
Coronaviruses Are Abundant and Genetically Diverse in West and Central African Bats, including Viruses Closely Related to Human Coronaviruses
 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
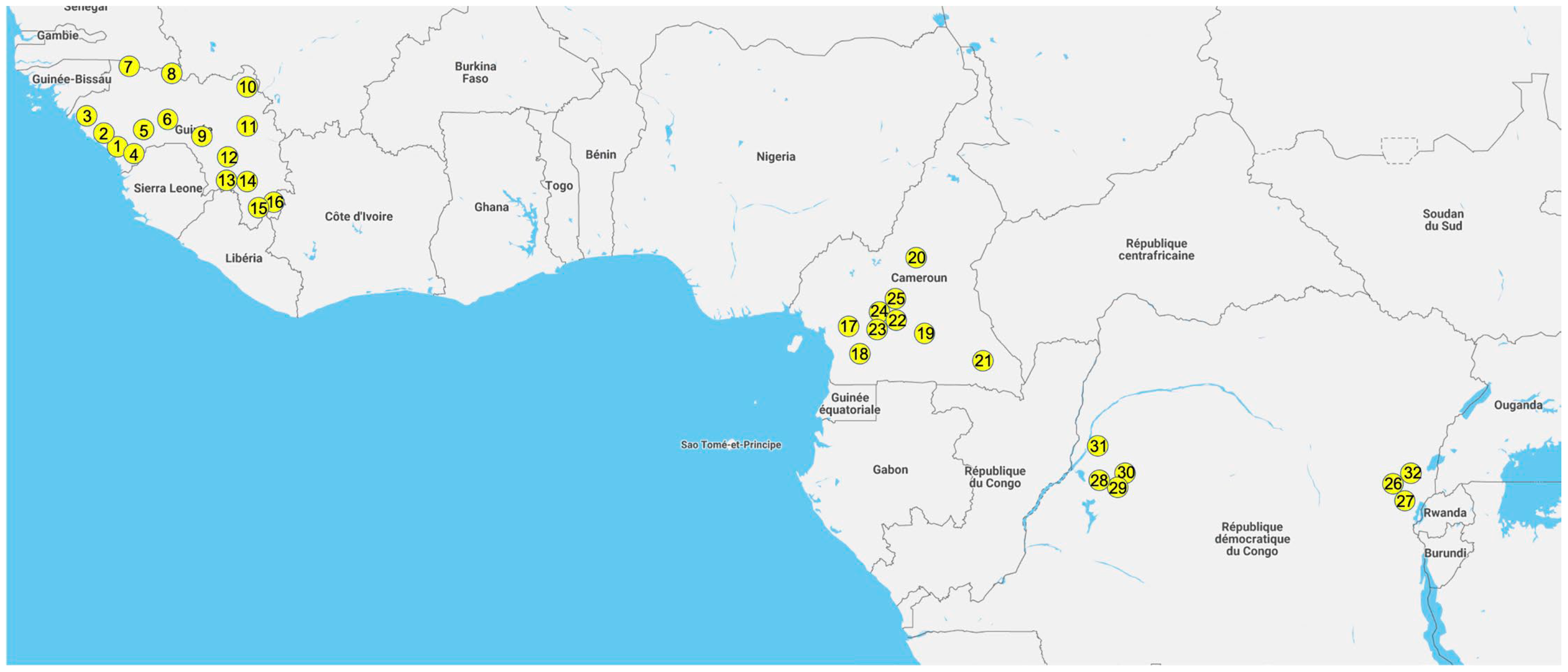
2.1. Study Sites and Sample Collection
2.2. Nucleic Acid Extraction and Screening for the Presence of Coronavirus RNA
2.3. Phylogenetic Analyses
2.4. Molecular Confirmation of Bat Species
2.5. Statistical Analyses
3. Results
3.1. Bat Samples
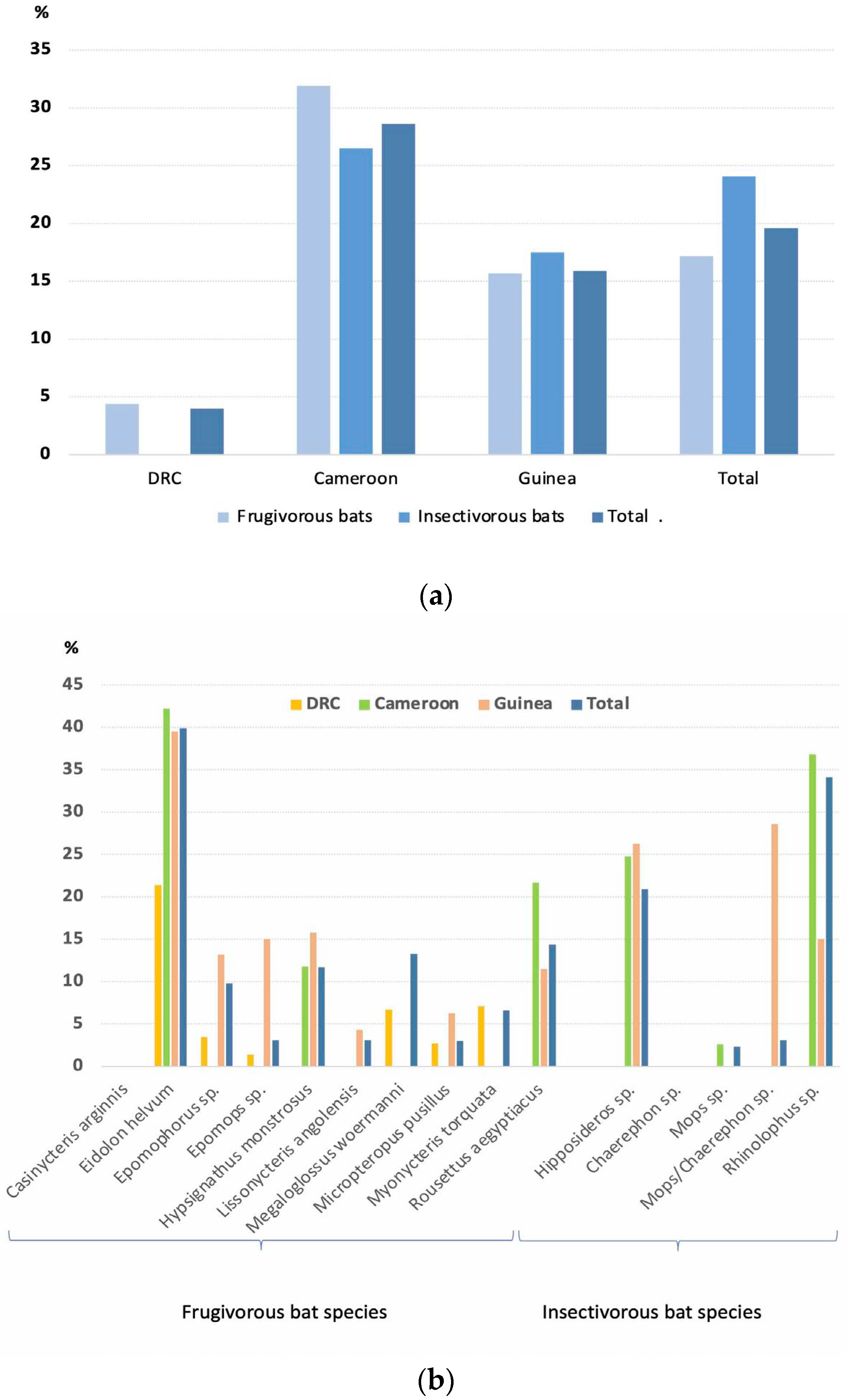
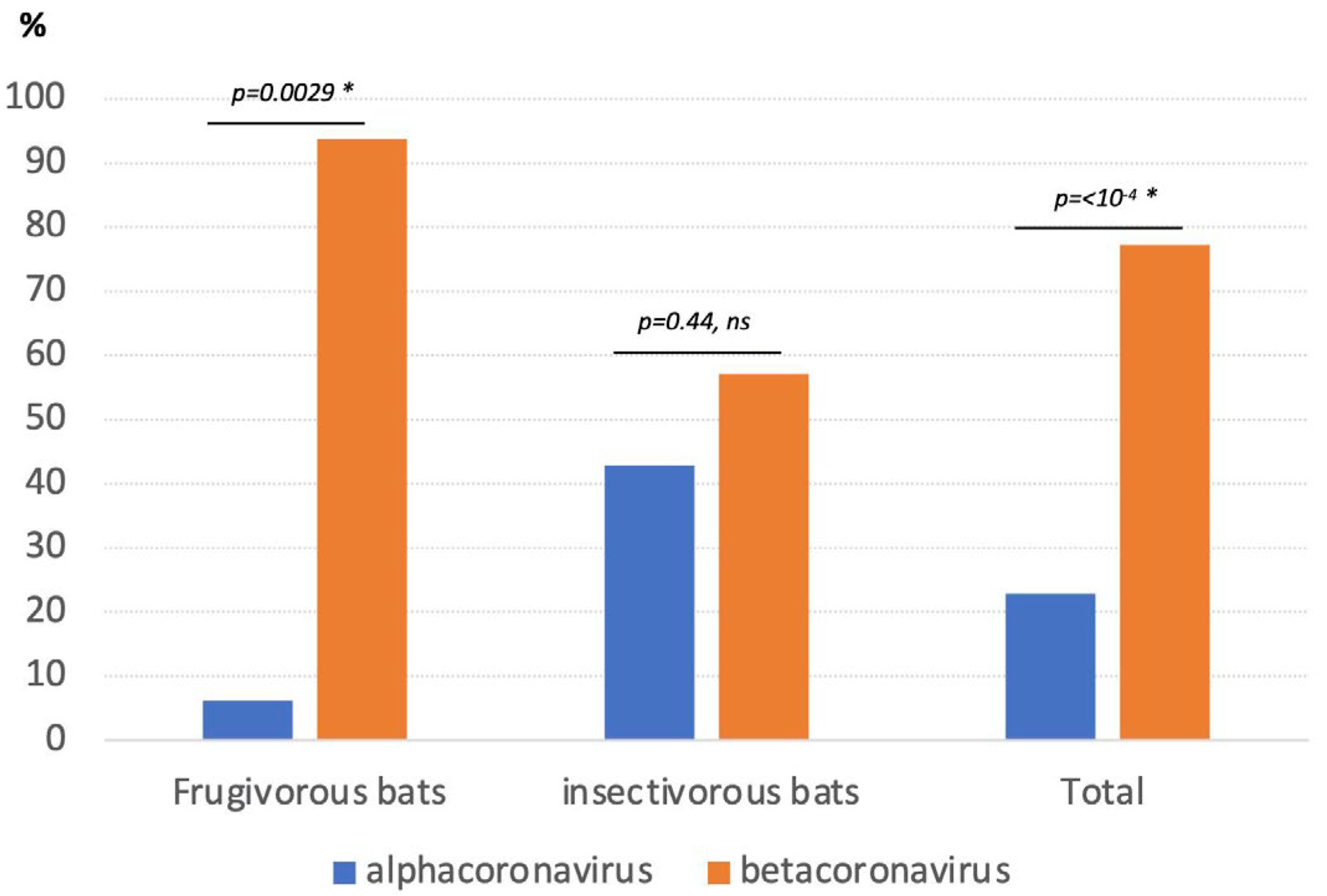
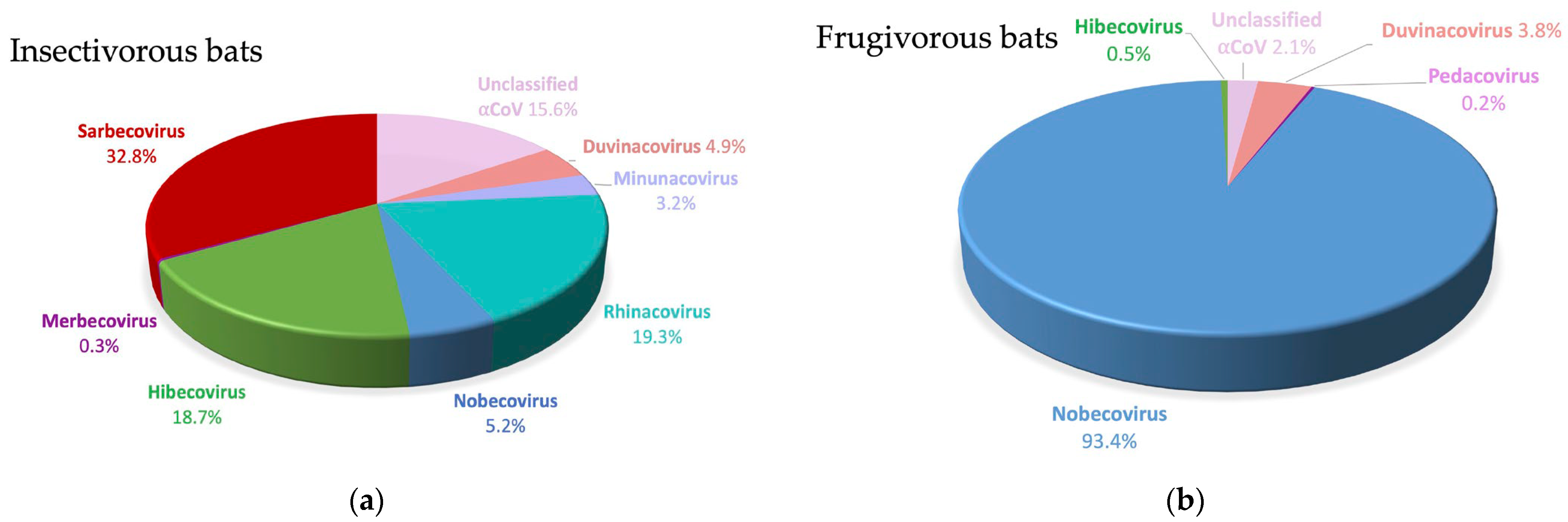
3.2. Detection and Genetic Diversity of Coronaviruses
3.3. Factors Associated with Detection of Coronaviruses in Bats
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 19 October 2022).
- Chisale, M.R.O.; Ramazanu, S.; Mwale, S.E.; Kumwenda, P.; Chipeta, M.; Kaminga, A.C.; Nkhata, O.; Nyambalo, B.; Chavura, E.; Mbakaya, B.C. Seroprevalence of anti-SARS-CoV-2 antibodies in Africa: A systematic review and meta-analysis. Rev. Med. Virol. 2022, 32, e2271. [Google Scholar] [CrossRef]
- Ndongo, F.A.; Guichet, E.; Mimbé, E.D.; Ndié, J.; Pelloquin, R.; Varloteaux, M.; Esemu, L.; Mpoudi-Etame, M.; Lamare, N.; Edoul, G.; et al. Rapid increase of community SARS-CoV-2 seroprevalence during second wave of COVID-19; Yaoundé; Cameroon. Emerg. Infect. Dis. 2022, 28, 1233–1236. [Google Scholar] [CrossRef]
- Nkuba, A.N.; Makiala, S.M.; Guichet, E.; Tshiminyi, P.M.; Bazitama, Y.M.; Yambayamba, M.K.; Kazenza, B.M.; Kabeya, T.M.; Matungulu, E.B.; Baketana, L.K.; et al. High prevalence of anti-Severe Acute Respiratory Syndrome Coronavirus 2 (Anti-SARS-CoV-2) antibodies after the first wave of Coronavirus Disease 2019 (COVID-19) in Kinshasa; Democratic Republic of the Congo: Results of a cross-sectional household-based survey. Clin. Infect. Dis. 2022, 74, 882–890. [Google Scholar] [CrossRef]
- Soumah, A.A.; Diallo, M.S.K.; Guichet, E.; Maxmlman, D.; Thaurignac, G.; Keita, A.K.; Bouillin, J.; Diallo, H.; Pelloquin, R.; Ayouba, A.; et al. High and rapid increase in seroprevalence for SARS-CoV-2 in Conakry; Guinea: Results from 3 successive cross-sectional surveys (ANRS COV16-ARIACOV). Open Forum Infect. Dis. 2022, 9, ofac152. [Google Scholar] [CrossRef]
- Worobey, M.; Levy, J.I.; Serrano, L.M.; Crits-Christoph, A.; Pekar, J.E.; Goldstein, S.A.; Rasmussen, A.L.; Kraemer, M.U.G.; Newman, C.; Koopmans, M.P.G.; et al. The Huanan seafood wholesale market in Wuhan was the early epicenter of the COVID-19 pandemic. Science 2022, 377, 951–959. [Google Scholar] [CrossRef]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology; Genetic Recombination; and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.W.; Yuan, S.; Yuen, K.S.; Fung, S.Y.; Chan, C.P.; Jin, D.Y. Zoonotic origins of human coronaviruses. Int. J. Biol. Sci. 2020, 16, 1686–1697. