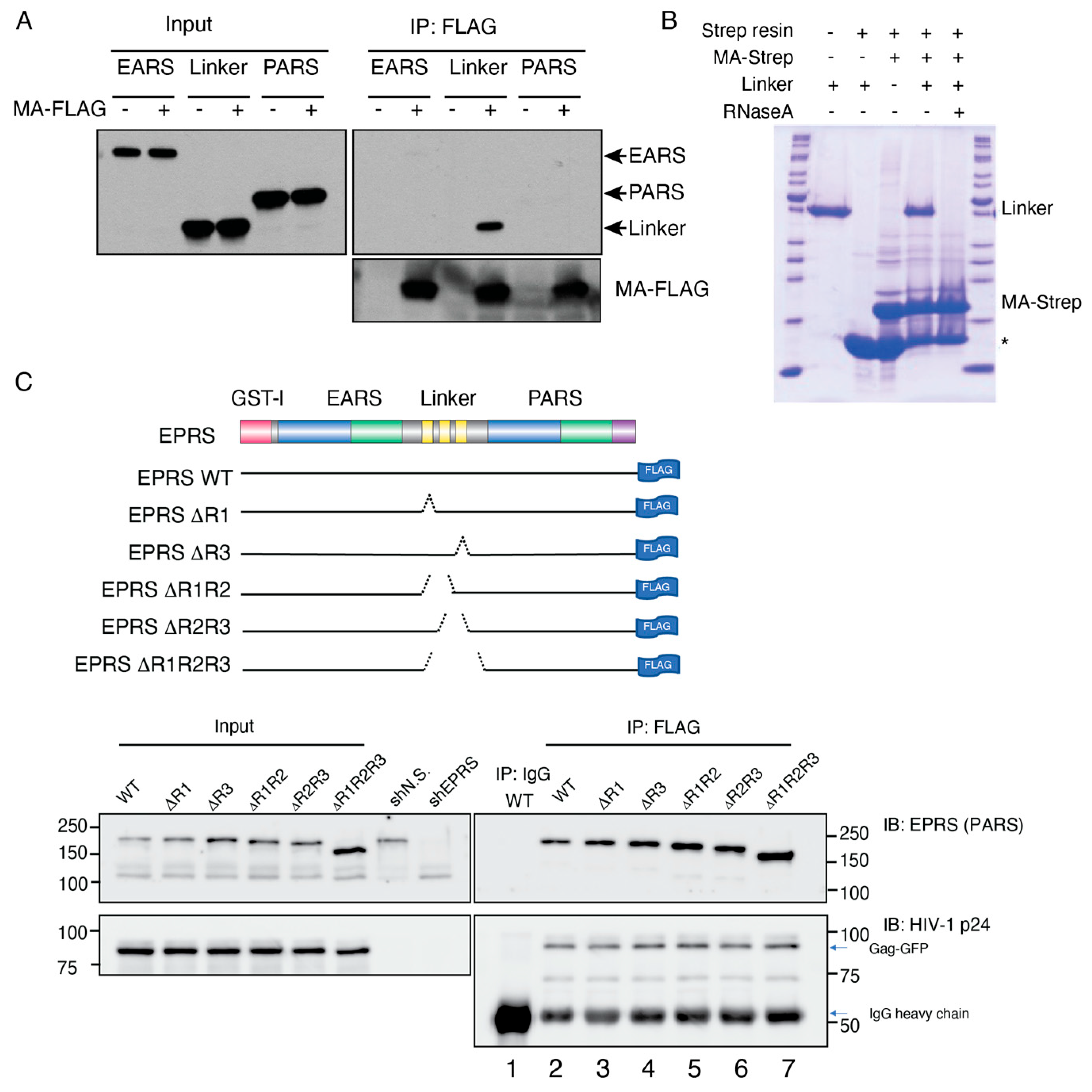
3.2. HIV-1 MA Interacts with the Linker Region of EPRS in an RNA-Dependent Manner

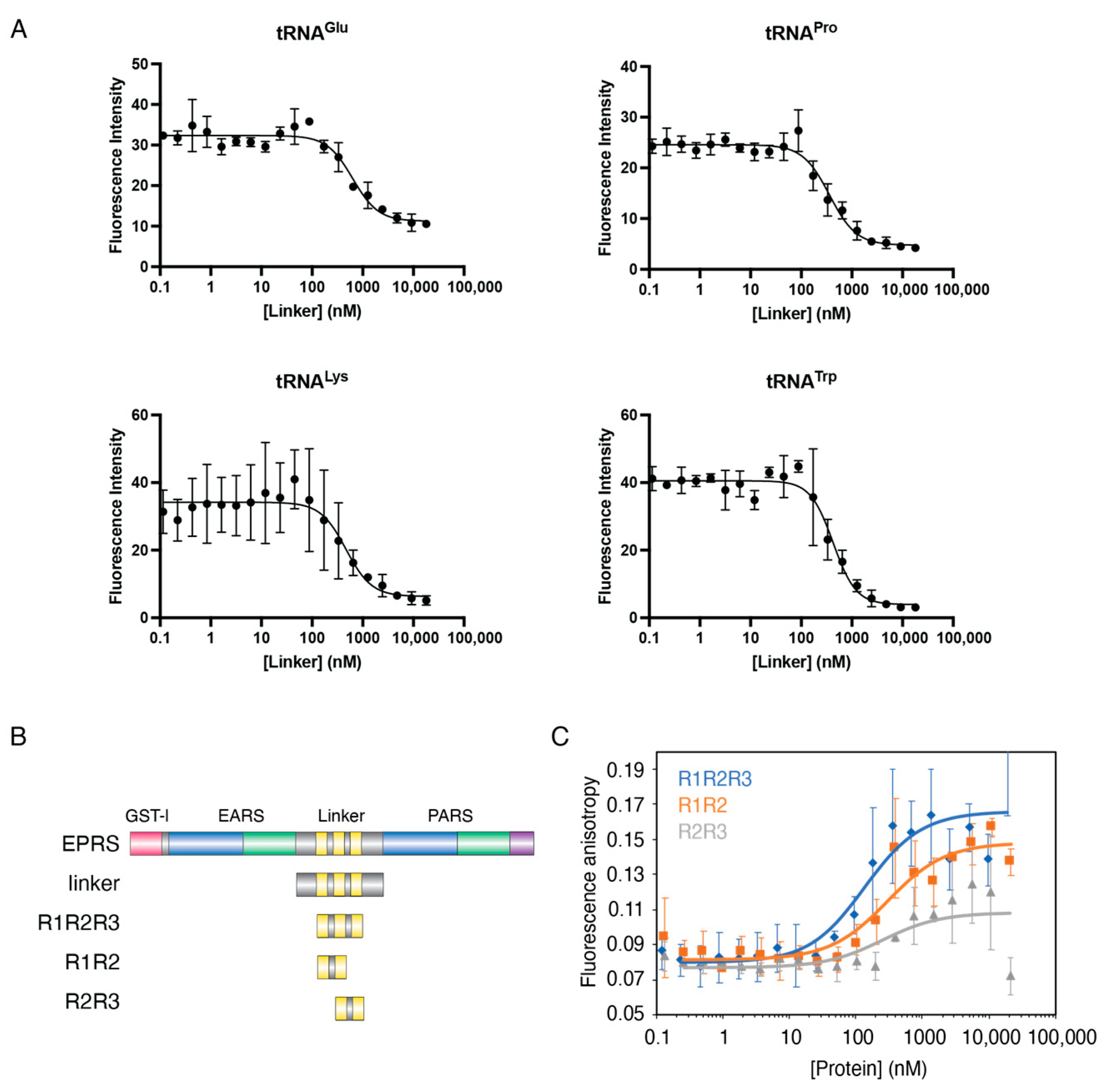
3.3. The Linker Domain of EPRS Interacts Promiscuously with tRNAs
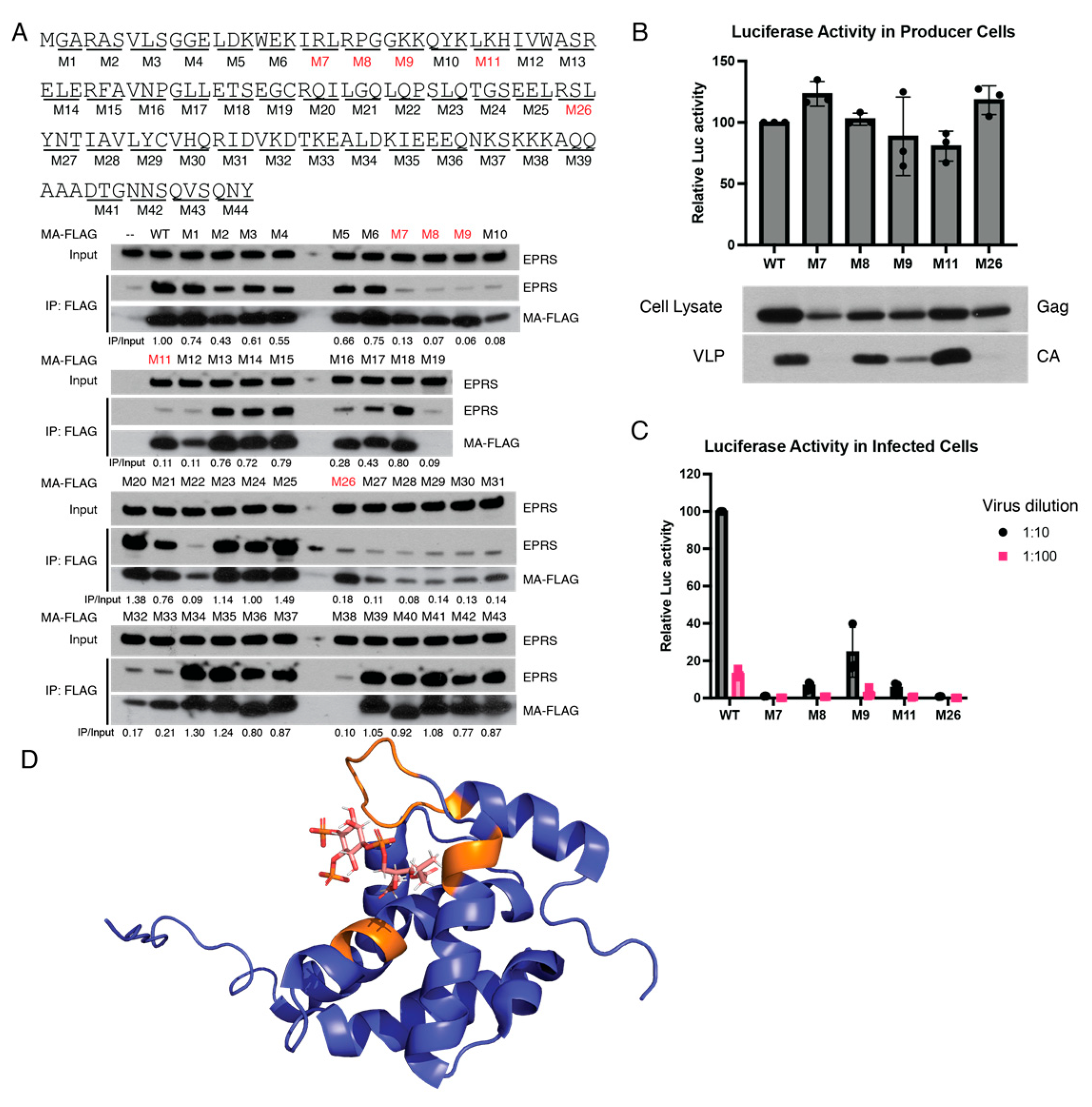
3.4. Identification of Amino Acid Residues in MA Critical for EPRS Interaction

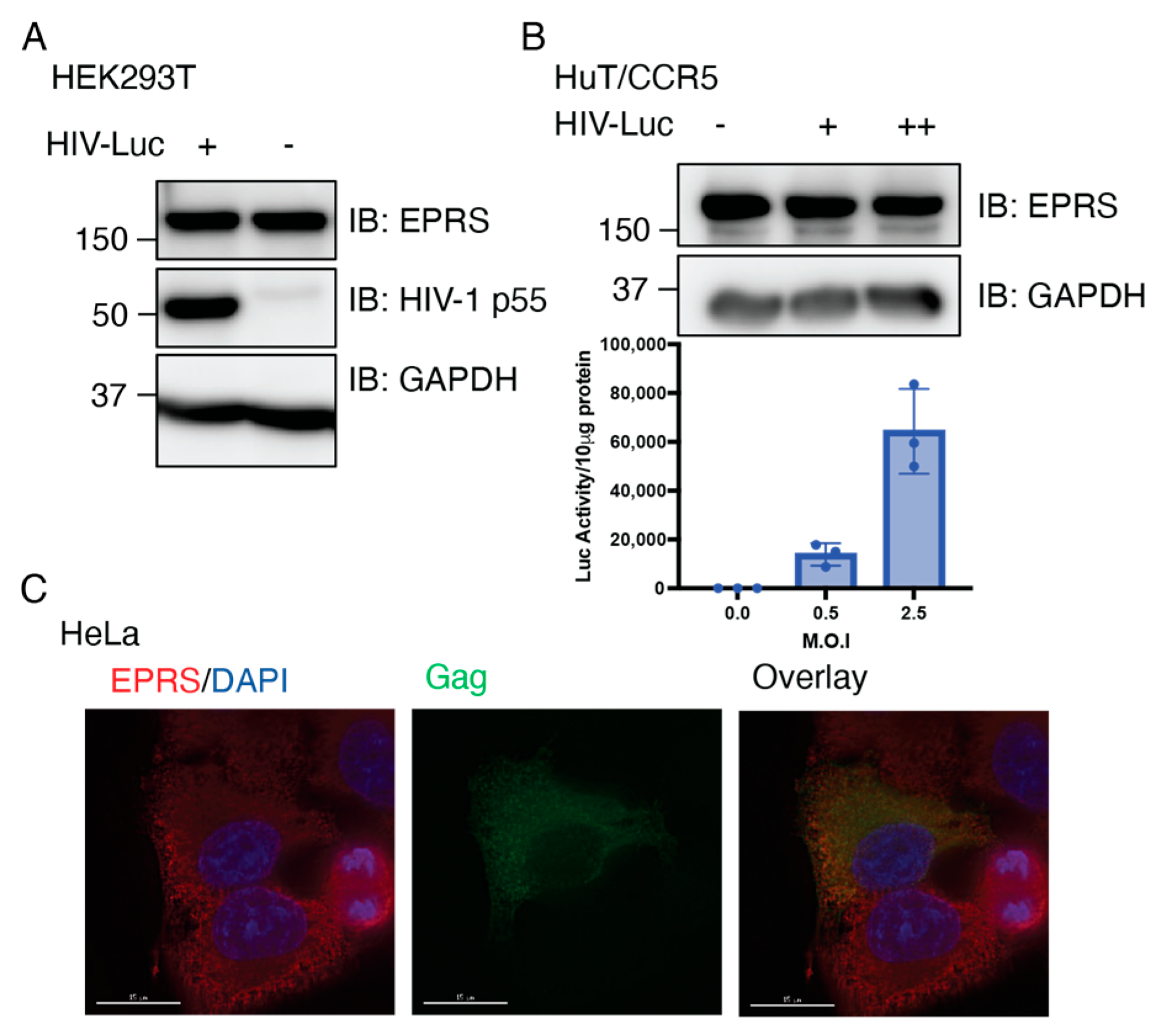
3.5. Single-Cycle HIV-1 Infection Does Not Alter the Expression of EPRS

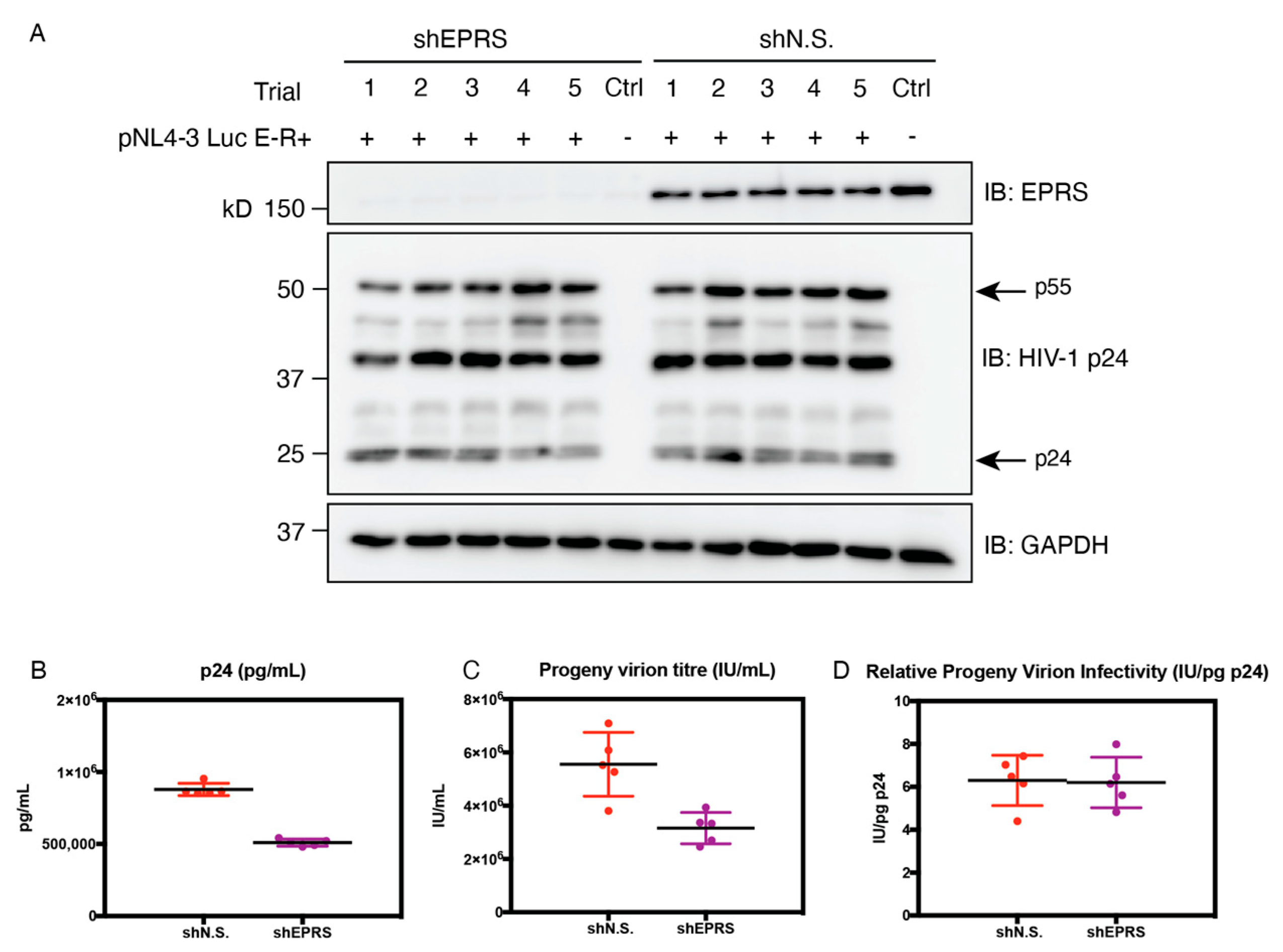
3.6. EPRS Knockdown Affects HIV-1 Gene Expression Due to Global Translational Defects

3.7. EPRS Knockdown Reduces Progeny Virion Production but Not Infectivity

4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HIV-1 | human immunodeficiency virus type 1 |
| PM | plasma membrane |
| MA | matrix |
| MSC | multi-aminoacyl-tRNA synthetase complex |
| ARS | aminoacyl-tRNA synthetase |
| EPRS | glutamyl-prolyl-tRNA synthetase |
| HDF | HIV-dependency factors |
| gRNA | genomic RNA |
| UTR | untranslated region |
| PBS | primer binding site |
| LysRS, KARS | lysyl-tRNA synthetase |
| CA | capsid |
| NC | nucleocapsid |
| HBR | highly basic region |
| PI(4,5)P2 | phosphatidylinositol-(4,5)-bisphosphate |
| MARS | methionyl-tRNA synthetase |
| LARS | leucyl-tRNA synthetase |
| IARS | isoleucyl-tRNA synthetase |
| DARS | aspartyl-tRNA synthetase |
| QARS | glutaminyl-tRNA synthetase |
| RARS | arginyl-tRNA synthetase |
| AIMP | ARS-interacting multifunctional protein |
| WARS | tryptophanyl-tRNA synthetase |
| HARS | histidyl-tRNA synthetase |
| IFN-g | interferon-g |
| GAIT | IFN-g-activated inhibition of translation |
| OLA1 | Obg-like ATPase 1 |
| NT | non-targeting |
| HIV-Luc/VSV-G | HIV-luciferase/vesicular stomatitis virus G |
| HuT/CCR5 | human CD4+ T cells expressing C-C chemokine receptor 5 |
References
- Friedrich, B.M.; Dziuba, N.; Li, G.; Endsley, M.A.; Murray, J.L.; Ferguson, M.R. Host Factors Mediating HIV-1 Replication. Virus Res. 2011, 161, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K.; et al. Global Landscape of HIV–Human Protein Complexes. Nature 2011, 481, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Börner, K.; Hermle, J.; Sommer, C.; Brown, N.P.; Knapp, B.; Glass, B.; Kunkel, J.; Torralba, G.; Reymann, J.; Beil, N.; et