

Development of Oncolytic Vectors Based on Human Adenovirus Type 6 for Cancer Treatment
 , , , and
, , , and 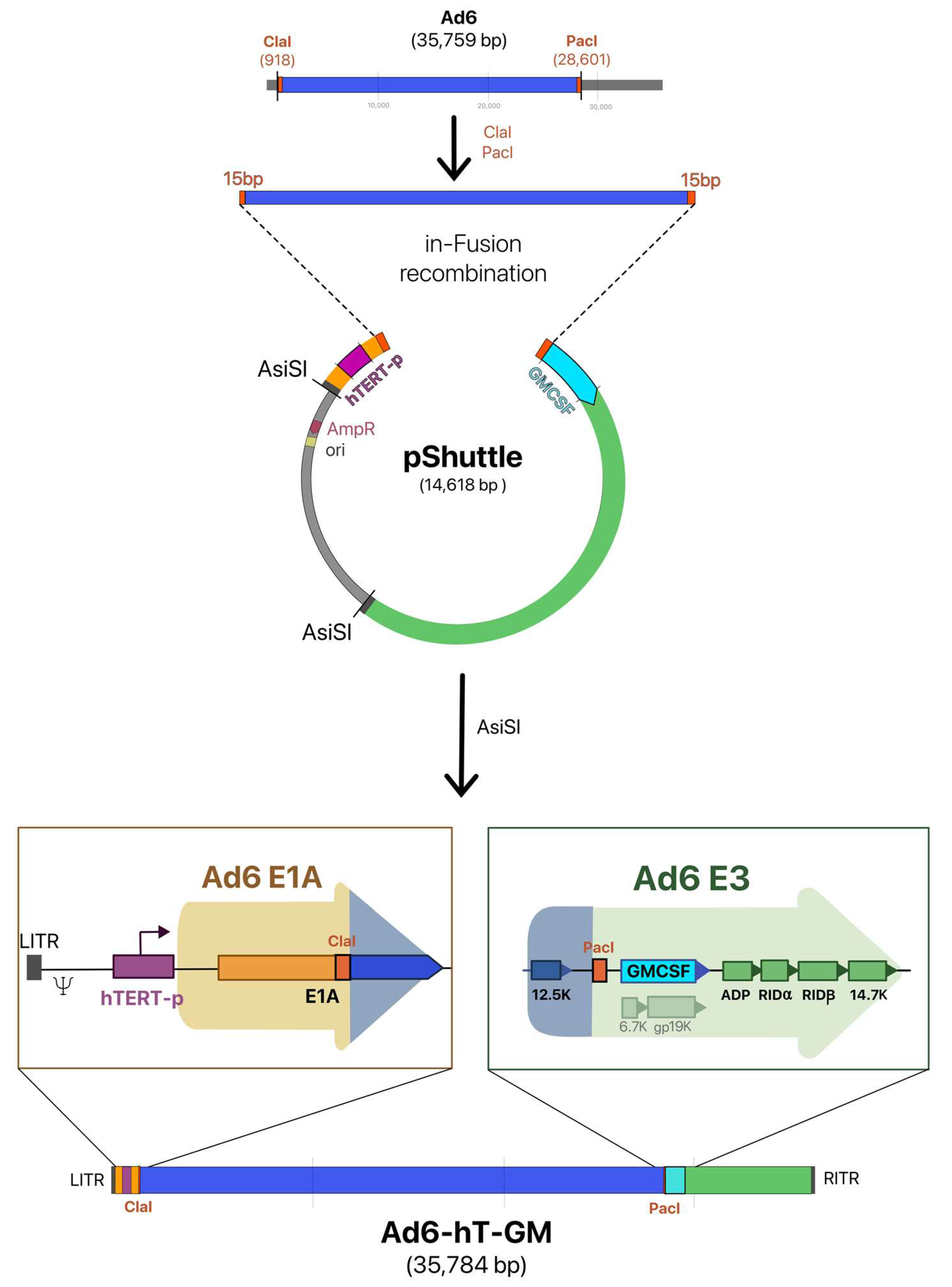{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Recombinant Ad6 Vectors
2.2. Cells Lines
2.3. Cell Viability Assays
2.4. In Vivo Study
2.5. Biological Activity of GM-CSF
2.6. ELISA Assay
2.7. RT-qPCR for Viral mRNA
2.8. Quantification of Ad by qPCR
2.9. Statistical Analyses
3. Results
3.1. Construction of the Recombinant Ad6-Based Vectors
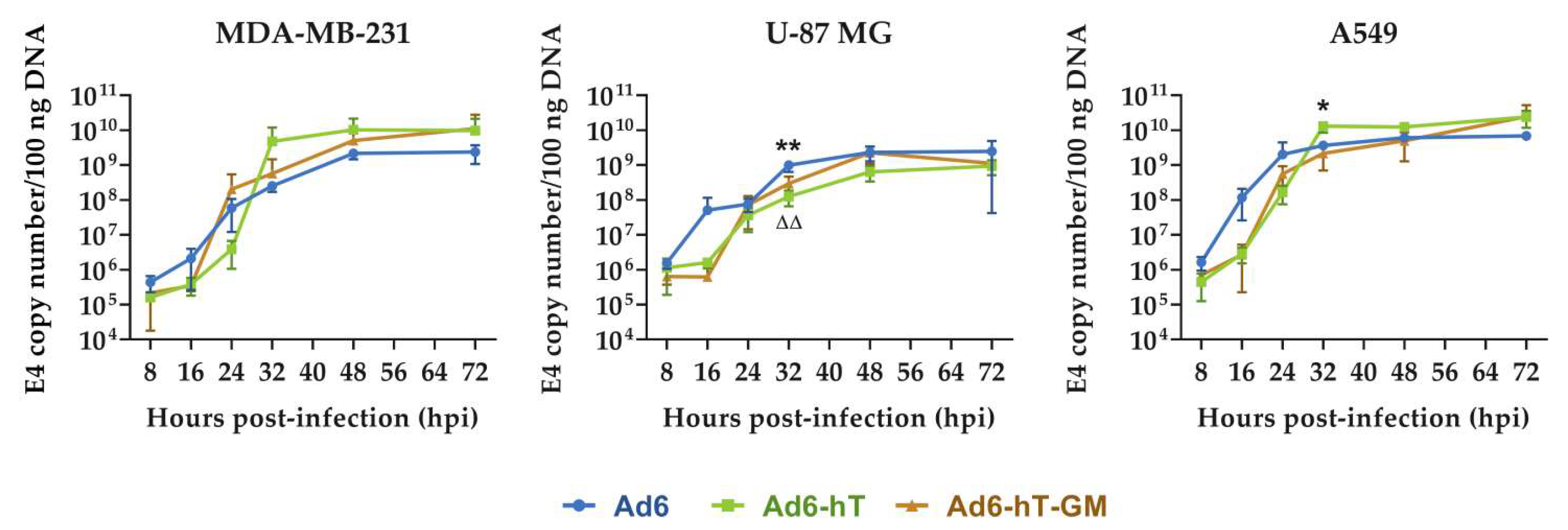
3.2. The Influence of hTERT-Promoter Insertion on Adenoviral Replication In Vitro
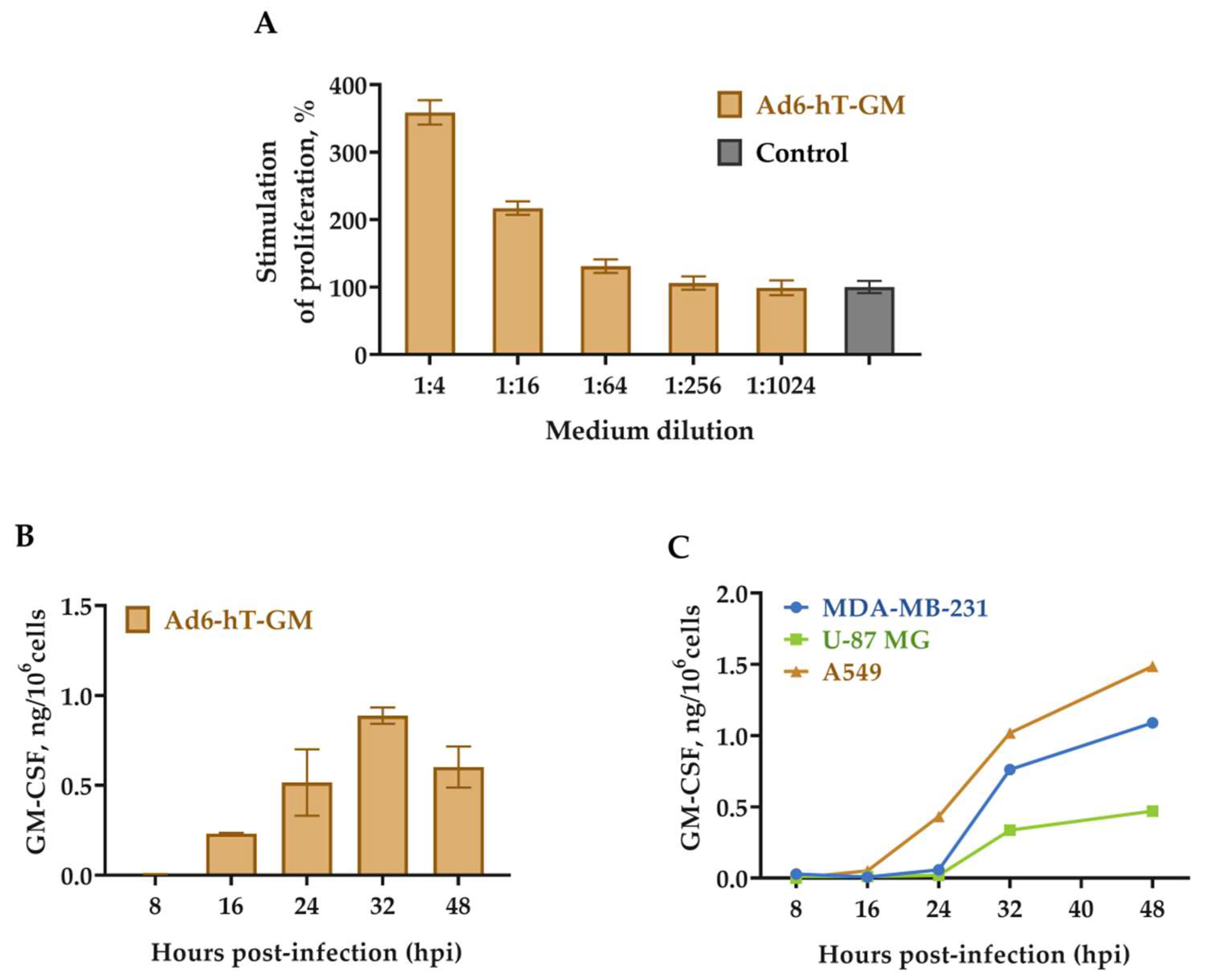
3.3. GM-CSF Expression in Ad6-hT-GM-Infected Cells
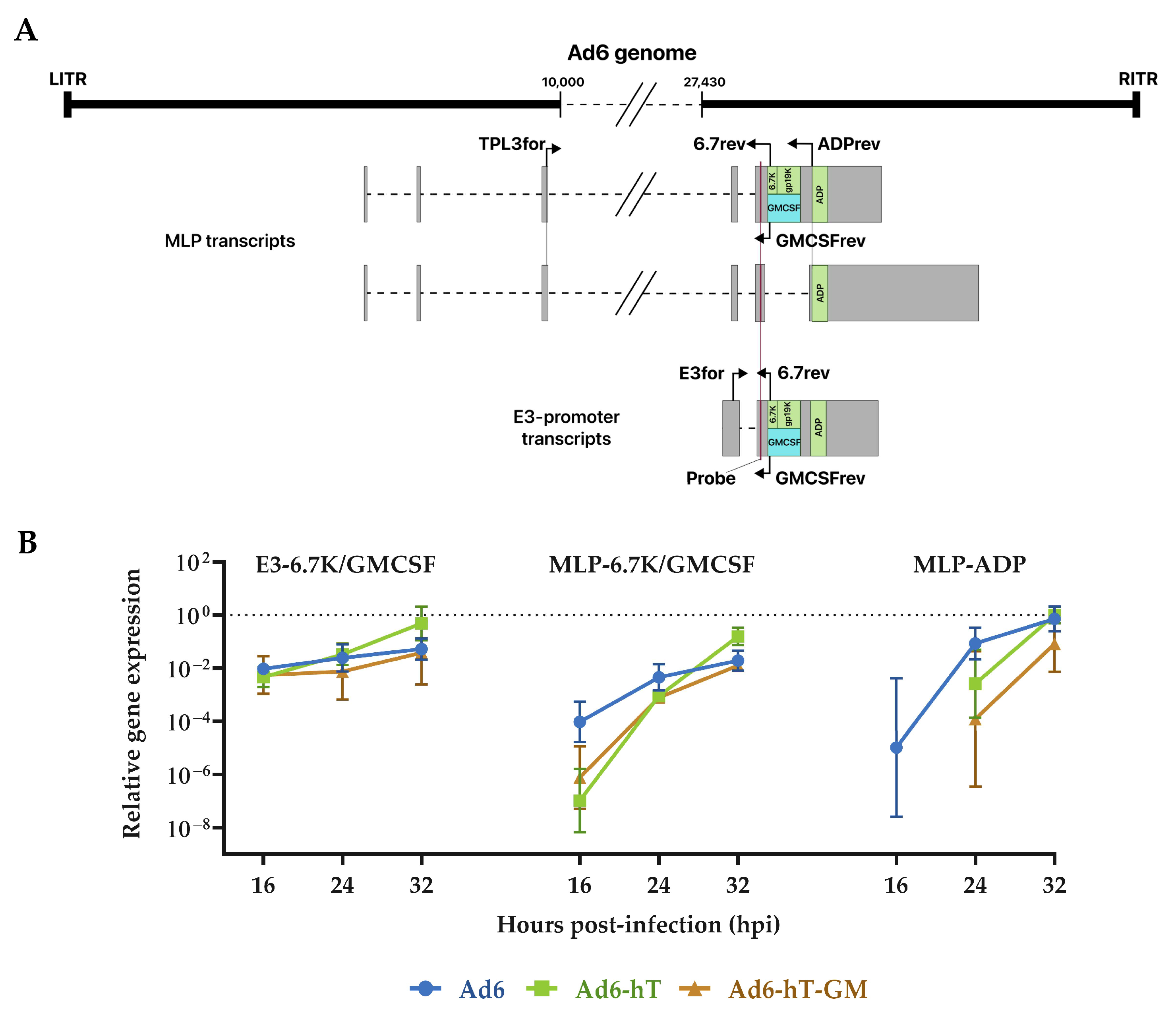
3.4. The Influence of Ad6 Genome Modification on the Expression of E3-Coded Genes
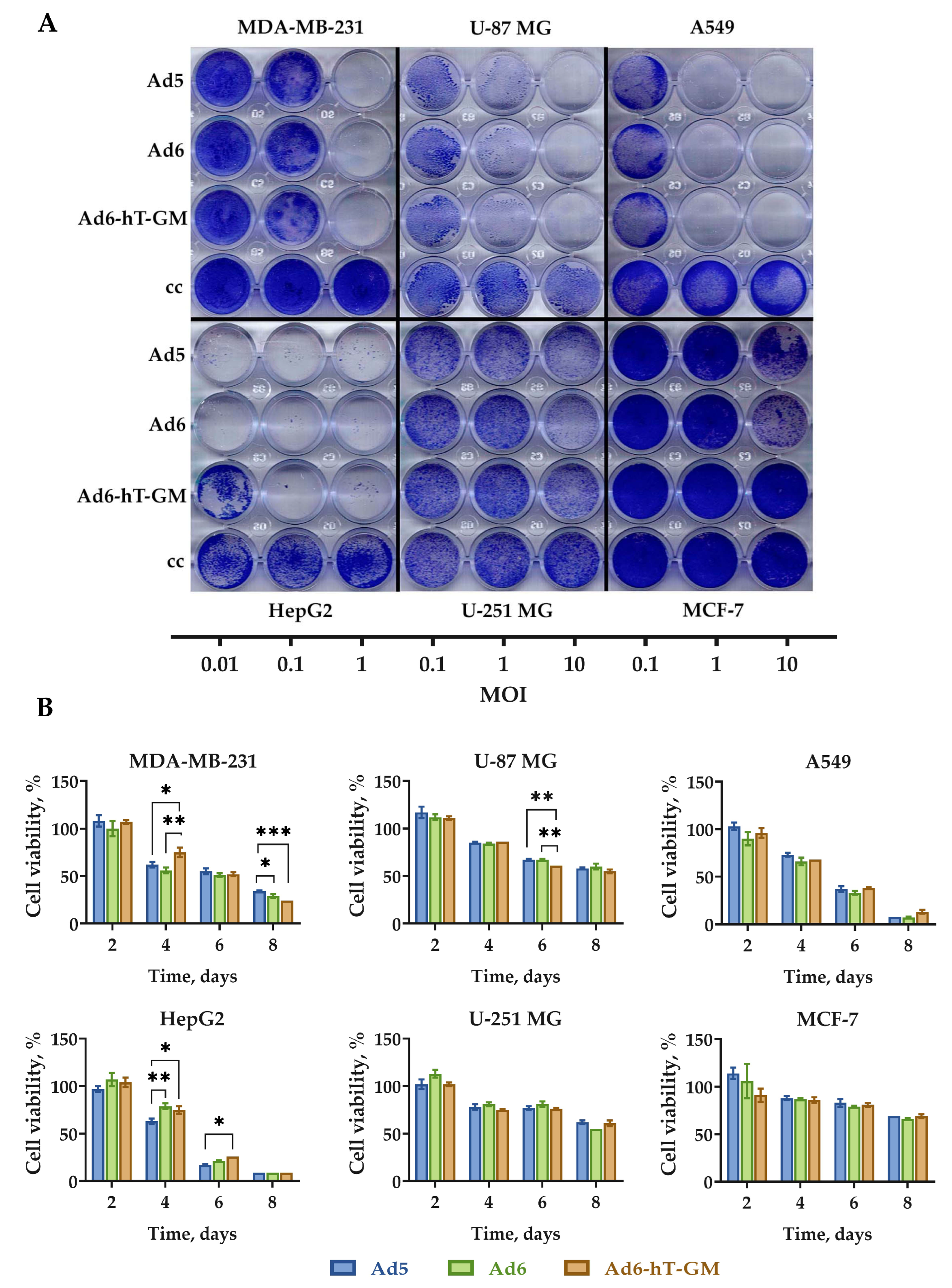
3.5. Oncolytic Efficacy of Ad6-hT-GM In Vitro
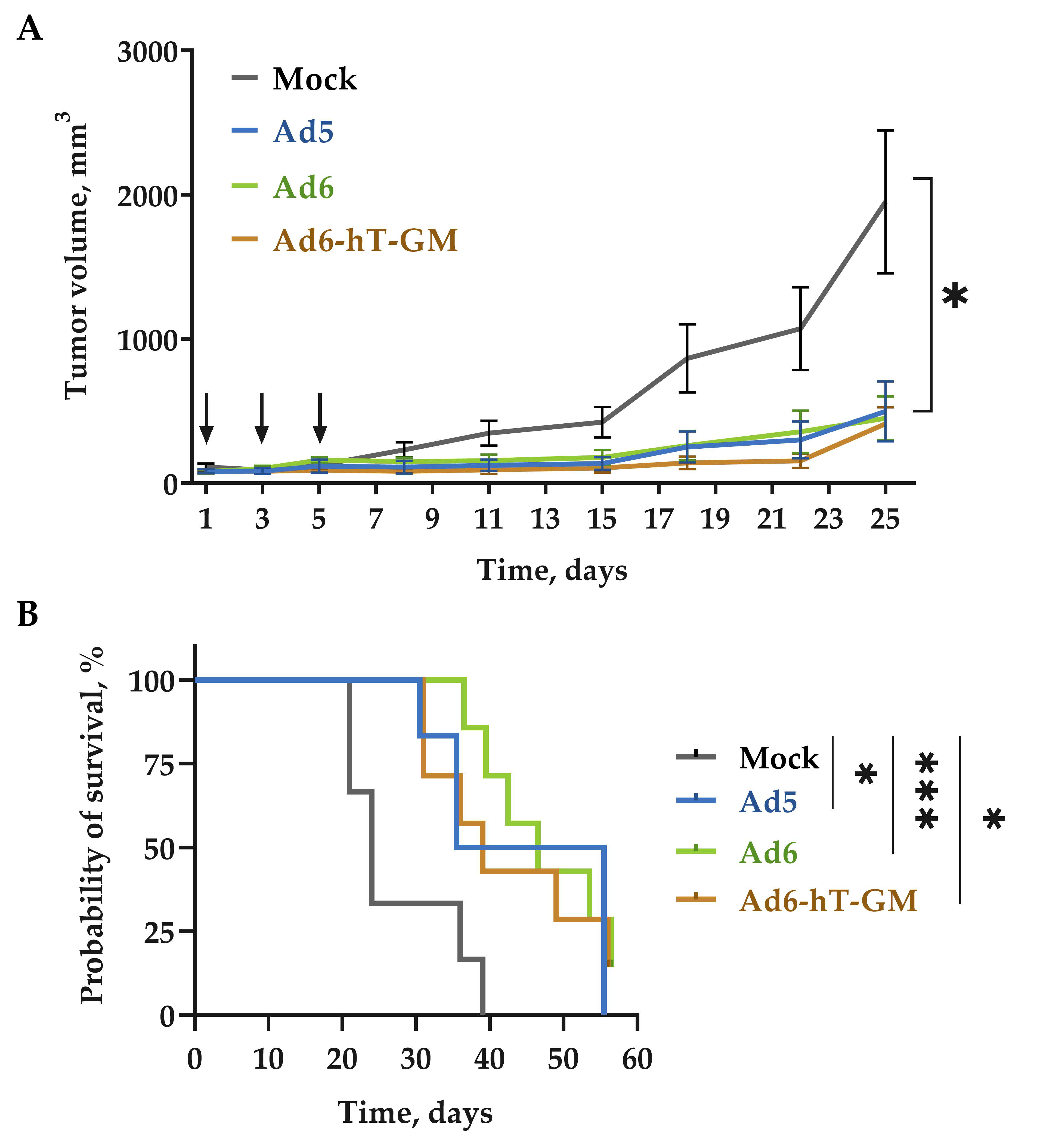
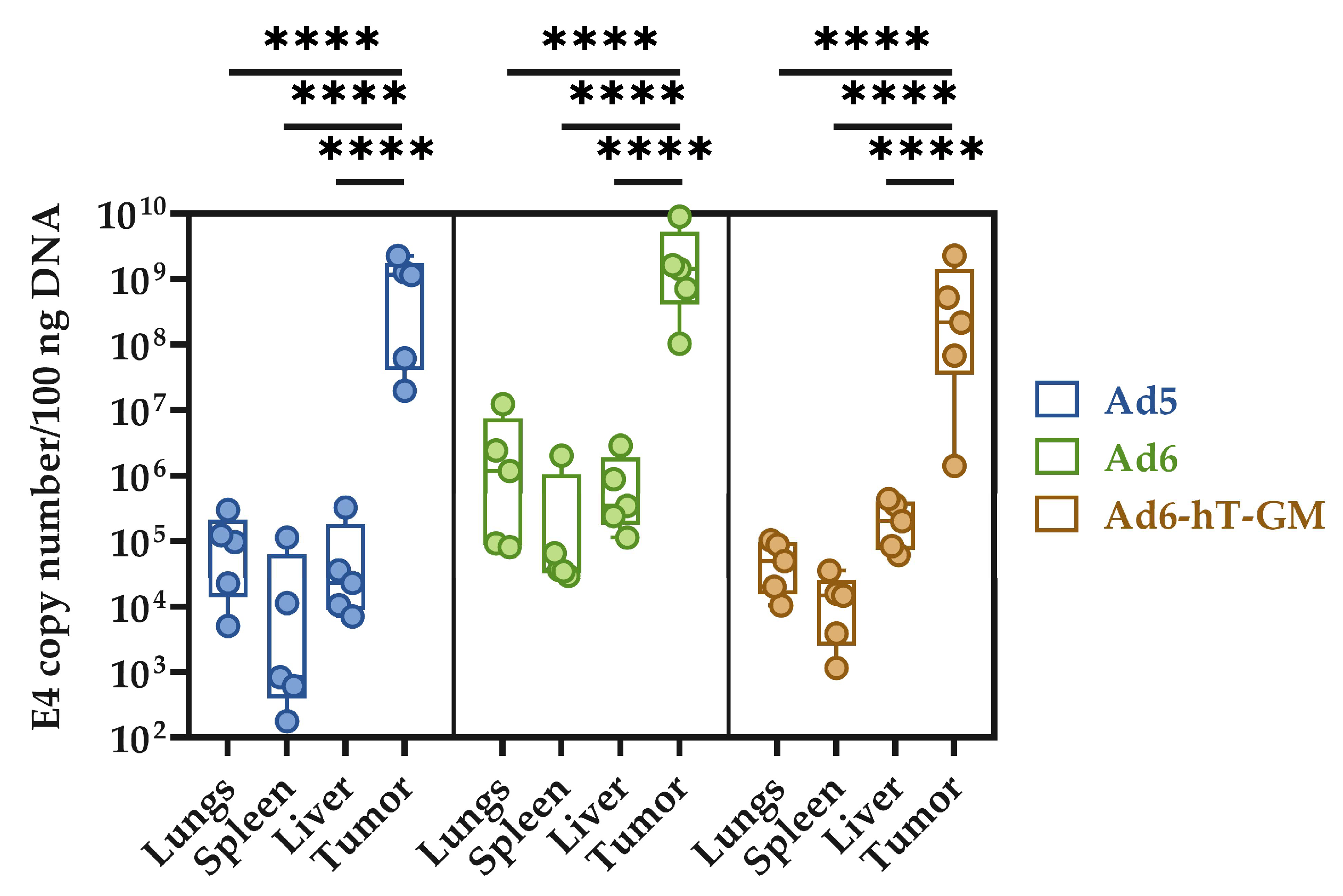
3.6. The Influence of Genome Modification on Ad-hT-GM Lytic Ability In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liang, M. Oncorine, the World First Oncolytic Virus Medicine and Its Update in China. Curr Cancer Drug Targets 2018, 18, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Aldrak, N.; Alsaab, S.; Algethami, A.; Bhere, D.; Wakimoto, H.; Shah, K.; Alomary, M.N.; Zaidan, N. Oncolytic Herpes Simplex Virus-Based Therapies for Cancer. Cells 2021, 10, 1541. [Google Scholar] [CrossRef] [PubMed]
- Mennechet, F.J.D.; Paris, O.; Ouoba, A.R.; Salazar Arenas, S.; Sirima, S.B.; Takoudjou Dzomo, G.R.; Diarra, A.; Traore, I.T.; Kania, D.; Eichholz, K.; et al. A Review of 65 Years of Human Adenovirus Seroprevalence. Expert Rev. Vaccines 2019, 18, 597–613. [Google Scholar] [CrossRef] [PubMed]
- Khare, R.; May, S.M.; Vetrini, F.; Weaver, E.A.; Palmer, D.; Rosewell, A.; Grove, N.; Ng, P.; Barry, M.A. Generation of a Kupffer Cell-Evading Adenovirus for Systemic and Liver-Directed Gene Transfer. Mol. Ther. 2011, 19, 1254–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alba, R.; Bradshaw, A.C.; Parker, A.L.; Bhella, D.; Waddington, S.N.; Nicklin, S.A.; van