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Guan, Y.J.; et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Tsoi, H.W.; Wong, B.H.; Wong, S.S.; Leung, S.Y.; Chan, K.H.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Luo, C.M.; Wang, N.; Yang, X.L.; Liu, H.Z.; Zhang, W.; Li, B.; Hu, B.; Peng, C.; Geng, Q.B.; Zhu, G.J.; et al. Discovery of novel bat coronaviruses in South China that use the same receptor as Middle East Respiratory Syndrome Coronavirus. J. Virol. 2018, 92, e00116-18. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.; Lau, S.K.; To, K.K.; Cheng, V.C.; Woo, P.C.; Yuen, K.Y. Middle East respiratory syndrome coronavirus: Another zoonotic betacoronavirus causing SARS-like disease. Clin. Microbiol. Rev. 2015, 28, 465–522. [Google Scholar] [CrossRef] [Green Version]
- Corman, V.M.; Ithete, N.L.; Richards, L.R.; Schoeman, M.C.; Preiser, W.; Drosten, C.; Drexler, J.F. Rooting the phylogenetic tree of Middle East Respiratory Syndrome Coronavirus by characterization of a conspecific virus from an African bat. J. Virol. 2014, 88, 11297–11303. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Lam, T.T.; Jia, N.; Zhang, Y.W.; Shum, M.H.; Jiang, J.F.; Zhu, H.C.; Tong, Y.G.; Shi, Y.X.; Ni, X.B.; Liao, Y.S.; et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020, 583, 282–285. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Pekar, J.E.; Magee, A.; Parker, E.; Moshiri, N.; Izhikevich, K.; Havens, J.L.; Gangavarapu, K.; Malpica Serrano, L.M.; Crits-Christoph, A.; Matteson, N.L.; et al. The molecular epidemiology of multiple zoonotic origins of SARS-CoV-2. Science 2022, 377, 960–966. [Google Scholar] [CrossRef]
- Anthony, S.J.; Johnson, C.K.; Greig, D.J.; Kramer, S.; Che, X.; Wells, H.; Hicks, A.L.; Joly, D.O.; Wolfe, N.D.; Daszak, P.; et al. Global patterns in coronavirus diversity. Virus Evol. 2017, 3, vex012. [Google Scholar] [CrossRef] [Green Version]
- Burgin, C.J.; Colella, J.P.; Kahn, P.L.; Upham, N.S. How many species of mammals are there? J. Mammal. 2018, 99, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Frick, W.F.; Kingston, T.; Flanders, J. A review of the major threats and challenges to global bat conservation. Ann. N. Y. Acad. Sci. 2019, 1469, 5–25. [Google Scholar] [CrossRef]
- Baudel, H.; De Nys, H.; Mpoudi Ngole, E.; Peeters, M.; Desclaux, A. Understanding Ebola virus and other zoonotic transmission risks through human-bat contacts: Exploratory study on knowledge; attitudes and practices in Southern Cameroon. Zoonoses Public Health 2019, 66, 288–295. [Google Scholar] [CrossRef]
- Mickleburgh, S.; Waylen, K.; Racey, P. Bats as bushmeat: A global review. Oryx 2009, 43, 217–234. [Google Scholar] [CrossRef] [Green Version]
