
SARS-CoV-2 Non-Structural Proteins and Their Roles in Host Immune Evasion

 , , and
, , and
Abstract
:1. Introduction
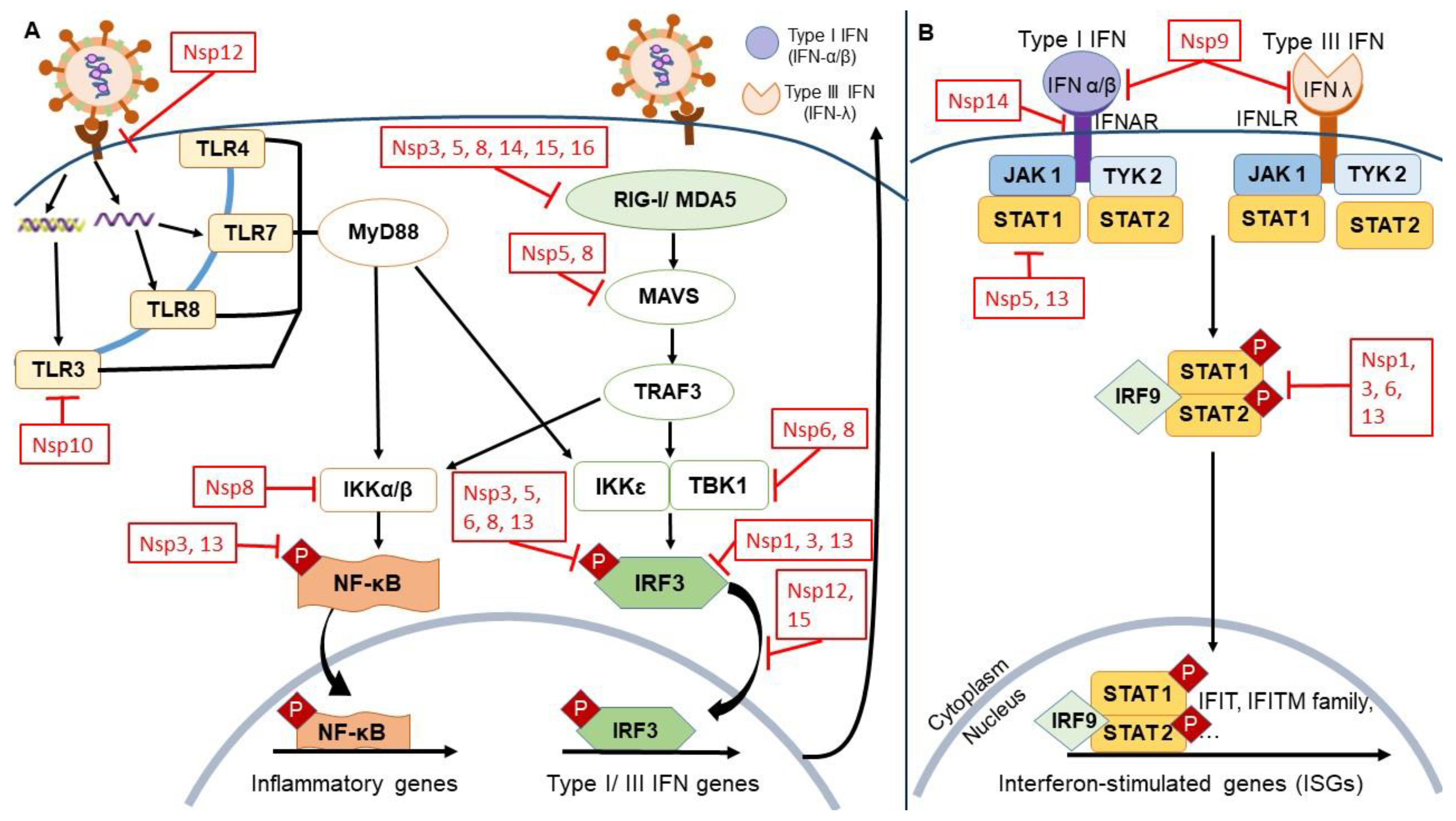
The Immune Signaling Pathways Associated with Viral Infection
2. SARS-CoV-2 NSPs and Their Multiple Mechanisms of Immune Escape
2.1. NSP1
2.2. NSP3
2.3. NSP5
2.4. NSP6
2.5. NSP7
2.6. NSP8
2.7. NSP9
2.8. NSP10
2.9. NSP12
2.10. NSP13
2.11. NSP14
2.12. NSP15
2.13. NSP16

{kind=link}
| NSP | Mode of Immune Escape | References |
|---|---|---|
| NSP1 |
| [30,32,33,34,35,37,38,39,40] |
| NSP3 |
| [51,54] |
| NSP5 |
| [67,68,69,73] |
| NSP6 |
| [42,43,73] |
| NSP7 |
| [43] |
| NSP8 |
| [43,95] |
| NSP9 |
| [33,103] |
| NSP10 |
| [120,125] |
| NSP12 |
| [43,137,138,139,141] |
| NSP13 |
| [151,153,154] |
| NSP14 |
| [158,161,162] |
| NSP15 |
| [131,139,178,179,180] |
| NSP16 |
| [33,192] |
| NSP | Drug Compound | Function | Ref. |
|---|---|---|---|
| NSP1 | Montelukast sodium hydrate |
| [44] |
| Mitoxantrone dihydrochloride |
| [45,46] | |
| NSP3 | GRL0617 |
| [57] |
| Sitagliptin |
| [60] | |
| Daclastavir |
| [60] | |
| NSP5 | Vinyl sulfone (2CN115) |
| [68] |
| Ivermectin |
| [74] | |
| NSP6 | 1α,25-dihydroxyvitamin D3 |
| [82] |
| Metformin |
| [82] | |
| Polydatin |
| [82] | |
| Dextromethorphan |
| [75] | |
| Haloperidol |
| [75] | |
| NSP7–12, NSP8–12 complex | Cepharanthine |
| [87,88] |
| Lonafarnib |
| [87,90] | |
| Nilotinib |
| [87,89] | |
| Filibuvir |
| [87,91] | |
| Olysio |
| [87,92] | |
| Saquinavir |
| [87,94] | |
| Tipranavir |
| [87,93] | |
| NSP9 | Fluspirilene |
| [110,196] |
| Troglitazone |
| [110,197] | |
| NSP10–16 complex | Tegobuvir |
| [198] |
| Siramesine |
| [199] | |
| Bemcentinib |
| [200,201] | |
| Sonidegib |
| [202] | |
| Itacitinib |
| [203] | |
| NSP13 | Cepharathine |
| [150,156] |
| NSP14 | Ritonavir |
| [163] |
| SAM analogs (DS0464, SS148) |
| [165,166] | |
| NSP15 | Epigallocatechin gallate, glycyrrhizic acid |
| [181,185] |
| NSP16 | Sinefungin |
| [118,204] |
| Maraviroc, raltegravir |
| [194,195] |
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Worldometer.info COVID-19 Coronavirus Pandemic. Available online: https://www.worldometers.info/coronavirus/ (accessed on 14 July 2022).
- Zheng, J. SARS-CoV-2: An Emerging Coronavirus That Causes a Global Threat. Int. J. Biol. Sci. 2020, 16, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Hui, D.S.; Perlman, S. Middle East Respiratory Syndrome. The Lancet 2015, 386, 995–1007. [Google Scholar] [CrossRef]
- Velavan, T.P.; Meyer, C.G. The COVID-19 Epidemic. Trop. Med. Int. Health 2020, 25, 278–280. [Google Scholar] [CrossRef]