al. From Experimental Setup to Bioinformatics: An RNAi Screening Platform to Identify Host Factors Involved in HIV-1 Replication. Biotechnol. J. 2010, 5, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Knoener, R.A.; Becker, J.T.; Scalf, M.; Sherer, N.M.; Smith, L.M. Elucidating the in Vivo Interactome of HIV-1 RNA by Hybridization Capture and Mass Spectrometry. Sci. Rep. 2017, 7, 16965. [Google Scholar] [CrossRef] [PubMed]
- König, R.; Zhou, Y.; Elleder, D.; Diamond, T.L.; Bonamy, G.M.C.; Irelan, J.T.; Chiang, C.-Y.; Tu, B.P.; De Jesus, P.D.; Lilley, C.E.; et al. Global Analysis of Host-Pathogen Interactions That Regulate Early-Stage HIV-1 Replication. Cell 2008, 135, 49–60. [Google Scholar] [CrossRef]
- Pache, L.; König, R.; Chanda, S.K. Identifying HIV-1 Host Cell Factors by Genome-Scale RNAi Screening. Methods 2011, 53, 3–12. [Google Scholar] [CrossRef]
- OhAinle, M.; Helms, L.; Vermeire, J.; Roesch, F.; Humes, D.; Basom, R.; Delrow, J.J.; Overbaugh, J.; Emerman, M. A Virus-Packageable CRISPR Screen Identifies Host Factors Mediating Interferon Inhibition of HIV. eLife 2018, 7, e39823. [Google Scholar] [CrossRef]
- Engeland, C.E.; Brown, N.P.; Börner, K.; Schümann, M.; Krause, E.; Kaderali, L.; Müller, G.A.; Kräusslich, H.-G. Proteome Analysis of the HIV-1 Gag Interactome. Virology 2014, 460–461, 194–206. [Google Scholar] [CrossRef]
- Hultquist, J.F.; Schumann, K.; Woo, J.M.; Manganaro, L.; McGregor, M.J.; Doudna, J.; Simon, V.; Krogan, N.J.; Marson, A. A Cas9 Ribonucleoprotein Platform for Functional Genetic Studies of HIV-Host Interactions in Primary Human T Cells. Cell Rep. 2016, 17, 1438–1452. [Google Scholar] [CrossRef]
- Lingappa, J.R.; Lingappa, V.R.; Reed, J.C. Addressing Antiretroviral Drug Resistance with Host-Targeting Drugs—First Steps towards Developing a Host-Targeting HIV-1 Assembly Inhibitor. Viruses 2021, 13, 451. [Google Scholar] [CrossRef]
- Jin, D.; Musier-Forsyth, K. Role of Host TRNAs and Aminoacyl-TRNA Synthetases in Retroviral Replication. J. Biol. Chem. 2019, 294, 5352–5364. [Google Scholar] [CrossRef]
- Kleiman, L. TRNALys3: The Primer TRNA for Reverse Transcription in HIV-1. IUBMB Life 2002, 53, 107–114. [Google Scholar] [CrossRef]
