
Genetic Diversity and Expanded Host Range of J Paramyxovirus Detected in Wild Small Mammals in China

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Next-Generation Sequencing (NGS)
2.3. Reverse Transcription-PCR (RT-PCR) Sequencing for JPV
2.4. Phylogenetic Analysis
2.5. Statistical Analysis
2.6. Nucleotide Sequence Accession Numbers
3. Results
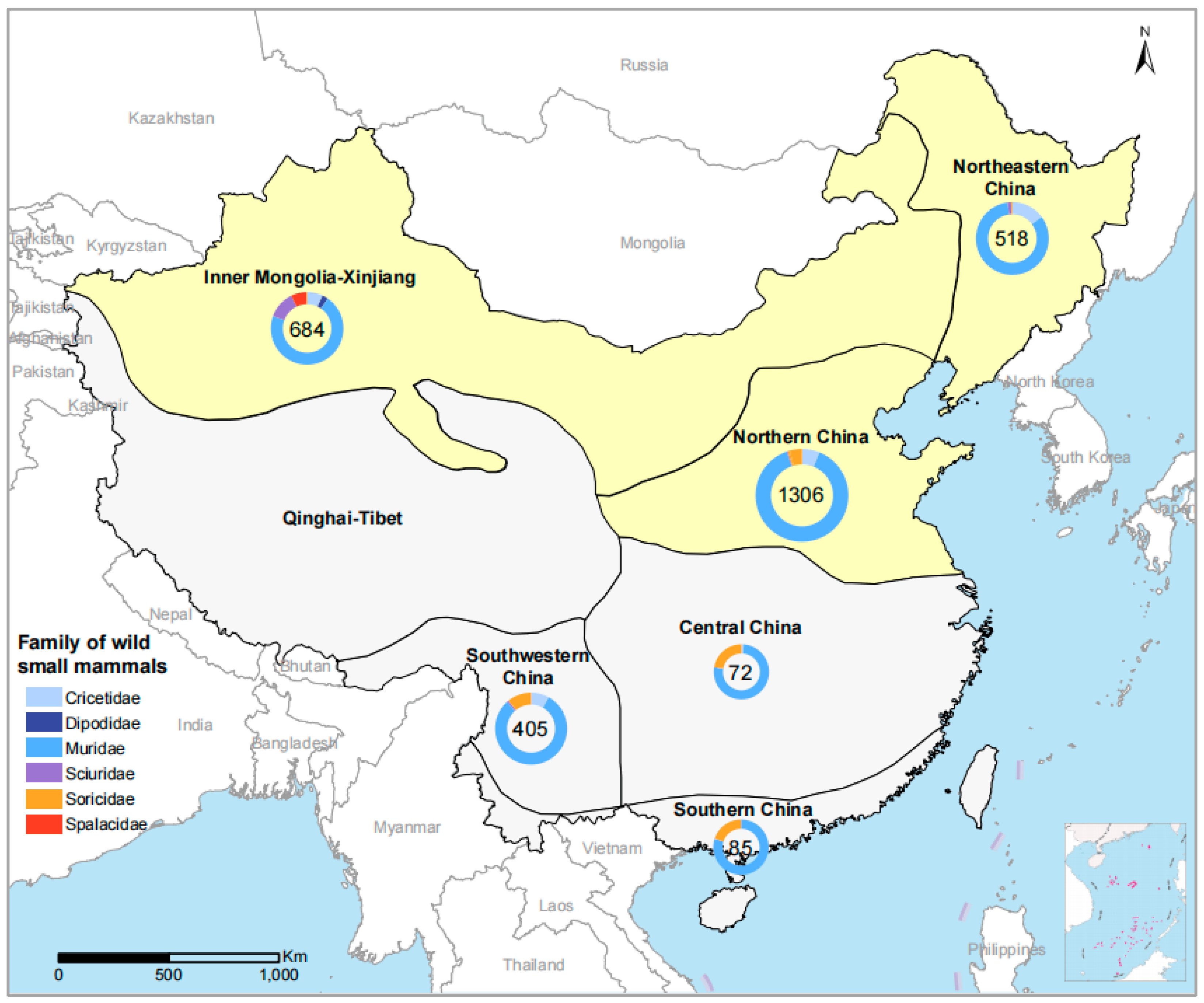
3.1. Study Site and Sample Collection
3.2. Identification of JPV in M. musculus by NGS
3.3. JPV Screening in Wild Small Mammals by RT-PCR
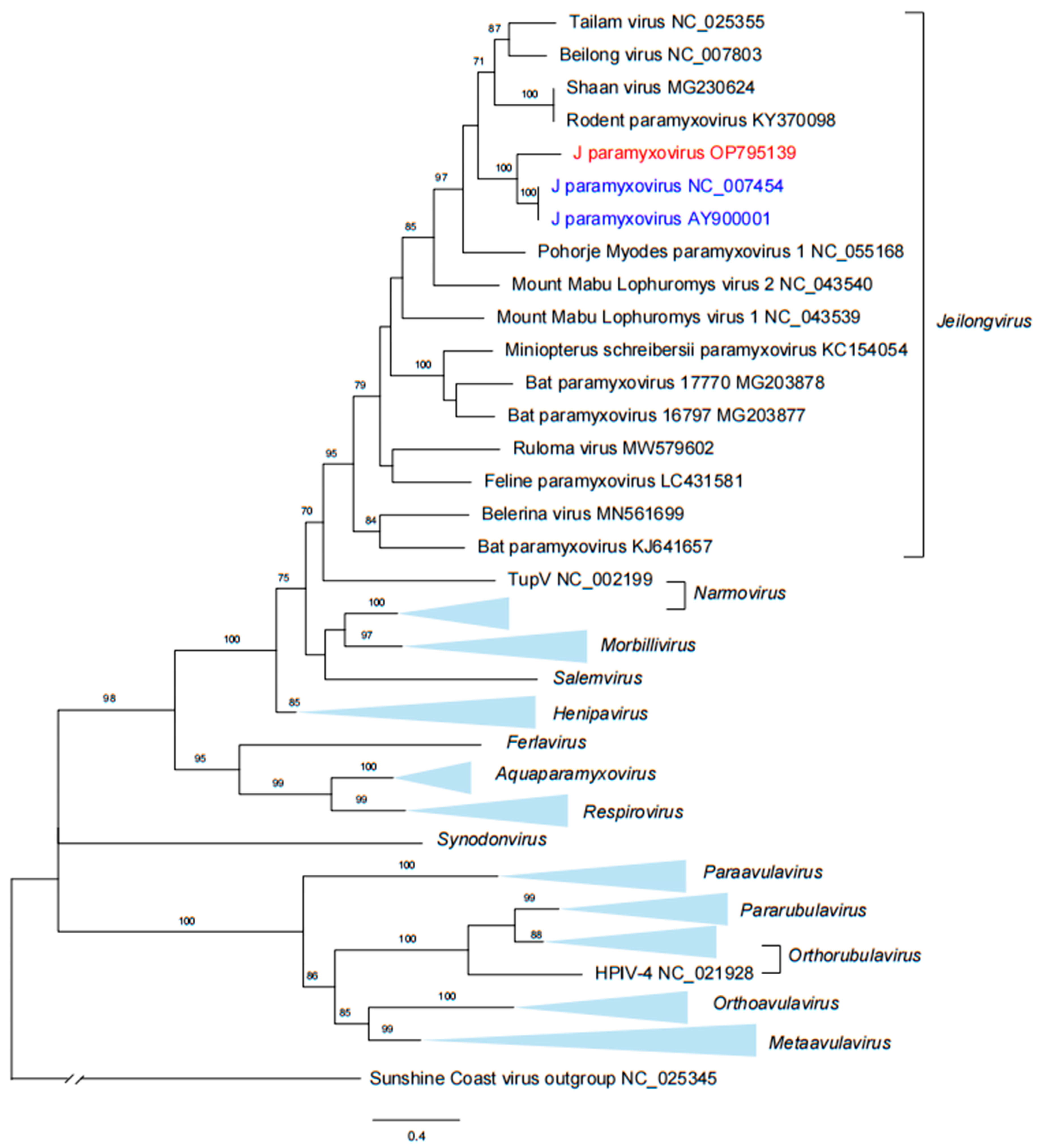
3.4. Phylogenetic Analysis of JPV Sequences
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thibault, P.A.; Watkinson, R.E.; Moreira-Soto, A.; Drexler, J.F.; Lee, B. Zoonotic Potential of Emerging Paramyxoviruses: Knowns and Unknowns. Adv. Virus Res. 2017, 98, 1–55. [Google Scholar] [PubMed] [Green Version]
- Donato, G.; Masucci, M.; De Luca, E.; Alibrandi, A.; De Majo, M.; Berjaoui, S.; Martino, C.; Mangano, C.; Lorusso, A.; Pennisi, M.G. Feline Morbillivirus in Southern Italy: Epidemiology, Clinico-Pathological Features and Phylogenetic Analysis in Cats. Viruses 2021, 13, 1449. [Google Scholar] [CrossRef] [PubMed]
- Darold, G.M.; Alfieri, A.A.; Araujo, J.P.J.; da Cruz, T.F.; Bertti, K.; da Silva, G.C.P.; Amude, A.M.; Muraro, L.S.; Lavorente, F.L.P.; Lunardi, M. High genetic diversity of paramyxoviruses infecting domestic cats in Western Brazil. Transbound. Emerg. Dis. 2021, 68, 3453–3462. [Google Scholar] [CrossRef] [PubMed]
- Jack, P.J.; Boyle, D.B.; Eaton, B.T.; Wang, L.F. The complete genome sequence of J virus reveals a unique genome structure in the family Paramyxoviridae. J. Virol. 2005, 79, 10690–10700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Yu, M.; Zhang, H.; Magoffin, D.E.; Jack, P.J.; Hyatt, A.; Wang, H.Y.; Wang, L.F. Beilong virus, a novel paramyxovirus with the largest genome of non-segmented negative-stranded RNA viruses. Virology 2006, 346, 219–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016, 10, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Lu, L.; Du, J.; Yang, L.; Ren, X.; Liu, B.; Jiang, J.; Yang, J.; Dong, J.; Sun, L.; et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 2018, 6, 178. [Google Scholar] [CrossRef]