- He, F.; Deng, Y.; Li, W. Coronavirus Disease 2019: What We Know? J. Med. Virol. 2020, 92, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The Proximal Origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Sharma, A.; Ahmad Farouk, I.; Lal, S.K. COVID-19: A Review on the Novel Coronavirus Disease Evolution, Transmission, Detection, Control and Prevention. Viruses 2021, 13, 202. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. In Coronaviruses; Maier, H.J., Bickerton, E., Britton, P., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; Volume 1282, pp. 1–23. ISBN 978-149-392-437-0. [Google Scholar]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 Genome, Structure, Evolution, Pathogenesis and Therapies: Structural Genomics Approach. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Low, Z.Y.; Yip, A.J.W.; Sharma, A.; Lal, S.K. SARS Coronavirus Outbreaks Past and Present—A Comparative Analysis of SARS-CoV-2 and Its Predecessors. Virus Genes 2021, 57, 307–317. [Google Scholar] [CrossRef]
- Astuti, I.; Ysrafil. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An Overview of Viral Structure and Host Response. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 407–412. [Google Scholar] [CrossRef]
- Boson, B.; Legros, V.; Zhou, B.; Siret, E.; Mathieu, C.; Cosset, F.-L.; Lavillette, D.; Denolly, S. The SARS-CoV-2 Envelope and Membrane Proteins Modulate Maturation and Retention of the Spike Protein, Allowing Assembly of Virus-like Particles. J. Biol. Chem. 2021, 296, 100111. [Google Scholar] [CrossRef] [PubMed]
- Surjit, M.; Lal, S.K. The SARS-CoV Nucleocapsid Protein: A Protein with Multifarious Activities. Infect. Genet. Evol. 2008, 8, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Low, Z.Y.; Wen Yip, A.J.; Chow, V.T.K.; Lal, S.K. The Suppressor of Cytokine Signalling Family of Proteins and Their Potential Impact on COVID-19 Disease Progression. Rev. Med. Virol. 2021, 32, e2300. [Google Scholar] [CrossRef] [PubMed]
- Yip, A.J.W.; Low, Z.Y.; Chow, V.T.K.; Lal, S.K. Repurposing Molnupiravir for COVID-19: The Mechanisms of Antiviral Activity. Viruses 2022, 14, 1345. [Google Scholar] [CrossRef] [PubMed]
- Low, Z.Y.; Farouk, I.A.; Lal, S.K. Drug Repositioning: New Approaches and Future Prospects for Life-Debilitating Diseases and the COVID-19 Pandemic Outbreak. Viruses 2020, 12, 1058. [Google Scholar] [CrossRef] [PubMed]
- Low, Z.Y.; Yip, A.J.W.; Lal, S.K. Repositioning Ivermectin for COVID-19 Treatment: Molecular Mechanisms of Action against SARS-CoV-2 Replication. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2022, 1868, 166294. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. COVID-19 Advice for the Public: Getting Vaccinated. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/covid-19-vaccines/advice (accessed on 10 August 2022).