Rooijen, N.; Custers, J.; Goudsmit, J.; Barouch, D.H.; et al. Identification of Coagulation Factor (F)X Binding Sites on the Adenovirus Serotype 5 Hexon: Effect of Mutagenesis on FX Interactions and Gene Transfer. Blood 2009, 114, 965–971. [Google Scholar] [CrossRef] [Green Version]
- Bradley, R.R.; Maxfield, L.F.; Lynch, D.M.; Iampietro, M.J.; Borducchi, E.N.; Barouch, D.H. Adenovirus Serotype 5-Specific Neutralizing Antibodies Target Multiple Hexon Hypervariable Regions. J. Virol. 2012, 86, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Coughlan, L.; Bradshaw, A.C.; Parker, A.L.; Robinson, H.; White, K.; Custers, J.; Goudsmit, J.; van Roijen, N.; Barouch, D.H.; Nicklin, S.A.; et al. Ad5:Ad48 Hexon Hypervariable Region Substitutions Lead to Toxicity and Increased Inflammatory Responses Following Intravenous Delivery. Mol. Ther. 2012, 20, 2268–2281. [Google Scholar] [CrossRef] [Green Version]
- Khare, R.; Hillestad, M.L.; Xu, Z.; Byrnes, A.P.; Barry, M.A. Circulating Antibodies and Macrophages as Modulators of Adenovirus Pharmacology. J. Virol. 2013, 87, 3678–3686. [Google Scholar] [CrossRef] [Green Version]
- Capone, S.; Meola, A.; Ercole, B.B.; Vitelli, A.; Pezzanera, M.; Ruggeri, L.; Davies, M.E.; Tafi, R.; Santini, C.; Luzzago, A.; et al. A Novel Adenovirus Type 6 (Ad6)-Based Hepatitis C Virus Vector That Overcomes Preexisting Anti-Ad5 Immunity and Induces Potent and Broad Cellular Immune Responses in Rhesus Macaques. J. Virol. 2006, 80, 1688–1699. [Google Scholar] [CrossRef] [Green Version]
- Shashkova, E.v.; May, S.M.; Barry, M.A. Characterization of Human Adenovirus Serotypes 5, 6, 11, and 35 as Anticancer Agents. Virology 2009, 394, 311–320. [Google Scholar] [CrossRef]
- Chen, C.Y.; Weaver, E.A.; Khare, R.; May, S.M.; Barry, M.A. Mining the Adenovirus Virome for Oncolytics against Multiple Solid Tumor Types. Cancer Gene 2011, 18, 744–750. [Google Scholar] [CrossRef]
- Nguyen, T.V.; Heller, G.J.; Barry, M.E.; Crosby, C.M.; Turner, M.A.; Barry, M.A. Evaluation of Polymer Shielding for Adenovirus Serotype 6 (Ad6) for Systemic Virotherapy against Human Prostate Cancers. Mol. Oncolytics 2016, 3, 15021. [Google Scholar] [CrossRef] [Green Version]