- Berkhout, B. The Primer Binding Site on the RNA Genome of Human and Simian Immunodeficiency Viruses Is Flanked by an Upstream Hairpin Structure. Nucleic Acids Res. 1997, 25, 4013–4017. [Google Scholar] [CrossRef]
- Jiang, M.; Mak, J.; Ladha, A.; Cohen, E.; Klein, M.; Rovinski, B.; Kleiman, L. Identification of TRNAs Incorporated into Wild-Type and Mutant Human Immunodeficiency Virus Type 1. J. Virol. 1993, 67, 3246–3253. [Google Scholar] [CrossRef]
- Pavon-Eternod, M.; Wei, M.; Pan, T.; Kleiman, L. Profiling Non-Lysyl TRNAs in HIV-1. RNA 2010, 16, 267–273. [Google Scholar] [CrossRef]
- Kleiman, L.; Jones, C.P.; Musier-Forsyth, K. Formation of the TRNALys Packaging Complex in HIV-1. FEBS Lett. 2009, 584, 359–365. [Google Scholar] [CrossRef]
- Cen, S.; Khorchid, A.; Javanbakht, H.; Gabor, J.; Stello, T.; Shiba, K.; Musier-Forsyth, K.; Kleiman, L. Incorporation of Lysyl-TRNA Synthetase into Human Immunodeficiency Virus Type 1. J. Virol. 2001, 75, 5043–5048. [Google Scholar] [CrossRef]
- Duchon, A.A.; St. Gelais, C.; Titkemeier, N.; Hatterschide, J.; Wu, L.; Musier-Forsyth, K. HIV-1 Exploits a Dynamic Multi-Aminoacyl-TRNA Synthetase Complex to Enhance Viral Replication. J. Virol. 2017, 91, e01240-17. [Google Scholar] [CrossRef]
- Kobbi, L.; Octobre, G.; Dias, J.; Comisso, M.; Mirande, M. Association of Mitochondrial Lysyl-TRNA Synthetase with HIV-1 GagPol Involves Catalytic Domain of the Synthetase and Transframe and Integrase Domains of Pol. J. Mol. Biol. 2011, 410, 875–886. [Google Scholar] [CrossRef]
- Jones, C.P.; Datta, S.A.K.; Rein, A.; Rouzina, I.; Musier-Forsyth, K. Matrix Domain Modulates HIV-1 Gag’s Nucleic Acid Chaperone Activity via Inositol Phosphate Binding. J. Virol. 2011, 85, 1594–1603. [Google Scholar] [CrossRef] [Green Version]
- Sumner, C.; Ono, A. Relationship between HIV-1 Gag Multimerization and Membrane Binding. Viruses 2022, 14, 622. [Google Scholar] [CrossRef] [PubMed]
- Marie, V.; Gordon, M.L. The HIV-1 Gag Protein Displays Extensive Functional and Structural Roles in Virus Replication and Infectivity. Int. J. Mol. Sci. 2022, 23, 7569. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Parent, L.J.; Wills, J.W.; Resh, M.D. Identification of a Membrane-Binding Domain within the Amino-Terminal Region of Human Immunodeficiency Virus Type 1 Gag Protein Which Interacts with Acidic Phospholipids. J. Virol. 1994, 68, 2556–2569. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.P.; Worthylake, D.; Bancroft, D.P.; Christensen, A.M.; Sundquist, W.I. Crystal Structures of the Trimeric Human Immunodeficiency Virus Type 1 Matrix Protein: Implications for Membrane Association and Assembly. Proc. Natl. Acad. Sci. USA 1996, 93, 3099–3104. [Google Scholar] [CrossRef] [PubMed]
- Dalton, A.K.; Ako-Adjei, D.; Murray, P.S.; Murray, D.; Vogt, V.M. Electrostatic Interactions Drive Membrane Association of the Human Immunodeficiency Virus Type 1 Gag MA Domain. J. Virol. 2007, 81, 6434–6445. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Ablan, S.D.; Lockett, S.J.; Nagashima, K.; Freed, E.O. Phosphatidylinositol (4,5) Bisphosphate Regulates HIV-1 Gag Targeting to the Plasma Membrane. Proc. Natl. Acad. Sci. USA 2004, 101, 14889–14894. [Google Scholar] [CrossRef]
- Chukkapalli, V.; Hogue, I.B.; Boyko, V.; Hu, W.-S.; Ono, A. Interaction between the Human Immunodeficiency Virus Type 1 Gag Matrix Domain and Phosphatidylinositol-(4,5)-Bisphosphate Is Essential for Efficient Gag Membrane Binding. J. Virol. 2008, 82, 2405–2417. [Google Scholar] [CrossRef]
- Chukkapalli, V.; Oh, S.J.; Ono, A. Opposing Mechanisms Involving RNA and Lipids Regulate HIV-1 Gag Membrane Binding through the Highly Basic Region of the Matrix Domain. Proc. Natl. Acad. Sci. USA 2010, 107, 1600–1605. [Google Scholar] [CrossRef]
- Chukkapalli, V.; Inlora, J.; Todd, G.C.; Ono, A. Evidence in Support of RNA-Mediated Inhibition of Phosphatidylserine-Dependent HIV-1 Gag Membrane Binding in Cells. J. Virol. 2013, 87, 7155–7159. [Google Scholar] [CrossRef]
- Kutluay, S.B.; Zang, T.; Blanco-Melo, D.; Powell, C.; Jannain, D.; Errando, M.; Bieniasz, P.D. Global Changes in the RNA Binding Specificity of HIV-1 Gag Regulate Virion Genesis. Cell 2014, 159, 1096–1109. [Google Scholar] [CrossRef] [Green Version]
- Gaines, C.R.; Tkacik, E.; Rivera-Oven, A.; Somani, P.; Achimovich, A.; Alabi, T.; Zhu, A.; Getachew, N.; Yang, A.L.; McDonough, M.; et al. HIV-1 Matrix Protein Interactions with TRNA: Implications for Membrane Targeting. J. Mol. Biol. 2018, 430, 2113–2127. [Google Scholar] [CrossRef]
- Todd, G.C.; Duchon, A.; Inlora, J.; Olson, E.D.; Musier-Forsyth, K.; Ono, A. Inhibition of HIV-1 Gag–Membrane Interactions by Specific RNAs. RNA 2017, 23, 395–405. [Google Scholar] [CrossRef]