- Kamins, A.O.; Restif, O.; Ntiamoa-Baidu, Y.; Suu-Ire, R.; Hayman, D.T.; Cunningham, A.A.; Wood, J.L.; Rowcliffe, J.M. Uncovering the fruit bat bushmeat commodity chain and the true extent of fruit bat hunting in Ghana; West Africa. Biol. Conserv. 2011, 144, 3000–3008. [Google Scholar] [CrossRef] [Green Version]
- Anti, P.; Owusu, M.; Agbenyega, O.; Annan, A.; Badu, E.K.; Nkrumah, E.E.; Tschapka, M.; Oppong, S.; Adu-Sarkodie, Y.; Drosten, C. Human-bat interactions in rural West Africa. Emerg. Infect. Dis. 2015, 21, 1418–1421. [Google Scholar] [CrossRef] [Green Version]
- Geldenhuys, M.; Mortlock, M.; Epstein, J.H.; Pawęska, J.T.; Weyer, J.; Markotter, W. Overview of bat and wildlife coronavirus surveillance in Africa: A framework for global investigations. Viruses 2021, 13, 936. [Google Scholar] [CrossRef]
- De Nys, H.M.; Kingebeni, P.M.; Keita, A.K.; Butel, C.; Thaurignac, G.; Villabona-Arenas, C.J.; Lemarcis, T.; Geraerts, M.; Vidal, N.; Esteban, A.; et al. Survey of Ebola viruses in frugivorous and insectivorous bats in Guinea; Cameroon; and the Democratic Republic of the Congo; 2015-2017. Emerg. Infect. Dis. 2018, 24, 2228–2240. [Google Scholar] [CrossRef]
- Chu, D.K.; Leung, C.Y.; Gilbert, M.; Joyner, P.H.; Ng, E.M.; Tse, T.M.; Guan, Y.; Peiris, J.S.; Poon, L.L. Avian coronavirus in wild aquatic birds. J. Virol. 2011, 85, 12815–12820. [Google Scholar] [CrossRef] [Green Version]
- Lacroix, A.; Vidal, N.; Keita, A.K.; Thaurignac, G.; Esteban, A.; De Nys, H.; Diallo, R.; Toure, A.; Goumou, S.; Soumah, A.K.; et al. Wide diversity of coronaviruses in frugivorous and insectivorous bat species: A pilot study in Guinea; West Africa. Viruses 2020, 12, 855. [Google Scholar] [CrossRef]
- Wilkinson, D.A.; Joffrin, L.; Lebarbenchon, C.; Mavingui, P. Analysis of partial sequences of the RNA-dependent RNA polymerase gene as a tool for genus and subgenus classification of coronaviruses. J. Gen. Virol. 2020, 101, 1261–1269. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Hahn, M.W.; Lanfear, R. New methods to calculate concordance factors for phylogenomic datasets. Mol. Biol. Evol. 2020, 37, 2727–2733. [Google Scholar] [CrossRef] [PubMed]
- Raulino, R.; Thaurignac, G.; Keita, A.K.; Esteban, A.; Goumou, S.; Diallo, R.; Ndimbo-Kumugo, S.P.; Ndong Bass, I.; Mbala Kingebeni, P.; Toure, A.; et al. Seroprevalence of IgG antibodies against multiple arboviruses in bats from Cameroon; Guinea; and the Democratic Republic of Congo. Vector Borne Zoonotic Dis. 2022, 22, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, A.; Mbala Kingebeni, P.; Ndimbo Kumugo, S.P.; Lempu, G.; Butel, C.; Serrano, L.; Vidal, N.; Thaurignac, G.; Esteban, A.; Mukadi Bamuleka, D.; et al. Investigating the circulation of Ebola viruses in bats during the Ebola virus disease outbreaks in the Equateur and North Kivu provinces of the Democratic Republic of Congo from 2018. Pathogens 2021, 10, 557. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.M.; Kocher, T.D.; Wilson, A.C. Evolution of the Cytochrome b Gene of Mammals. J. Mol. Evol. 1991, 32, 128–144. [Google Scholar] [CrossRef] [PubMed]