- Romanenko, M.V.; Dolgova, E.V.; Osipov, I.D.; Ritter, G.S.; Sizova, M.S.; Proskurina, A.S.; Efremov, Y.R.; Bayborodin, S.I.; Potter, E.A.; Taranov, O.S.; et al. Oncolytic Effect of Adenoviruses Serotypes 5 and 6 Against U87 Glioblastoma Cancer Stem Cells. Anticancer Res 2019, 39, 6073–6086. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Tazawa, H.; Teraishi, F.; Kojima, T.; Watanabe, Y.; Uno, F.; Yano, S.; Urata, Y.; Kagawa, S.; Fujiwara, T. The HTERT Promoter Enhances the Antitumor Activity of an Oncolytic Adenovirus under a Hypoxic Microenvironment. PLoS ONE 2012, 7, e39292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.-W.; Chueh, H.-Y.; Tsai, C.-C.; Lin, C.-T.; Qiu, J.T. Novel GM-CSF-Based Vaccines: One Small Step in GM-CSF Gene Optimization, One Giant Leap for Human Vaccines. Hum. Vaccin. Immunother. 2016, 12, 3020–3028. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, S.; Kim, T.; Zager, J.; Bennett, J.; Ebright, M.; D’Angelica, M.; Fong, Y. Use of an Oncolytic Virus Secreting GM-CSF as Combined Oncolytic and Immunotherapy for Treatment of Colorectal and Hepatic Adenocarcinomas. Surgery 2007, 141, 520–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davydova, J.; Yamamoto, M. Oncolytic Adenoviruses: Design, Generation, and Experimental Procedures. Curr. Protoc. Hum. Genet. 2013, 78, 12–14. [Google Scholar] [CrossRef]
- R Core Team R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 31 December 2022).
- Tollefson, A.E.; Scaria, A.; Hermiston, T.W.; Ryerse, J.S.; Wold, L.J.; Wold, W.S. The Adenovirus Death Protein (E3-11.6K) Is Required at Very Late Stages of Infection for Efficient Cell Lysis and Release of Adenovirus from Infected Cells. J. Virol. 1996, 70, 2296–2306. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hallden, G.; Hill, R.; Anand, A.; Liu, T.-C.; Francis, J.; Brooks, G.; Lemoine, N.; Kirn, D. E3 Gene Manipulations Affect Oncolytic Adenovirus Activity in Immunocompetent Tumor Models. Nat. Biotechnol. 2003, 21, 1328–1335. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.S.; Ganesh, S.; Hawkins, L.; Idamakanti, N. Generation of Recombinant Adenovirus Using the Escherichia Coli BJ5183 Recombination System. In Adenovirus Methods and Protocols, Second Edition, Volume 1; Humana Press: New Jersey, NJ, USA, 2007; pp. 61–68. [Google Scholar]