- Sumner, C.; Kotani, O.; Liu, S.; Musier-Forsyth, K.; Sato, H.; Ono, A. Molecular Determinants in TRNA D-Arm Required for Inhibition of HIV-1 Gag Membrane Binding. J. Mol. Biol. 2022, 434, 167390. [Google Scholar] [CrossRef]
- Bou-Nader, C.; Muecksch, F.; Brown, J.B.; Gordon, J.M.; York, A.; Peng, C.; Ghirlando, R.; Summers, M.F.; Bieniasz, P.D.; Zhang, J. HIV-1 Matrix-TRNA Complex Structure Reveals Basis for Host Control of Gag Localization. Cell Host Microbe 2021, 29, 1421–1436.e7. [Google Scholar] [CrossRef]
- Mirande, M. The Aminoacyl-TRNA Synthetase Complex BT. In Macromolecular Protein Complexes: Structure and Function; Harris, J.R., Marles-Wright, J., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 505–522. ISBN 978-3-319-46503-6. [Google Scholar]
- Ray, P.S.; Arif, A.; Fox, P.L. Macromolecular Complexes as Depots for Releasable Regulatory Proteins. Trends Biochem. Sci. 2007, 32, 158–164. [Google Scholar] [CrossRef]
- Cui, H.; Kapur, M.; Diedrich, J.K.; Yates, J.R.; Ackerman, S.L.; Schimmel, P. Regulation of Ex-Translational Activities Is the Primary Function of the Multi-TRNA Synthetase Complex. Nucleic Acids Res. 2021, 49, 3603–3616. [Google Scholar] [CrossRef]
- Sampath, P.; Mazumder, B.; Seshadri, V.; Gerber, C.A.; Chavatte, L.; Kinter, M.; Ting, S.M.; Dignam, J.D.; Kim, S.; Driscoll, D.M.; et al. Noncanonical Function of Glutamyl-Prolyl-TRNA Synthetase: Gene-Specific Silencing of Translation. Cell 2004, 119, 195–208. [Google Scholar] [CrossRef]
- Lee, E.-Y.; Lee, H.-C.; Kim, H.-K.; Jang, S.Y.; Park, S.-J.; Kim, Y.-H.; Kim, J.H.; Hwang, J.; Kim, J.-H.; Kim, T.-H.; et al. Infection-Specific Phosphorylation of Glutamyl-Prolyl TRNA Synthetase Induces Antiviral Immunity. Nat. Immunol. 2016, 17, 1252–1262. [Google Scholar] [CrossRef]
- Jia, J.; Arif, A.; Ray, P.S.; Fox, P.L. WHEP Domains Direct Noncanonical Function of Glutamyl-Prolyl TRNA Synthetase in Translational Control of Gene Expression. Mol. Cell 2008, 29, 679–690. [Google Scholar] [CrossRef]
- Arif, A.; Jia, J.; Mukhopadhyay, R.; Willard, B.; Kinter, M.; Fox, P.L. Two-Site Phosphorylation of EPRS Coordinates Multimodal Regulation of Noncanonical Translational Control Activity. Mol. Cell 2009, 35, 164–180. [Google Scholar] [CrossRef] [Green Version]