- Noh, J.Y.; Jeong, D.G.; Yoon, S.W.; Kim, J.H.; Choi, Y.G.; Kang, S.Y.; Kim, H.K. Isolation and characterization of novel bat paramyxovirus B16-40 potentially belonging to the proposed genus Shaanvirus. Sci. Rep. 2018, 8, 12533. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Wong, B.H.; Wong, A.Y.; Poon, R.W.; Yuen, K.Y. Complete genome sequence of a novel paramyxovirus, Tailam virus, discovered in Sikkim rats. J. Virol. 2011, 85, 13473–13474. [Google Scholar] [CrossRef] [Green Version]
- Jun, M.H.; Karabatsos, N.; Johnson, R.H. A new mouse paramyxovirus (J virus). Aust. J. Exp. Biol. Med. Sci. 1977, 55, 645–647. [Google Scholar] [CrossRef]
- Li, Z.; Xu, J.; Chen, Z.; Gao, X.; Wang, L.F.; Basler, C.; Sakamoto, K.; He, B. The L gene of J paramyxovirus plays a critical role in viral pathogenesis. J. Virol. 2013, 87, 12990–12998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.J.; Zhang, X.A.; Fan, H.; Jiang, F.C.; Jin, M.Z.; Dai, K.; Wang, N.; Zhang, P.H.; Li, X.K.; Li, H.; et al. Distribution and characteristics of Beilong virus among wild rodents and shrews in China. Infect. Genet. Evol. 2020, 85, 104454. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, V.; Querouil, S.; Verheyen, E.; Verheyen, W.; Mboumba, J.F.; Dillen, M.; Colyn, M. Mitochondrial phylogeny of African wood mice, genus Hylomyscus (Rodentia, Muridae): Implications for their taxonomy and biogeography. Mol. Phylogenet. Evol. 2006, 38, 779–793. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.A.; Li, H.; Jiang, F.C.; Zhu, F.; Zhang, Y.F.; Chen, J.J.; Tan, C.W.; Anderson, D.E.; Fan, H.; Dong, L.Y.; et al. A Zoonotic Henipavirus in Febrile Patients in China. N. Engl. J. Med. 2022, 387, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Xing, Y.; Zhou, L.; Zhang, Y.; Wang, X. Geographical patterns based on faunal types of breeding birds and mammals in China. Integr. Zool. 2008, 3, 280–289. [Google Scholar] [CrossRef]