- Zhang, Z.; Zhang, X.; Newman, K.; Liu, X. MicroRNA Regulation of Oncolytic Adenovirus 6 for Selective Treatment of Castration-Resistant Prostate Cancer. Mol. Cancer 2012, 11, 2410–2418. [Google Scholar] [CrossRef]
- Turner, M.A.; Middha, S.; Hofherr, S.E.; Barry, M.A. Comparison of the Life Cycles of Genetically Distant Species C and Species D Human Adenoviruses Ad6 and Ad26 in Human Cells. J. Virol. 2015, 89, 12401–12417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, L.; Johnson, L.; Bauzon, M.; Nye, J.; Castro, D.; Kitzes, G.; Young, M.; Holt, J.; Trown, P.; Hermiston, T. Gene Delivery from the E3 Region of Replicating Human Adenovirus: Evaluation of the 6.7 K/Gp19 K Region. Gene 2001, 8, 1123–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, L.; Hermiston, T. Gene Delivery from the E3 Region of Replicating Human Adenovirus: Evaluation of the E3B Region. Gene 2001, 8, 1142–1148. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, L.; Hermiston, T. Gene Delivery from the E3 Region of Replicating Human Adenovirus: Evaluation of the ADP Region. Gene 2001, 8, 1132–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donovan-Banfield, I.; Turnell, A.S.; Hiscox, J.A.; Leppard, K.N.; Matthews, D.A. Deep Splicing Plasticity of the Human Adenovirus Type 5 Transcriptome Drives Virus Evolution. Commun. Biol. 2020, 3, 124. [Google Scholar] [CrossRef] [Green Version]
- Romanenko, M.; Osipov, I.; Netesov, S.V.; Davydova, J. Adenovirus Type 6: Subtle Structural Distinctions from Adenovirus Type 5 Result in Essential Differences in Properties and Perspectives for Gene Therapy. Pharmaceutics 2021, 13, 1641. [Google Scholar] [CrossRef] [PubMed]
- Doerner, J.; Sallard, E.; Zhang, W.; Solanki, M.; Liu, J.; Ehrke-Schulz, E.; Zirngibl, H.; Lieber, A.; Ehrhardt, A. Novel Group C Oncolytic Adenoviruses Carrying a MiRNA Inhibitor Demonstrate Enhanced Oncolytic Activity In Vitro and In Vivo. Mol. Cancer 2022, 21, 460–470. [Google Scholar] [CrossRef]