- US Food and Drug Administration Comirnaty and Pfizer-BioNTech COVID-19 Vaccine. Available online: https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/comirnaty-and-pfizer-biontech-covid-19-vaccine (accessed on 10 August 2022).
- Centers for Disease Control and Prevention About Variants. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/delta-variant.html (accessed on 10 August 2022).
- Park, A.; Iwasaki, A. Type I and Type III Interferons—Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A. A Virological View of Innate Immune Recognition. Annu. Rev. Microbiol. 2012, 66, 177–196. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The Nuclear Factor NF-ΚB Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed]
- Platanitis, E.; Demiroz, D.; Schneller, A.; Fischer, K.; Capelle, C.; Hartl, M.; Gossenreiter, T.; Müller, M.; Novatchkova, M.; Decker, T. A Molecular Switch from STAT2-IRF9 to ISGF3 Underlies Interferon-Induced Gene Transcription. Nat. Commun. 2019, 10, 2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovanessian, A.G. On the Discovery of Interferon-Inducible, Double-Stranded RNA Activated Enzymes: The 2′–5′oligoadenylate Synthetases and the Protein Kinase PKR. Cytokine Growth Factor Rev. 2007, 18, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Mengist, H.M.; Kombe Kombe, A.J.; Mekonnen, D.; Abebaw, A.; Getachew, M.; Jin, T. Mutations of SARS-CoV-2 Spike Protein: Implications on Immune Evasion and Vaccine-Induced Immunity. Semin. Immunol. 2021, 55, 101533. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.S.; Johnson, M.A.; Herrmann, T.; Geralt, M.; Wüthrich, K. Novel β-Barrel Fold in the Nuclear Magnetic Resonance Structure of the Replicase Nonstructural Protein 1 from the Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2007, 81, 3151–3161. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.K.; Green, T.J.; Petit, C.M. Structure of Nonstructural Protein 1 from SARS-CoV-2. J. Virol. 2021, 95, e02019-20. [Google Scholar] [CrossRef] [PubMed]
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.-A.; Leibundgut, M.; Thiel, V.; Mühlemann, O.; Ban, N. SARS-CoV-2 Nsp1 Binds the Ribosomal MRNA Channel to Inhibit Translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Semper, C.; Watanabe, N.; Savchenko, A. Structural Characterization of Nonstructural Protein 1 from SARS-CoV-2. iScience 2021, 24, 101903. [Google Scholar] [CrossRef]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural Basis for Translational Shutdown and Immune Evasion by the Nsp1 Protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef]
- Banerjee, A.K.; Blanco, M.R.; Bruce, E.A.; Honson, D.D.; Chen, L.M.; Chow, A.; Bhat, P.; Ollikainen, N.; Quinodoz, S.A.; Loney, C.; et al. SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell 2020, 183, 1325–1339.e21. [Google Scholar] [CrossRef]
- Zhang, K.; Miorin, L.; Makio, T.; Dehghan, I.; Gao, S.; Xie, Y.; Zhong, H.; Esparza, M.; Kehrer, T.; Kumar, A.; et al. Nsp1 Protein of SARS-CoV-2 Disrupts the MRNA Export Machinery to Inhibit Host Gene Expression. Sci. Adv. 2021, 7, eabe7386. [Google Scholar] [CrossRef] [PubMed]
- Gaglia, M.M.; Covarrubias, S.; Wong, W.; Glaunsinger, B.A. A Common Strategy for Host RNA Degradation by Divergent Viruses. J. Virol. 2012, 86, 9527–9530. [Google Scholar] [CrossRef] [Green Version]