- Gomez Roman, R.; Wang, L.F.; Lee, B.; Halpin, K.; de Wit, E.; Broder, C.C.; Rahman, M.; Kristiansen, P.; Saville, M. Nipah@20: Lessons Learned from Another Virus with Pandemic Potential. mSphere 2020, 5, e00602-20. [Google Scholar] [CrossRef]
- Epstein, J.H.; Anthony, S.J.; Islam, A.; Kilpatrick, A.M.; Ali Khan, S.; Balkey, M.D.; Ross, N.; Smith, I.; Zambrana-Torrelio, C.; Tao, Y.; et al. Nipah virus dynamics in bats and implications for spillover to humans. Proc. Natl. Acad. Sci. USA 2020, 117, 29190–29201. [Google Scholar] [CrossRef] [PubMed]
- Eaton, B.T.; Broder, C.C.; Middleton, D.; Wang, L.F. Hendra and Nipah viruses: Different and dangerous. Nat. Rev. Microbiol. 2006, 4, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Audsley, M.D.; Marsh, G.A.; Lieu, K.G.; Tachedjian, M.; Joubert, D.A.; Wang, L.F.; Jans, D.A.; Moseley, G.W. The immune evasion function of J and Beilong virus V proteins is distinct from that of other paramyxoviruses, consistent with their inclusion in the proposed genus Jeilongvirus. J. Gen. Virol. 2016, 97, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Wong, A.Y.P.; Wong, B.H.L.; Lam, C.S.F.; Fan, R.Y.Y.; Lau, S.K.P.; Yuen, K.Y. Comparative genome and evolutionary analysis of naturally occurring Beilong virus in brown and black rats. Infect. Genet. Evol. 2016, 45, 311–319. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eco-Climate Regions | Year | Species (No. of Positive/No. Tested) | No. Total Tested | No. (%) of Positive |
|---|---|---|---|---|
| Northern | 1306 | 15 (1.15) | ||
| Shandong | 2018 | Apodemus agrarius (1/115), Mus musculus (9/286), Rattus norvegicus (0/134), Tscherskia triton (0/42), Suncus murinus (0/13), Crocidura lasiura (0/20), Crocidura shantungensis (0/7) and Crocidura tanakae (0/15) | 632 | 10 (1.58) |