- Nguyen, T.V.; Crosby, C.M.; Heller, G.J.; Mendel, Z.I.; Barry, M.E.; Barry, M.A. Oncolytic Adenovirus Ad657 for Systemic Virotherapy against Prostate Cancer. Oncolytic Virother. 2018, 7, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesney, J.A.; Ribas, A.; Long, G.V.; Kirkwood, J.M.; Dummer, R.; Puzanov, I.; Hoeller, C.; Gajewski, T.F.; Gutzmer, R.; Rutkowski, P.; et al. Randomized, Double-Blind, Placebo-Controlled, Global Phase III Trial of Talimogene Laherparepvec Combined with Pembrolizumab for Advanced Melanoma. J. Clin. Oncol. 2022. [Google Scholar] [CrossRef]
- Samson, A.; West, E.J.; Carmichael, J.; Scott, K.J.; Turnbull, S.; Kuszlewicz, B.; Dave, R.V.; Peckham-Cooper, A.; Tidswell, E.; Kingston, J.; et al. Neoadjuvant Intravenous Oncolytic Vaccinia Virus Therapy Promotes Anticancer Immunity in Patients. Cancer Immunol. Res. 2022, 10, 745–756. [Google Scholar] [CrossRef]
- Bett, A.J.; Prevec, L.; Graham, F.L. Packaging Capacity and Stability of Human Adenovirus Type 5 Vectors. J. Virol. 1993, 67, 5911–5921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Alemany, R.; Yamamoto, M.; Curiel, D.T. The Presence of the Adenovirus E3 Region Improves the Oncolytic Potency of Conditionally Replicative Adenoviruses. Clin. Cancer Res. 2002, 8, 3348–3359. [Google Scholar] [PubMed]
- Armstrong, L.; Arrington, A.; Han, J.; Gavrikova, T.; Brown, E.; Yamamoto, M.; Vickers, S.M.; Davydova, J. Generation of a Novel, Cyclooxygenase-2–Targeted, Interferon-Expressing, Conditionally Replicative Adenovirus for Pancreatic Cancer Therapy. Am. J. Surg. 2012, 204, 741–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davydova, J.; Gavrikova, T.; Brown, E.J.; Luo, X.; Curiel, D.T.; Vickers, S.M.; Yamamoto, M. In Vivo Bioimaging Tracks Conditionally Replicative Adenoviral Replication and Provides an Early Indication of Viral Antitumor Efficacy. Cancer Sci. 2010, 101, 