- Tidu, A.; Janvier, A.; Schaeffer, L.; Sosnowski, P.; Kuhn, L.; Hammann, P.; Westhof, E.; Eriani, G.; Martin, F. The Viral Protein Nsp1 Acts as a Ribosome Gatekeeper for Shutting down Host Translation and Fostering SARS-CoV-2 Translation. RNA 2021, 27, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Peng, L.; Park, J.J.; Hu, Y.; Devarkar, S.C.; Dong, M.B.; Shen, Q.; Wu, S.; Chen, S.; Lomakin, I.B.; et al. Nonstructural Protein 1 of SARS-CoV-2 Is a Potent Pathogenicity Factor Redirecting Host Protein Synthesis Machinery toward Viral RNA. Mol. Cell 2020, 80, 1055–1066.e6. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, C.P.; Grosely, R.; Johnson, A.G.; Wang, J.; Fernández, I.S.; Puglisi, J.D. Dynamic Competition between SARS-CoV-2 Nsp1 and MRNA on the Human Ribosome Inhibits Translation Initiation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017715118. [Google Scholar] [CrossRef] [PubMed]
- Wathelet, M.G.; Orr, M.; Frieman, M.B.; Baric, R.S. Severe Acute Respiratory Syndrome Coronavirus Evades Antiviral Signaling: Role of Nsp1 and Rational Design of an Attenuated Strain. J. Virol. 2007, 81, 11620–11633. [Google Scholar] [CrossRef]
- Tanaka, T.; Kamitani, W.; DeDiego, M.L.; Enjuanes, L.; Matsuura, Y. Severe Acute Respiratory Syndrome Coronavirus Nsp1 Facilitates Efficient Propagation in Cells through a Specific Translational Shutoff of Host MRNA. J. Virol. 2012, 86, 11128–11137. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and Evasion of Type I Interferon Responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.-C.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.-Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef]
- Afsar, M.; Narayan, R.; Akhtar, M.N.; Rahil, H.; Eswarappa, S.M.; Tripathi, S.; Hussain, T. Drug Targeting Nsp1-Ribosomal Complex Shows Antiviral Activity against SARS-CoV-2. eLife 2021, 11, e74877. [Google Scholar] [CrossRef]
- Kumar, P.; Bhardwaj, T.; Giri, R. Mitoxantrone Dihydrochloride, an FDA Approved Drug, Binds with SARS-CoV-2 Nsp1 C-Terminal. RSC Adv. 2022, 12, 5648–5655. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, C.Z.; Swaroop, M.; Xu, M.; Wang, L.; Lee, J.; Wang, A.Q.; Pradhan, M.; Hagen, N.; Chen, L.; et al. Heparan Sulfate Assists SARS-CoV-2 in Cell Entry and Can Be Targeted by Approved Drugs In Vitro. Cell Discov. 2020, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Neuman, B.W. Bioinformatics and Functional Analyses of Coronavirus Nonstructural Proteins Involved in the Formation of Replicative Organelles. Antivir. Res. 2016, 135, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39, 198–216. [Google Scholar] [CrossRef] [PubMed]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; El Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity Profiling and Crystal Structures of Inhibitor-Bound SARS-CoV-2 Papain-like Protease: A Framework for Anti–COVID-19 Drug Design. Sci. Adv. 2020, 6, eabd4596. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, M.; Kitade, Y.; Fayez, M.; Venugopala, K.N.; Ibrahim, A. The Emerging SARS-CoV-2 Papain-like Protease: Its Relationship with Recent Coronavirus Epidemics. J. Med. Virol. 2021, 93, 1581–1588. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like Protease Regulates SARS-CoV-2 Viral Spread and Innate Immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Frieman, M.; Ratia, K.; Johnston, R.E.; Mesecar, A.D.; Baric, R.S. Severe Acute Respiratory Syndrome Coronavirus Papain-like Protease Ubiquitin-like Domain and Catalytic Domain Regulate Antagonism of IRF3 and NF-ΚB Signalling. J. Virol. 2009, 83, 6689–6705. [Google Scholar] [CrossRef]
- Clementz, M.A.; Chen, Z.; Banach, B.S.; Wang, Y.; Sun, L.; Ratia, K.; Baez-Santos, Y.M.; Wang, J.; Takayama, J.; Ghosh, A.K.; et al. Deubiquitinating and Interferon Antagonism Activities of Coronavirus Papain-like Proteases. J. Virol. 2010, 84, 4619–4629. [Google Scholar] [CrossRef]
- Malakhova, O.A.; Zhang, D.-E. ISG15 Inhibits Nedd4 Ubiquitin E3 Activity and Enhances the Innate Antiviral Response. J. Biol. Chem. 2008, 283, 8783–8787. [Google Scholar] [CrossRef]