| Henan | 2018, 2021 | Apodemus agrarius (0/54), Apodemus draco (0/4), Callosciurus erythraeus (0/5), Crocidura tanakae (0/6), Episoriculus fumidus (0/2), Mus musculus (4/60), Niviventer andersoni (0/2), Niviventer confucianus (0/5), Niviventer niviventer (0/18), Rattus norvegicus (0/24), Rattus tanezumi (0/285) and Suncus murinus (0/1) | 466 | 4 (0.86) |
| Beijing | 2018 | Allocricetulus eversmanni (0/6), Apodemus agrarius (0/51), Apodemus draco (0/56), Apodemus peninsulae (1/40), Cricetulus longicaudatus (0/28), Mus musculus (0/3), Myodes rufocanus (0/3), Niviventer confucianus (0/19), Suncus murinus (0/1) and Tscherskia triton (0/1) | 208 | 1 (0.48) |
| Inner Mongolia-Xinjiang | 684 | 4 (0.58) | ||
| Inner Mongolia | 2018, 2019, 2021 | Allactaga sibirica (0/11), Cricetulus barabensis (0/2), Cricetulus migratorius (0/37), Dipus sagitta (0/5), Meriones meridianus (0/57), Meriones unguiculatus (0/181), Mus musculus (0/4), Myospalax aspalax (0/2), Myospalax psilurus (0/45), Phodopus roborovskii (0/4), Rattus pyctoris (0/1) and Spermophilus dauricus (0/28) | 377 | 0 (0) |
| Xinjiang | 2017–2019 | Meriones libycus (0/42), Rhombomys opimus (0/74), Apodemus sylvaticus (0/15), Mus musculus (4/89), Spermophilus undulatus (0/25), Suncus murinus (0/2), Microtus oeconomus (0/1), Cricetulyus migratorius (0/4), Rattus norvegicus (0/20), Spermophilus erythrogenys (0/31), Allactaga sibirica (0/2) and Meriones tamariscinus (0/2) | 307 | 4 (1.30) |
| Northeastern | 518 | 2 (0.39) | ||
| Heilongjiang | 2017, 2021 | Apodemus agrarius (1/170), Apodemus peninsulae (0/12), Microtus fortis (1/18), Microtus maximowiczii (0/1), Mus musculus (0/2), Myodes rufocanus (0/4), Myodes rutilus (0/5), Rattus norvegicus (0/110), Tamias sibiricus (0/7) and Tscherskia triton (0/1) | 330 | 2 (0.61) |
| Jilin | 2017 | Apodemus peninsulae (0/8), Crocidura tanakae (0/2), Myodes rufocanus (0/1), Myodes rutilus (0/47), Sorex isodon (0/1) and Tamias sibiricus (0/1) | 60 | 0 (0) |
| Liaoning | 2017, 2018 | Mus musculus (0/8) and Rattus norvegicus (0/120) | 128 | 0 (0) |
| Southwestern | 405 | 0 (0) | ||