- Arif, A.; Jia, J.; Moodt, R.A.; DiCorleto, P.E.; Fox, P.L. Phosphorylation of Glutamyl-Prolyl TRNA Synthetase by Cyclin-Dependent Kinase 5 Dictates Transcript-Selective Translational Control. Proc. Natl. Acad. Sci. USA 2011, 108, 1415–1420. [Google Scholar] [CrossRef] [PubMed]
- Sampath, P.; Mazumder, B.; Seshadri, V.; Fox, P.L. Transcript-Selective Translational Silencing by Gamma Interferon Is Directed by a Novel Structural Element in the Ceruloplasmin MRNA 3′ Untranslated Region. Mol. Cell. Biol. 2003, 23, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.; March, P.E.; Lee, R.; Tillett, D. Site-Directed, Ligase-Independent Mutagenesis (SLIM): A Single-Tube Methodology Approaching 100% Efficiency in 4 H. Nucleic Acids Res. 2004, 32, e174. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.; Tillett, D.; Dawes, I.W.; March, P.E. Site-Directed, Ligase-Independent Mutagenesis (SLIM) for Highly Efficient Mutagenesis of Plasmids Greater than 8kb. J. Microbiol. Methods 2008, 73, 195–198. [Google Scholar] [CrossRef]
- Zhu, Y.; Luo, S.; Sabo, Y.; Wang, C.; Tong, L.; Goff, S.P. Heme Oxygenase 2 Binds Myristate to Regulate Retrovirus Assembly and TLR4 Signaling. Cell Host Microbe 2017, 21, 220–230. [Google Scholar] [CrossRef]
- Milligan, J.F.; Uhlenbeck, O.C. Synthesis of Small RNAs Using T7 RNA Polymerase. RNA Process. Part A Gen. Methods 1989, 180, 51–62. [Google Scholar]
- Rye-McCurdy, T.; Rouzina, I.; Musier-Forsyth, K. Fluorescence Anisotropy-Based Salt-Titration Approach to Characterize Protein–Nucleic Acid Interactions. In RNA Remodeling Proteins: Methods and Protocols; Boudvillain, M., Ed.; Humana Press: New York, NY, USA, 2015; pp. 385–402. ISBN 978-1-4939-2214-7. [Google Scholar]
- Longo, P.A.; Kavran, J.M.; Kim, M.-S.; Leahy, D.J. Transient Mammalian Cell Transfection with Polyethylenimine (PEI). Methods Enzymol. 2013, 529, 227–240. [Google Scholar] [CrossRef]
- Studier, F.W. Protein Production by Auto-Induction in High-Density Shaking Cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef]
- Wu, L.; Martin, T.D.; Vazeux, R.; Unutmaz, D.; KewalRamani, V.N. Functional Evaluation of DC-SIGN Monoclonal Antibodies Reveals DC-SIGN Interactions with ICAM-3 Do Not Promote Human Immunodeficiency Virus Type 1 Transmission. J. Virol. 2002, 76, 5905–5914. [Google Scholar] [CrossRef]