474–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, M.G.; Eidenschink, B.B.; Iguchi, E.; Zakharkin, S.O.; LaRocca, C.J.; Tolosa, E.J.; Truty, M.J.; Jacobsen, K.; Fernandez-Zapico, M.E.; Davydova, J. Cancer Imaging and Therapy Utilizing a Novel NIS-Expressing Adenovirus: The Role of Adenovirus Death Protein Deletion. Mol. Oncolytics 2021, 20, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Tollefson, A.E.; Ryerse, J.S.; Scaria, A.; Hermiston, T.W.; Wold, W.S.M. The E3-11.6-KDa Adenovirus Death Protein (ADP) Is Required for Efficient Cell Death: Characterization of Cells Infected WithadpMutants. Virology 1996, 220, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Doronin, K.; Toth, K.; Kuppuswamy, M.; Krajcsi, P.; Tollefson, A.E.; Wold, W.S.M. Overexpression of the ADP (E3-11.6K) Protein Increases Cell Lysis and Spread of Adenovirus. Virology 2003, 305, 378–387. [Google Scholar] [CrossRef]
- Elsing, A.; Burgert, H.-G. The Adenovirus E3/10.4K–14.5K Proteins down-Modulate the Apoptosis Receptor Fas/Apo-1 by Inducing Its Internalization. Proc. Natl. Acad. Sci. USA 1998, 95, 10072–10077. [Google Scholar] [CrossRef] [Green Version]
- Benedict, C.A.; Norris, P.S.; Prigozy, T.I.; Bodmer, J.-L.; Mahr, J.A.; Garnett, C.T.; Martinon, F.; Tschopp, J.; Gooding, L.R.; Ware, C.F. Three Adenovirus E3 Proteins Cooperate to Evade Apoptosis by Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Receptor-1 and -2. J. Biol. Chem. 2001, 276, 3270–3278. [Google Scholar] [CrossRef] [Green Version]
- McSharry, B.P.; Burgert, H.-G.; Owen, D.P.; Stanton, R.J.; Prod’homme, V.; Sester, M.; Koebernick, K.; Groh, V.; Spies, T.; Cox, S.; et al. Adenovirus E3/19K Promotes Evasion of NK Cell Recognition by Intracellular Sequestration of the NKG2D Ligands Major Histocompatibility Complex Class I Chain-Related Proteins A and B. J. Virol. 2008, 82, 4585–4594. [Google Scholar] [CrossRef]
- Sester, M.; Koebernick, K.; Owen, D.; Ao, M.; Bromberg, Y.; May, E.; Stock, E.; Andrews, L.; Groh, V.; Spies, T.; et al. Conserved Amino Acids within the Adenovirus 2 E3/19K Protein Differentially Affect Downregulation of MHC Class I and MICA/B Proteins. J. Immunol. 