| Yunnan | 2013, 2015, 2016 | Suncus murinus (0/4), Rattus tanezumi (0/144), Crocidura tanakae (0/5), Crocidura lasiura (0/4), Rattus yunnanensis (0/9), Rattus steini (0/7), Tamiops swinhoei (0/3), Niviventer coxingi (0/7), Apodemus ilex (0/80), Anourosorex squamipes (0/15), Eothenomys eleusis (0/8), Sorex bedfordiae (0/8), Blarinella quadraticauda (0/4), Niviventer andersoni (0/4), Episoriculus caudatus (0/3), Melomys burtoni (0/2), Rattus brunneusculus (0/7), Berylmys bowersi (0/1), Mus pahari (0/6), Apodemus chevrieri (0/56), Eothenomys cachinus (0/1), Crocidura horsfieldii (0/1), Chiropodomys gliroides (0/1), Micromys minutus (0/1), Eothenomys miletus (0/16) and Eothenomys proditor (0/8) | 405 | 0 (0) |
| Central | 72 | 0 (0) | ||
| Zhejiang | 2018, 2012 | Apodemus agrarius (0/31), Crocidura lasiura (0/9), Crocidura shantungensis (0/5), Microtus fortis (0/1), Niviventer confucianus (0/9), Niviventer fulvescens (0/15) and Sorex caecutiens (0/2) | 72 | 0 (0) |
| Southern | 85 | 0 (0) | ||
| Guangdong | 2017, 2021 | Bandicota indica (0/14), Crocidura tanakae (0/3), Rattus andamanensis (0/11), Rattus norvegicus (0/38), Rattus tanezumi (0/5) and Suncus murinus (0/14) | 85 | 0 (0) |
| Total | 3070 | 21 (0.68) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Zhang, J.; Wang, Y.; Tian, F.; Zhang, X.; Wang, G.; Li, S.; Ding, H.; Hu, Z.; Liu, W.; et al. Genetic Diversity and Expanded Host Range of J Paramyxovirus Detected in Wild Small Mammals in China. Viruses 2023, 15, 49. https://doi.org/10.3390/v15010049
Zhang Y, Zhang J, Wang Y, Tian F, Zhang X, Wang G, Li S, Ding H, Hu Z, Liu W, et al. Genetic Diversity and Expanded Host Range of J Paramyxovirus Detected in Wild Small Mammals in China. Viruses. 2023; 15(1):49. https://doi.org/10.3390/v15010049
Chicago/Turabian StyleZhang, Yunfa, Jingtao Zhang, Yuna Wang, Feng Tian, Xiaolong Zhang, Gang Wang, Shuang Li, Heng Ding, Zhenyu Hu, Wei Liu, and et al. 2023. "Genetic Diversity and Expanded Host Range of J Paramyxovirus Detected in Wild Small Mammals in China" Viruses 15, no. 1: 49. https://doi.org/10.3390/v15010049