- van der Kuyl, A.C.; Kuiken, C.L.; Dekker, J.T.; Goudsmit, J. Phylogeny of African monkeys based upon mitochondrial 12S rRNA sequences. J. Mol. Evol. 1995, 40, 173–180. [Google Scholar] [CrossRef]
- Nesi, N.; Nakouné, E.; Cruaud, C.; Hassanin, A. DNA barcoding of African fruit bats (Mammalia; Pteropodidae). The mitochondrial genome does not provide a reliable discrimination between Epomophorus gambianus and Micropteropus pusillus. Comptes R. Biol. 2011, 334, 544–554. [Google Scholar] [CrossRef]
- Ntumvi, N.F.; Ndze, V.N.; Gillis, A.; Le Doux Diffo, J.; Tamoufe, U.; Takuo, J.M.; Mouiche, M.M.M.; Nwobegahay, J.; LeBreton, M.; Rimoin, A.W.; et al. Wildlife in Cameroon harbor diverse coronaviruses; including many closely related to human coronavirus 229E. Virus Evol. 2022, 8, veab110. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Wang, M.; Lau, S.K.; Xu, H.; Poon, R.W.; Guo, R.; Wong, B.H.; Gao, K.; Tsoi, H.W.; Huang, Y.; et al. Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J. Virol. 2007, 81, 1574–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, H.L.; Letko, M.; Lasso, G.; Ssebide, B.; Nziza, J.; Byarugaba, D.K.; Navarrete-Macias, I.; Liang, E.; Cranfield, M.; Han, B.A.; et al. The evolutionary history of ACE2 usage within the coronavirus subgenus Sarbecovirus. Virus Evol. 2021, 7, veab007. [Google Scholar] [CrossRef]
- Kumakamba, C.; Niama, F.R.; Muyembe, F.; Mombouli, J.V.; Kingebeni, P.M.; Nina, R.A.; Lukusa, I.N.; Bounga, G.; N’Kawa, F.; Nkoua, C.G.; et al. Coronavirus surveillance in wildlife from two Congo basin countries detects RNA of multiple species circulating in bats and rodents. PLoS ONE 2021, 16, e0236971. [Google Scholar] [CrossRef]
- Hu, B.; Zeng, L.P.; Yang, X.L.; Ge, X.Y.; Zhang, W.; Li, B.; Xie, J.Z.; Shen, X.R.; Zhang, Y.Z.; Wang, N.; et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13, e1006698. [Google Scholar] [CrossRef]
- Yu, P.; Hu, B.; Shi, Z.L.; Cui, J. Geographical structure of bat SARS-related coronaviruses. Infect. Genet. Evol. 2019, 69, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.C.P.; Li, X.; Lau, S.K.P.; Woo, P.C.Y. Global epidemiology of bat coronaviruses. Viruses 2019, 11, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ar Gouilh, M.; Puechmaille, S.J.; Diancourt, L.; Vandenbogaert, M.; Serra-Cobo, J.; Lopez Roïg, M.; Brown, P.; Moutou, F.; Caro, V.; Vabret, A.; et al. SARS-CoV related Betacoronavirus and diverse Alphacoronavirus members found in western old-world. Virology 2018, 517, 88–97. [Google Scholar] [CrossRef]