- Durfee, L.A.; Lyon, N.; Seo, K.; Huibregtse, J.M. The ISG15 Conjugation System Broadly Targets Newly Synthesized Proteins: Implications for the Antiviral Function of ISG15. Mol. Cell 2010, 38, 722–732. [Google Scholar] [CrossRef]
- Woods, M.W.; Kelly, J.N.; Hattlmann, C.J.; Tong, J.G.; Xu, L.S.; Coleman, M.D.; Quest, G.R.; Smiley, J.R.; Barr, S.D. Human HERC5 Restricts an Early Stage of HIV-1 Assembly by a Mechanism Correlating with the ISGylation of Gag. Retrovirology 2011, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Cao, D.; Li, J.; et al. Structural Basis for the Inhibition of the Papain-like Protease of SARS-CoV-2 by Small Molecules. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like Receptors: Their Regulation and Roles in RNA Sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Liu, G.; Lee, J.-H.; Parker, Z.M.; Acharya, D.; Chiang, J.J.; van Gent, M.; Riedl, W.; Davis-Gardner, M.E.; Wies, E.; Chiang, C.; et al. ISG15-Dependent Activation of the Sensor MDA5 Is Antagonized by the SARS-CoV-2 Papain-like Protease to Evade Host Innate Immunity. Nat. Microbiol. 2021, 6, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.S.; Jose, J. Identification of SARS-CoV-2 Inhibitors Targeting Mpro and PLpro Using in-Cell-Protease Assay. Commun. Biol. 2022, 5, 169. [Google Scholar] [CrossRef]
- Roe, M.K.; Junod, N.A.; Young, A.R.; Beachboard, D.C.; Stobart, C.C. Targeting Novel Structural and Functional Features of Coronavirus Protease Nsp5 (3CLpro, Mpro) in the Age of COVID-19. J. Gen. Virol. 2021, 102, 1558. [Google Scholar] [CrossRef]
- Stobart, C.C.; Sexton, N.R.; Munjal, H.; Lu, X.; Molland, K.L.; Tomar, S.; Mesecar, A.D.; Denison, M.R. Chimeric Exchange of Coronavirus Nsp5 Proteases (3CLpro) Identifies Common and Divergent Regulatory Determinants of Protease Activity. J. Virol. 2013, 87, 12611–12618. [Google Scholar] [CrossRef]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 2021, 9, 622898. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Shi, J.; Song, J. The Catalysis of the SARS 3C-like Protease Is under Extensive Regulation by Its Extra Domain. FEBS J. 2006, 273, 1035–1045. [Google Scholar] [CrossRef]
- Shi, J.; Sivaraman, J.; Song, J. Mechanism for Controlling the Dimer-Monomer Switch and Coupling Dimerization to Catalysis of the Severe Acute Respiratory Syndrome Coronavirus 3C-like Protease. J. Virol. 2008, 82, 4620–4629. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ma, L.; Zhuang, Z.; Cai, S.; Zhao, Z.; Zhou, L.; Zhang, J.; Wang, P.-H.; Zhao, J.; Cui, J. Main Protease of SARS-CoV-2 Serves as a Bifunctional Molecule in Restricting Type I Interferon Antiviral Signaling. Signal Transduct. Target. Ther. 2020, 5, 221. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qin, C.; Rao, Y.; Ngo, C.; Feng, J.J.; Zhao, J.; Zhang, S.; Wang, T.-Y.; Carriere, J.; Savas, A.C.; et al. SARS-CoV-2 Nsp5 Demonstrates Two Distinct Mechanisms Targeting RIG-I and MAVS To Evade the Innate Immune Response. mBio 2021, 12, e02335-21. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Sun, L.; Jiang, X.; Chen, X.; Hou, F.; Adhikari, A.; Xu, M.; Chen, Z.J. Reconstitution of the RIG-I Pathway Reveals a Signaling Role of Unanchored Polyubiquitin Chains in Innate Immunity. Cell 2010, 141, 315–330. [Google Scholar] [CrossRef]
- Shemesh, M.; Aktepe, T.E.; Deerain, J.M.; McAuley, J.L.; Audsley, M.D.; David, C.T.; Purcell, D.F.J.; Urin, V.; Hartmann, R.; Moseley, G.W.; et al. SARS-CoV-2 Suppresses IFNβ Production Mediated by Nsp1, 5, 6, 15, ORF6 and ORF7b but Does Not Suppress the Effects of Added Interferon. PLoS Pathog. 2021, 17, e1009800. [Google Scholar] [CrossRef]
- Li, W.; Qiao, J.; You, Q.; Zong, S.; Peng, Q.; Liu, Y.; Hu, S.; Liu, W.; Li, S.; Shu, X.; et al. SARS-CoV-2 Nsp5 Activates NF-ΚB Pathway by Upregulating SUMOylation of MAVS. Front. Immunol. 2021, 12, 750969. [Google Scholar] [CrossRef]