- Saad, J.S.; Miller, J.; Tai, J.; Kim, A.; Ghanam, R.H.; Summers, M.F. Structural Basis for Targeting HIV-1 Gag Proteins to the Plasma Membrane for Virus Assembly. Proc. Natl. Acad. Sci. USA 2006, 103, 11364–11369. [Google Scholar] [CrossRef]
- Golumbeanu, M.; Desfarges, S.; Hernandez, C.; Quadroni, M.; Rato, S.; Mohammadi, P.; Telenti, A.; Beerenwinkel, N.; Ciuffi, A. Proteo-Transcriptomic Dynamics of Cellular Response to HIV-1 Infection. Sci. Rep. 2019, 9, 213. [Google Scholar] [CrossRef] [Green Version]
- Thornhill, D.; Murakami, T.; Ono, A. Rendezvous at Plasma Membrane: Cellular Lipids and TRNA Set up Sites of HIV-1 Particle Assembly and Incorporation of Host Transmembrane Proteins. Viruses 2020, 12, 842. [Google Scholar] [CrossRef]
- Giegé, R.; Sissler, M.; Florentz, C. Universal Rules and Idiosyncratic Features in TRNA Identity. Nucleic Acids Res. 1998, 26, 5017–5035. [Google Scholar] [CrossRef]
- Beuning, P.J.; Musier-Forsyth, K. Transfer RNA Recognition by Aminoacyl-TRNA Synthetases. Biopolymers 1999, 52, 1–28. [Google Scholar] [CrossRef]
- Cahuzac, B.; Berthonneau, E.; Birlirakis, N.; Guittet, E.; Mirande, M. A Recurrent RNA-Binding Domain Is Appended to Eukaryotic Aminoacyl-TRNA Synthetases. EMBO J. 2000, 19, 445–452. [Google Scholar] [CrossRef]
- Rho, S.B.; Lee, K.H.; Kim, J.W.; Shiba, K.; Jo, Y.J.; Kim, S. Interaction between Human TRNA Synthetases Involves Repeated Sequence Elements. Proc. Natl. Acad. Sci. USA 1996, 93, 10128–10133. [Google Scholar] [CrossRef]
- Stapulionis, R.; Deutscher, M.P. A Channeled TRNA Cycle during Mammalian Protein Synthesis. Proc. Natl. Acad. Sci. USA 1995, 92, 7158–7161. [Google Scholar] [CrossRef]
- El Mekdad, H.; Boutant, E.; Karnib, H.; Biedma, M.E.; Sharma, K.K.; Malytska, I.; Laumond, G.; Roy, M.; Réal, E.; Paillart, J.-C.; et al. Characterization of the Interaction between the HIV-1 Gag Structural Polyprotein and the Cellular Ribosomal Protein L7 and Its Implication in Viral Nucleic Acid Remodeling. Retrovirology 2016, 13, 54. [Google Scholar] [CrossRef]
- Deng, Y.; Hammond, J.A.; Pauszek, R.; Ozog, S.; Chai, I.; Rabuck-Gibbons, J.; Lamichhane, R.; Henderson, S.C.; Millar, D.P.; Torbett, B.E.; et al. Discrimination between Functional and Non-Functional Cellular Gag Complexes Involved in HIV-1 Assembly. J. Mol. Biol. 2021, 433, 166842. [Google Scholar] [CrossRef]