- Lecis, R.; Mucedda, M.; Pidinchedda, E.; Pittau, M.; Alberti, A. Molecular identification of Betacoronavirus in bats from Sardinia (Italy): First detection and phylogeny. Virus Genes 2019, 55, 60–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drexler, J.F.; Gloza-Rausch, F.; Glende, J.; Corman, V.M.; Muth, D.; Goettsche, M.; Seebens, A.; Niedrig, M.; Pfefferle, S.; Yordanov, S.; et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol. 2010, 84, 11336–11349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rihtaric, D.; Hostnik, P.; Steyer, A.; Grom, J.; Toplak, I. Identification of SARS-like coronaviruses in horseshoe bats (Rhinolophus hipposideros) in Slovenia. Arch. Virol. 2010, 155, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Lelli, D.; Papetti, A.; Sabelli, C.; Rosti, E.; Moreno, A.; Boniotti, M.B. Detection of coronaviruses in bats of various species in Italy. Viruses 2013, 5, 2679–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markotter, W.; Geldenhuys, M.; Jansen van Vuren, P.; Kemp, A.; Mortlock, M.; Mudakikwa, A.; Nel, L.; Nziza, J.; Paweska, J.; Weyer, J. Paramyxo- and coronaviruses in Rwandan bats. Trop. Med. Infect. Dis. 2019, 4, 99. [Google Scholar] [CrossRef] [Green Version]
- Chidoti, V.; De Nys, H.; Pinarello, V.; Mashura, G.; Missé, D.; Guerrini, L.; Pfukenyi, D.; Cappelle, J.; Chiweshe, N.; Ayouba, A.; et al. Longitudinal survey of coronavirus circulation and diversity in insectivorous bat colonies in Zimbabwe. Viruses 2022, 14, 781. [Google Scholar] [CrossRef]
- Lau, S.K.; Li, K.S.; Tsang, A.K.; Shek, C.T.; Wang, M.; Choi, G.K.; Guo, R.; Wong, B.H.; Poon, R.W.; Lam, C.S.; et al. Recent transmission of a novel alphacoronavirus; bat coronavirus HKU10; from Leschenault’s rousettes to pomona leaf-nosed bats: First evidence of interspecies transmission of coronavirus between bats of different suborders. J. Virol. 2012, 86, 11906–11918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montecino-Latorre, D.; Goldstein, T.; Kelly, T.R.; Wolking, D.J.; Kindunda, A.; Kongo, G.; Bel-Nono, S.O.; Kazwala, R.R.; Suu-Ire, R.D.; Barker, C.M.; et al. Seasonal shedding of coronavirus by straw-colored fruit bats at urban roosts in Africa. PLoS ONE 2022, 17, e0274490. [Google Scholar] [CrossRef] [PubMed]
- Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Göttsche, M.; Panning, M.; Drexler, J.F.; Petersen, N.; Annan, A.; Grywna, K.; Müller, M.; et al. Detection and prevalence patterns of group I coronaviruses in bats; northern Germany. Emerg. Infect. Dis. 2008, 14, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Wegner, T.; Tateno, A.F.; Zerbinati, R.M.; Gloza-Rausch, F.; Seebens, A.; Müller, M.A.; Drosten, C. Amplification of emerging viruses in a bat colony. Emerg. Infect. Dis. 2011, 17, 449–456. [Google Scholar] [CrossRef]