- Bensussen, A.; Álvarez-Buylla, E.R.; Díaz, J. SARS-CoV-2 Nsp5 Protein Causes Acute Lung Inflammation, a Dynamical Mathematical Model. Front. Syst. Biol. 2021, 1, 764155. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.C.J.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-Chymotrypsin like Protease (3CLPro) Inhibitors as Potential Anti-SARS-CoV-2 Agents. Commun. Biol. 2021, 4, 93. [Google Scholar] [CrossRef]
- Pandey, P.; Prasad, K.; Prakash, A.; Kumar, V. Insights into the Biased Activity of Dextromethorphan and Haloperidol towards SARS-CoV-2 NSP6: In Silico Binding Mechanistic Analysis. J. Mol. Med. 2020, 98, 1659–1673. [Google Scholar] [CrossRef]
- Thomas, S. Mapping the Nonstructural Transmembrane Proteins of Severe Acute Respiratory Syndrome Coronavirus 2. J. Comput. Biol. 2021, 28, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe Acute Respiratory Syndrome Coronavirus Nonstructural Proteins 3, 4, and 6 Induce Double-Membrane Vesicles. mBio 2013, 4, e00524-13. [Google Scholar] [CrossRef] [PubMed]
- Van der Hoeven, B.; Oudshoorn, D.; Koster, A.J.; Snijder, E.J.; Kikkert, M.; Bárcena, M. Biogenesis and Architecture of Arterivirus Replication Organelles. Virus Res. 2016, 220, 70–90. [Google Scholar] [CrossRef] [PubMed]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus Nsp6 Proteins Generate Autophagosomes from the Endoplasmic Reticulum via an Omegasome Intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of Innate Immune Adaptor Proteins MAVS, STING, and TRIF Induces IRF3 Activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef]
- Onoguchi, K.; Yoneyama, M.; Takemura, A.; Akira, S.; Taniguchi, T.; Namiki, H.; Fujita, T. Viral Infections Activate Types I and III Interferon Genes through a Common Mechanism. J. Biol. Chem. 2007, 282, 7576–7581. [Google Scholar] [CrossRef]
- Sun, X.; Liu, Y.; Huang, Z.; Xu, W.; Hu, W.; Yi, L.; Liu, Z.; Chan, H.; Zeng, J.; Liu, X.; et al. SARS-CoV-2 Non-Structural Protein 6 Triggers NLRP3-Dependent Pyroptosis by Targeting ATP6AP1. Cell Death Differ. 2022, 29, 1240–1254. [Google Scholar] [CrossRef]
- Courouble, V.V.; Dey, S.K.; Yadav, R.; Timm, J.; Harrison, J.J.E.K.; Ruiz, F.X.; Arnold, E.; Griffin, P.R. Revealing the Structural Plasticity of SARS-CoV-2 Nsp7 and Nsp8 Using Structural Proteomics. J. Am. Soc. Mass Spectrom. 2021, 32, 1618–1630. [Google Scholar] [CrossRef]
- Wilamowski, M.; Hammel, M.; Leite, W.; Zhang, Q.; Kim, Y.; Weiss, K.L.; Jedrzejczak, R.; Rosenberg, D.J.; Fan, Y.; Wower, J.; et al. Transient and Stabilized Complexes of Nsp7, Nsp8, and Nsp12 in SARS-CoV-2 Replication. Biophys. J. 2021, 120, 3152–3165. [Google Scholar] [CrossRef]
- Biswal, M.; Diggs, S.; Xu, D.; Khudaverdyan, N.; Lu, J.; Fang, J.; Blaha, G.; Hai, R.; Song, J. Two Conserved Oligomer Interfaces of NSP7 and NSP8 Underpin the Dynamic Assembly of SARS-CoV-2 RdRP. Nucleic Acids Res. 2021, 49, 5956–5966. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV Nsp12 Polymerase Bound to Nsp7 and Nsp8 Co-Factors. Nat. Commun. 2019, 10, 2342. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Z.; Liu, C.; Guo, Y.; He, Z.; Huang, X.; Jia, X.; Yang, T. SARS-CoV-2 and SARS-CoV: Virtual Screening of Potential Inhibitors Targeting RNA-dependent RNA Polymerase Activity (Nsp12). J. Med. Virol. 2021, 93, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Rogosnitzky, M.; Okediji, P.; Koman, I. Cepharanthine: A Review of the Antiviral Potential of a Japanese-Approved Alopecia Drug in COVID-19. Pharmacol. Rep. 2020, 72, 1509–1516. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Sugiyama, M.; Akiyama, Y.; Ando, Y.; Sasaki, Y. The Small-Molecule Tyrosine Kinase Inhibitor Nilotinib Is a Potent Noncompetitive Inhibitor of the SN-38 Glucuronidation by Human UGT1A1. Cancer Chemother. Pharmacol. 2011, 67, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Shappell, H.; Massaro, J.; D’Agostino, R.B.; Brazier, J.; Campbell, S.E.; Kleinman, M.E.; Kieran, M.W. Association of Lonafarnib Treatment vs. No Treatment with Mortality Rate in Patients with Hutchinson-Gilford Progeria Syndrome. JAMA 2018, 319, 1687. [Google Scholar] [CrossRef]