2010, 184, 255–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortolanza, S.; Bunuales, M.; Alzuguren, P.; Lamas, O.; Aldabe, R.; Prieto, J.; Hernandez-Alcoceba, R. Deletion of the E3-6.7K/Gp19K Region Reduces the Persistence of Wild-Type Adenovirus in a Permissive Tumor Model in Syrian Hamsters. Cancer Gene 2009, 16, 703–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenthe, J.; Eriksson, E.; Milenova, I.; Moreno, R.; Alemany, R.; Loskog, A. 516. A Novel Oncolytic Adenovirus Expressing Tumor Microenvironment Stimulators to Evoke and Facilitate Anti-Tumor Immune Responses. Mol. Ther. 2016, 24, S206. [Google Scholar] [CrossRef]
- Koski, A.; Kangasniemi, L.; Escutenaire, S.; Pesonen, S.; Cerullo, V.; Diaconu, I.; Nokisalmi, P.; Raki, M.; Rajecki, M.; Guse, K.; et al. Treatment of Cancer Patients With a Serotype 5/3 Chimeric Oncolytic Adenovirus Expressing GMCSF. Mol. Ther. 2010, 18, 1874–1884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Fu, J.; Ehrhardt, A. Novel Vector Construction Based on Alternative Adenovirus Types via Homologous Recombination. Hum. Gene Methods 2018, 29, 124–134. [Google Scholar] [CrossRef]
- Abbink, P.; Kirilova, M.; Boyd, M.; Mercado, N.; Li, Z.; Nityanandam, R.; Nanayakkara, O.; Peterson, R.; Larocca, R.A.; Aid, M.; et al. Rapid Cloning of Novel Rhesus Adenoviral Vaccine Vectors. J. Virol. 2018, 92, e01924-17. [Google Scholar] [CrossRef] [Green Version]
- Miciak, J.J.; Hirshberg, J.; Bunz, F. Seamless Assembly of Recombinant Adenoviral Genomes from High-Copy Plasmids. PLoS ONE 2018, 13, e0199563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falchetti, M.L.; Fiorenzo, P.; Mongiardi, M.P.; Petrucci, G.; Montano, N.; Maira, G.; Pierconti, F.; Larocca, L.M.; Levi, A.; Pallini, R. Telomerase Inhibition Impairs Tumor Growth in Glioblastoma Xenografts. Neurol. Res. 2006, 28, 532–537. [Google Scholar] [CrossRef]
- Kondo, Y.; Kondo, S.; Tanaka, Y.; Haqqi, T.; Barna, B.P.; Cowell, J.K. Inhibition of Telomerase Increases the Susceptibility of Human Malignant Glioblastoma Cells to Cisplatin-Induced Apoptosis. Oncogene 1998, 16, 2243–2248. [Google Scholar] [CrossRef] [Green Version]