- Annan, A.; Baldwin, H.J.; Corman, V.M.; Klose, S.M.; Owusu, M.; Nkrumah, E.E.; Badu, E.K.; Anti, P.; Agbenyega, O.; Meyer, B.; et al. Human betacoronavirus 2c EMC/2012-related viruses in bats; Ghana and Europe. Emerg. Infect. Dis. 2013, 19, 456–459. [Google Scholar] [CrossRef]
- Sabir, J.S.; Lam, T.T.; Ahmed, M.M.; Li, L.; Shen, Y.; Abo-Aba, S.E.; Qureshi, M.I.; Abu-Zeid, M.; Zhang, Y.; Khiyami, M.A.; et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 2016, 351, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Maganga, G.D.; Pinto, A.; Mombo, I.M.; Madjitobaye, M.; Mbeang Beyeme, A.M.; Boundenga, L.; Ar Gouilh, M.; N’Dilimabaka, N.; Drexler, J.F.; Drosten, C.; et al. Genetic diversity and ecology of coronaviruses hosted by cave-dwelling bats in Gabon. Sci. Rep. 2020, 10, 7314. [Google Scholar] [CrossRef]
- Kumar, N.; Kaushik, R.; Tennakoon, C.; Uversky, V.N.; Mishra, A.; Sood, R.; Srivastava, P.; Tripathi, M.; Zhang, K.Y.J.; Bhatia, S. Evolutionary signatures governing the codon usage bias in coronaviruses and their implications for viruses infecting various bat species. Viruses 2021, 13, 1847. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AlphaCoronaviruses | BetaCoronaviruses | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unclass. CoV | DecaCoV | DuvinaCoV | MinunaCoV | PedaCoV | RhinaCoV | NobeCoV Eidolon | NobeCoV Epomophorus | NobeCoV Lissonycteris | NobeCoV Rousettus (HKU9) | NobeCoV Rousettus | NobeCoV Megaloglossus | HibeCoV | MerbeCoV | SarbeCoV | |
| Eidolon helvum | 2 | - | - | - | - | - | 233 | 3 | - | - | - | - | - | - | - |
| Epomophorus sp. a | 2 | - | - | - | - | - | 2 | 50 | - | 1 | - | - | - | - | - |
| Epomops sp. b | - | - | - | - | - | - | - | 5 | - | - | - | - | - | - | - |
| Hypsignathus monstrosus | - | - | - | - | - | - | 6 | 2 | - | 1 | - | - | 1 | - | - |
| Lissonycteris angolensis | - | - | - | - | 1 | - | - | - | 1 | - | - | - | - | - | - |
| Megaloglossus woermanni | - | - | - | - | - | - | - | - | - | - | - | 2 | - | - | - |
| Micropteropus pusillus | - | - | - | - | - | - | - | 5 | - | - | - | - | - | - | - |
| Myonycteris torquata | - | - | - | - | - | - | - | - | 6 | - | - | - | - | - | - |
| Nanonycteris sp. c | - | - | - | - | - | - | - | 1 | - | - | - | - | - | - | - |
| Rousettus aegyptiacus | 5 | 16 | - | - | - | - | 3 | - | - | 64 | 8 | - | 1 | - | - |
| Total frugivorous | 9 | 16 | - | - | 1 | - | 244 | 66 | 7 | 66 | 8 | 2 | 2 | - | - |
| Coleura afra | - | - | 1 | - | - | - | - | - | - | - | - | - | - | - | - |
| Hipposideros sp. c | - | - | 16 | - | - | - | 1 | - | 1 | 1 | - | - | 34 | - | - |
| Miniopterus sp. c | - | - | - | 7 | - | - | - | - | - | - | - | - | - | - | - |
| Mops sp. c | - | - | - | 3 | - | - | 3 | - | - | - | - | - | - | - | - |