- Beaulieu, P.L. Filibuvir, a Non-Nucleoside NS5B Polymerase Inhibitor for the Potential Oral Treatment of Chronic HCV Infection. IDrugs Investig. Drugs J. 2010, 13, 938–948. [Google Scholar]
- Meewan, I.; Zhang, X.; Roy, S.; Ballatore, C.; O’Donoghue, A.J.; Schooley, R.T.; Abagyan, R. Discovery of New Inhibitors of Hepatitis C Virus NS3/4A Protease and Its D168A Mutant. ACS Omega 2019, 4, 16999–17008. [Google Scholar] [CrossRef]
- Mehandru, S.; Markowitz, M. Tipranavir: A Novel Non-Peptidic Protease Inhibitor for the Treatment of HIV Infection. Expert Opin. Investig. Drugs 2003, 12, 1821–1828. [Google Scholar] [CrossRef]
- Bandiera, E.; Todeschini, P.; Romani, C.; Zanotti, L.; Erba, E.; Colmegna, B.; Bignotti, E.; Santin, A.D.; Sartori, E.; Odicino, F.E.; et al. The HIV-Protease Inhibitor Saquinavir Reduces Proliferation, Invasion and Clonogenicity in Cervical Cancer Cell Lines. Oncol. Lett. 2016, 12, 2493–2500. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, X.; Wang, F.; Wang, P.; Kuang, E.; Li, X. Suppression of MDA5-Mediated Antiviral Immune Responses by NSP8 of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Egloff, M.-P.; Ferron, F.; Campanacci, V.; Longhi, S.; Rancurel, C.; Dutartre, H.; Snijder, E.J.; Gorbalenya, A.E.; Cambillau, C.; Canard, B. The Severe Acute Respiratory Syndrome-Coronavirus Replicative Protein Nsp9 Is a Single-Stranded RNA-Binding Subunit Unique in the RNA Virus World. Proc. Natl. Acad. Sci. USA 2004, 101, 3792–3796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutton, G.; Fry, E.; Carter, L.; Sainsbury, S.; Walter, T.; Nettleship, J.; Berrow, N.; Owens, R.; Gilbert, R.; Davidson, A.; et al. The Nsp9 Replicase Protein of SARS-Coronavirus, Structure and Functional Insights. Structure 2004, 12, 341–353. [Google Scholar] [CrossRef]
- El-Kamand, S.; Du Plessis, M.; Breen, N.; Johnson, L.; Beard, S.; Kwan, A.H.; Richard, D.J.; Cubeddu, L.; Gamsjaeger, R. A Distinct ssDNA/RNA Binding Interface in the Nsp9 Protein from SARS-CoV-2. Proteins Struct. Funct. Bioinform. 2022, 90, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Littler, D.R.; Gully, B.S.; Colson, R.N.; Rossjohn, J. Crystal Structure of the SARS-CoV-2 Non-Structural Protein 9, Nsp9. iScience 2020, 23, 101258. [Google Scholar] [CrossRef] [PubMed]
- Buchko, G.W.; Zhou, M.; Craig, J.K.; Van Voorhis, W.C.; Myler, P.J. Backbone Chemical Shift Assignments for the SARS-CoV-2 Non-Structural Protein Nsp9: Intermediate (Ms–Μs) Dynamics in the C-Terminal Helix at the Dimer Interface. Biomol. NMR Assign. 2021, 15, 107–116. [Google Scholar] [CrossRef]
- Slanina, H.; Madhugiri, R.; Bylapudi, G.; Schultheiß, K.; Karl, N.; Gulyaeva, A.; Gorbalenya, A.E.; Linne, U.; Ziebuhr, J. Coronavirus Replication–Transcription Complex: Vital and Selective NMPylation of a Conserved Site in Nsp9 by the NiRAN-RdRp Subunit. Proc. Natl. Acad. Sci. USA 2021, 118, e2022310118. [Google Scholar] [CrossRef]
- Yan, L.; Ge, J.; Zheng, L.; Zhang, Y.; Gao, Y.; Wang, T.; Huang, Y.; Yang, Y.; Gao, S.; Li, M.; et al. Cryo-EM Structure of an Extended SARS-CoV-2 Replication and Transcription Complex Reveals an Intermediate State in Cap Synthesis. Cell 2021, 184, 184–193.e10. [Google Scholar] [CrossRef]