- Barros, M.; Heinrich, F.; Datta, S.A.K.; Rein, A.; Karageorgos, I.; Nanda, H.; Lösche, M. Membrane Binding of HIV-1 Matrix Protein: Dependence on Bilayer Composition and Protein Lipidation. J. Virol. 2016, 90, 4544–4555. [Google Scholar] [CrossRef]
- Murphy, R.E.; Samal, A.B.; Vlach, J.; Mas, V.; Prevelige, P.E.; Saad, J.S. Structural and Biophysical Characterizations of HIV-1 Matrix Trimer Binding to Lipid Nanodiscs Shed Light on Virus Assembly. J. Biol. Chem. 2019, 294, 18600–18612. [Google Scholar] [CrossRef] [PubMed]
- Carlson, L.-A.; Bai, Y.; Keane, S.C.; Doudna, J.A.; Hurley, J.H. Reconstitution of Selective HIV-1 RNA Packaging in Vitro by Membrane-Bound Gag Assemblies. eLife 2016, 5, e14663. [Google Scholar] [CrossRef] [PubMed]
- Arif, A.; Yao, P.; Terenzi, F.; Jia, J.; Ray, P.S.; Fox, P.L. The GAIT Translational Control System. WIREs RNA 2018, 9, e1441. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Fischer, B.; Frese, C.K.; Horos, R.; Alleaume, A.-M.; Foehr, S.; Curk, T.; Krijgsveld, J.; Hentze, M.W. Comprehensive Identification of RNA-Binding Domains in Human Cells. Mol. Cell 2016, 63, 696–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | RNA | Kd (nM) | Method |
|---|---|---|---|
| Linker | Human tRNALys3 | 589 ± 152 | Fluorescence quenching |
| Bovine tRNATrp | 511 ± 119 | ||
| Human tRNAGlu | 886 ± 203 | ||
| Human tRNAPro | 449 ± 196 | ||
| R1R2R3 | 43 ± 35 | Fluorescence anisotropy | |
| R1R2 | Human tRNALys3 | 209 ± 58 | |
| R2R3 | >10 µM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, D.; Zhu, Y.; Schubert, H.L.; Goff, S.P.; Musier-Forsyth, K. HIV-1 Gag Binds the Multi-Aminoacyl-tRNA Synthetase Complex via the EPRS Subunit. Viruses 2023, 15, 474. https://doi.org/10.3390/v15020474
Jin D, Zhu Y, Schubert HL, Goff SP, Musier-Forsyth K. HIV-1 Gag Binds the Multi-Aminoacyl-tRNA Synthetase Complex via the EPRS Subunit. Viruses. 2023; 15(2):474. https://doi.org/10.3390/v15020474
Chicago/Turabian StyleJin, Danni, Yiping Zhu, Heidi L. Schubert, Stephen P. Goff, and Karin Musier-Forsyth. 2023. "HIV-1 Gag Binds the Multi-Aminoacyl-tRNA Synthetase Complex via the EPRS Subunit" Viruses 15, no. 2: 474. https://doi.org/10.3390/v15020474