| Mops/Chaerephon sp. c | - | - | - | - | - | - | 2 | 2 | - | - | - | - | - | - | - |
| Nycteris sp. c | - | - | - | - | - | - | - | - | - | - | - | - | - | 1 | - |
| Rhinolophus sp. c | 54 | - | - | 1 | - | 67 | 5 | - | - | 2 | 1 | - | 31 | - | 114 |
| Total insectivorous | 54 | - | 17 | 11 | - | 67 | 11 | 2 | 1 | 3 | 1 | - | 65 | 1 | 114 |
| TOTAL | 63 | 16 | 17 | 11 | 1 | 67 | 255 | 68 | 8 | 69 | 9 | 2 | 67 | 1 | 114 |
| F | F | M | M | p−Values | |
|---|---|---|---|---|---|
| n+/N | % Pos | n+/N | % Pos | χ2 Test | |
| Frugivorous bats | |||||
| Family PTEROPODIDAE | |||||
| Casinycteris arginnis | 0/13 | 0.0 | 0/11 | 0.0 | na d |
| Eidolon helvum | 122/335 | 36.4 | 170/404 | 42.1 | 0.0197 * |
| Epomophorus sp. a | 43/447 | 9.6 | 27/286 | 9.4 | 0.96, ns e |
| Epomops sp. b | 3/155 | 1.9 | 5/102 | 4.9 | na |
| Hypsignathus monstrosus | 10/65 | 15.4 | 3/38 | 7.9 | na |
| Lissonycteris angolensis | 1/39 | 2.6 | 0/25 | 0.0 | na |
| Megaloglossus woermanni | 1/5 | 20.0 | 1/10 | 10.0 | na |
| Micropteropus pusillus | 2/80 | 2.5 | 3/85 | 3.5 | na |
| Myonycteris torquata | 2/38 | 5.3 | 4/53 | 7.5 | na |
| Nanonycteris sp. c | 1/1 | 100 | 0/2 | 0.0 | na |
| Rousettus aegyptiacus | 68/472 | 14.4 | 46/319 | 14.4 | 0.99, ns |
| Scotonycteris bergmansi | 0/1 | 0.0 | - | - | na |
| Subtotal frugivorous bats | 243/1651 | 14.7 | 259/1335 | 19.4 | 0.0007 * |
| Insectivorous bats | |||||
| Family EMBALLONURIDAE | |||||
| Coleura afra | 1/1 | 100 | - | - | na |
| Family HIPPOSIDERIDAE | |||||
| Hipposideros sp. c | 19/125 | 15.2 | 41/164 | 25.0 | 0.04 * |
| Family MINIOPTERIDAE | |||||
| Miniopterus sp. c | 2/2 | 100 | 6/6 | 100 | na |
| Family MOLOSSIDAE | |||||
| Chaerephon sp. c | 0/65 | 0.0 | 0/51 | 0.0 | na |
| Mops sp. | 4/131 | 3.1 | 2/118 | 1.7 | na |
| Mops/Chaerephon sp. c | 4/13 | 7.7 | 0/14 | 0.0 | na |
| Family NYCTERIDAE | |||||
| Nycteris sp. c | 0/2 | 0.0 | 1/3 | 33.3 | na |
| Family RHINOLOPHIDAE | |||||
| Rhinolophus sp. c | 174/485 | 35.9 | 130/411 | 31.6 | 0.2052, ns |
| Family VESPERTILIONIDAE | |||||
| Myotis sp. c | 0/2 | 0.0 | 0/2 | 0.0 | na |
| Scotophilus sp. c | 0/3 | 0.0 | - | - | na |
| Subtotal insectivorous bats | 204/829 | 24.6 | 180/769 | 23.4 | 0.57, ns |
| TOTAL | 447/2480 | 18.0 | 439/2104 | 20.9 | 0.0152 * |
| Adult | Adult | Juvenile | Juvenile | p−Values | |
|---|---|---|---|---|---|
| n+/N | % Pos | n+/N | % Pos | χ2 Test | |
| Frugivorous bats | |||||
| Family PTEROPODIDAE | |||||
| Casinycteris arginnis | 0/24 | 0.0 | na d | na | na |
| Eidolon helvum | 211/574 | 36.4 | 81/158 | 84.2 | <10−4 * |
| Epomophorus sp. a | 76/693 | 10.9 | 2/37 | 5.4 | na |
| Epomops sp. b | 8/250 | 3.2 | 0/6 | 0.0 | na |
| Hypsignathus monstrosus | 14/95 | 14.7 | 2/8 | 25 | na |
| Lissonycteris angolensis | 2/62 | 3.2 | 0/2 | 0.0 | na |
| Megaloglossus woermanni | 2/15 | 13.3 | na | na | na |