- Ogretmen, B.; Schady, D.; Usta, J.; Wood, R.; Kraveka, J.M.; Luberto, C.; Birbes, H.; Hannun, Y.A.; Obeid, L.M. Role of Ceramide in Mediating the Inhibition of Telomerase Activity in A549 Human Lung Adenocarcinoma Cells. J. Biol. Chem. 2001, 276, 24901–24910. [Google Scholar] [CrossRef]
- Cukusic, A.; Ivankovic, M.; Skrobot, N.; Ferenac, M.; Gotic, I.; Matijasic, M.; Polancec, D.; Rubelj, I. Spontaneous Senescence in the MDA-MB-231 Cell Line. Cell Prolif 2006, 39, 205–216. [Google Scholar] [CrossRef]
- Mach, N.; Gao, J.; Schaffarczyk, L.; Janz, S.; Ehrke-Schulz, E.; Dittmar, T.; Ehrhardt, A.; Zhang, W. Spectrum-Wide Exploration of Human Adenoviruses for Breast Cancer Therapy. Cancers 2020, 12, 1403. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.; Hazini, A.; Seymour, L.W. Tackling HLA Deficiencies Head on with Oncolytic Viruses. Cancers 2021, 13, 719. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, Z.; Tian, H.; Qi, M.; Zhai, Z.; Li, S.; Li, R.; Zhang, H.; Wang, W.; Fu, S.; et al. Biodistribution and Safety Assessment of Bladder Cancer Specific Recombinant Oncolytic Adenovirus in Subcutaneous Xenografts Tumor Model in Nude Mice. Curr. Gene Ther. 2012, 12, 67–76. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osipov, I.D.; Vasikhovskaia, V.A.; Zabelina, D.S.; Kutseikin, S.S.; Grazhdantseva, A.A.; Kochneva, G.V.; Davydova, J.; Netesov, S.V.; Romanenko, M.V. Development of Oncolytic Vectors Based on Human Adenovirus Type 6 for Cancer Treatment. Viruses 2023, 15, 182. https://doi.org/10.3390/v15010182
Osipov ID, Vasikhovskaia VA, Zabelina DS, Kutseikin SS, Grazhdantseva AA, Kochneva GV, Davydova J, Netesov SV, Romanenko MV. Development of Oncolytic Vectors Based on Human Adenovirus Type 6 for Cancer Treatment. Viruses. 2023; 15(1):182. https://doi.org/10.3390/v15010182
Chicago/Turabian StyleOsipov, Ivan D., Valeriia A. Vasikhovskaia, Daria S. Zabelina, Sergei S. Kutseikin, Antonina A. Grazhdantseva, Galina V. Kochneva, Julia Davydova, Sergey V. Netesov, and Margarita V. Romanenko. 2023. "Development of Oncolytic Vectors Based on Human Adenovirus Type 6 for Cancer Treatment" Viruses 15, no. 1: 182. https://doi.org/10.3390/v15010182