| Micropteropus pusillus | 5/160 | 3.1 | 0/2 | 0.0 | na |
| Myonycteris torquata | 6/86 | 6.7 | na | na | na |
| Nanonycteris sp. c | 1/3 | 33.3 | na | na | na |
| Rousettus aegyptiacus | 88/700 | 12.6 | 26/91 | 28.6 | 0.0001 * |
| Scotonycteris bergmansi | 0/1 | 0.0 | na | na | na |
| Subtotal frugivorous bats | 413/2667 | 15.5 | 111/304 | 36.5 | <10−4 * |
| Insectivorous bats | |||||
| Family EMBALLONURIDAE | |||||
| Coleura afra | 1/1 | 100 | na | na | na |
| Family HIPPOSIDERIDAE | |||||
| Hipposideros sp. c | 59/286 | 20.6 | 1/3 | 33.3 | na |
| Family MINIOPTERIDAE | |||||
| Miniopterus sp. c | 8/8 | 100 | na | na | na |
| Family MOLOSSIDAE | |||||
| Chaerephon sp. c | 0/89 | 0.0 | 0/25 | 0.0 | na |
| Mops sp. c | 4/181 | 2.2 | 0/31 | 0.0 | na |
| Mops/Chaerephon sp. c | 4/27 | 14.8 | na | na | na |
| Family NYCTERIDAE | |||||
| Nycteris sp. c | 1/5 | 20.0 | na | na | na |
| Family RHINOLOPHIDAE | |||||
| Rhinolophus sp. c | 283/868 | 32.6 | 21/28 | 75.0 | <10−4 * |
| Family VESPERTILIONIDAE | |||||
| Myotis sp. c | 0/4 | 0.0 | na | na | na |
| Scotophilus sp. c | 0/3 | 0.0 | na | na | na |
| Subtotal insectivorous bats | 360/1472 | 24.5 | 22/87 | 25.3 | 0.9627, ns e |
| Total | 773/4139 | 18.7 | 133/391 | 34.0 | <10−4 |
| Status | n+/N Tested | % Pos | p−Value |
|---|---|---|---|
| Adult females, gestation | 11/47 | 23.4% | 0.0882, ns |
| Adult females, no gestation | 57/269 | 21.2% | |
| Adult females, lactation | 10/85 | 11.8% | 0.0349 * |
| Adult females, no lactation | 93/410 | 22.7% | |
| Subadult females | 150/389 | 38.6% | 0.8833, ns |
| Juvenile females | 58/146 | 39.7% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meta Djomsi, D.; Lacroix, A.; Soumah, A.K.; Kinganda Lusamaki, E.; Mesdour, A.; Raulino, R.; Esteban, A.; Ndong Bass, I.; Mba Djonzo, F.A.; Goumou, S.; et al. Coronaviruses Are Abundant and Genetically Diverse in West and Central African Bats, including Viruses Closely Related to Human Coronaviruses. Viruses 2023, 15, 337. https://doi.org/10.3390/v15020337
Meta Djomsi D, Lacroix A, Soumah AK, Kinganda Lusamaki E, Mesdour A, Raulino R, Esteban A, Ndong Bass I, Mba Djonzo FA, Goumou S, et al. Coronaviruses Are Abundant and Genetically Diverse in West and Central African Bats, including Viruses Closely Related to Human Coronaviruses. Viruses. 2023; 15(2):337. https://doi.org/10.3390/v15020337
Chicago/Turabian StyleMeta Djomsi, Dowbiss, Audrey Lacroix, Abdoul Karim Soumah, Eddy Kinganda Lusamaki, Asma Mesdour, Raisa Raulino, Amandine Esteban, Innocent Ndong Bass, Flaubert Auguste Mba Djonzo, Souana Goumou, and et al. 2023. "Coronaviruses Are Abundant and Genetically Diverse in West and Central African Bats, including Viruses Closely Related to Human Coronaviruses" Viruses 15, no. 2: 337. https://doi.org/10.3390/v15020337