- Makiyama, K.; Hazawa, M.; Kobayashi, A.; Lim, K.; Voon, D.C.; Wong, R.W. Nsp9 of SARS-CoV-2 Attenuates Nuclear Transport by Hampering Nucleoporin 62 Dynamics and Functions in Host Cells. Biochem. Biophys. Res. Commun. 2022, 586, 137–142. [Google Scholar] [CrossRef]
- Deming, D.J.; Graham, R.L.; Denison, M.R.; Baric, R.S. Processing of Open Reading Frame 1a Replicase Proteins Nsp7 to Nsp10 in Murine Hepatitis Virus Strain A59 Replication. J. Virol. 2007, 81, 10280–10291. [Google Scholar] [CrossRef]
- Parida, P.K.; Paul, D.; Chakravorty, D. Nature’s Therapy for COVID-19: Targeting the Vital Non-Structural Proteins (Nsp) from SARS-CoV-2 with Phytochemicals from Indian Medicinal Plants. Phytomedicine Plus 2021, 1, 100002. [Google Scholar] [CrossRef]
- Wu, S.; Tian, C.; Liu, P.; Guo, D.; Zheng, W.; Huang, X.; Zhang, Y.; Liu, L. Effects of SARS-CoV-2 Mutations on Protein Structures and Intraviral Protein–Protein Interactions. J. Med. Virol. 2021, 93, 2132–2140. [Google Scholar] [CrossRef] [PubMed]
- Miknis, Z.J.; Donaldson, E.F.; Umland, T.C.; Rimmer, R.A.; Baric, R.S.; Schultz, L.W. Severe Acute Respiratory Syndrome Coronavirus Nsp9 Dimerization Is Essential for Efficient Viral Growth. J. Virol. 2009, 83, 3007–3018. [Google Scholar] [CrossRef] [PubMed]
- Chandel, V.; Sharma, P.P.; Raj, S.; Choudhari, R.; Rathi, B.; Kumar, D. Structure-Based Drug Repurposing for Targeting Nsp9 Replicase and Spike Proteins of Severe Acute Respiratory Syndrome Coronavirus 2. J. Biomol. Struct. Dyn. 2022, 40, 249–262. [Google Scholar] [CrossRef]
- Khan, M.T.; Irfan, M.; Ahsan, H.; Ahmed, A.; Kaushik, A.C.; Khan, A.S.; Chinnasamy, S.; Ali, A.; Wei, D.-Q. Structures of SARS-CoV-2 RNA-Binding Proteins and Therapeutic Targets. Intervirology 2021, 64, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Gurjar, V.; Ahmed, M.Z.; Alqahtani, A.S.; Qamar, I.; Singh, N. Exploring Potential Inhibitor of SARS-CoV2 Replicase from FDA Approved Drugs Using In Silico Drug Discovery Methods. J. Biomol. Struct. Dyn. 2021, 40, 5507–5514. [Google Scholar] [CrossRef]
- Bouvet, M.; Lugari, A.; Posthuma, C.C.; Zevenhoven, J.C.; Bernard, S.; Betzi, S.; Imbert, I.; Canard, B.; Guillemot, J.-C.; Lécine, P.; et al. Coronavirus Nsp10, a Critical Co-Factor for Activation of Multiple Replicative Enzymes. J. Biol. Chem. 2014, 289, 25783–25796. [Google Scholar] [CrossRef]
- Joseph, J.S.; Saikatendu, K.S.; Subramanian, V.; Neuman, B.W.; Brooun, A.; Griffith, M.; Moy, K.; Yadav, M.K.; Velasquez, J.; Buchmeier, M.J.; et al. Crystal Structure of Nonstructural Protein 10 from the Severe Acute Respiratory Syndrome Coronavirus Reveals a Novel Fold with Two Zinc-Binding Motifs. J. Virol. 2006, 80, 7894–7901. [Google Scholar] [CrossRef]
- Su, D.; Lou, Z.; Sun, F.; Zhai, Y.; Yang, H.; Zhang, R.; Joachimiak, A.; Zhang, X.C.; Bartlam, M.; Rao, Z. Dodecamer Structure of Severe Acute Respiratory Syndrome Coronavirus Nonstructural Protein Nsp10. J. Virol. 2006, 80, 7902–7908. [Google Scholar] [CrossRef]
- Rogstam, A.; Nyblom, M.; Christensen, S.; Sele, C.; Talibov, V.O.; Lindvall, T.; Rasmussen, A.A.; André, I.; Fisher, Z.; Knecht, W.; et al. Crystal Structure of Non-Structural Protein 10 from Severe Acute Respiratory Syndrome Coronavirus-2. Int. J. Mol. Sci. 2020, 21, 7375. [Google Scholar] [CrossRef]
- Horova, V.; Landova, B.; Hodek, J.; Chalupsky, K.; Krafcikova, P.; Chalupska, D.; Duchoslav, V.; Weber, J.; Boura, E.; Klima, M. Localization of SARS-CoV-2 Capping Enzymes Revealed by an Antibody against the Nsp10 Subunit. Viruses 2021, 13, 1487. [Google Scholar] [CrossRef]
- Jiang, Y.; Tong, K.; Yao, R.; Zhou, Y.; Lin, H.; Du, L.; Jin, Y.; Cao, L.; Tan, J.; Zhang, X.-D.; et al. Genome-Wide Analysis of Protein–Protein Interactions and Involvement of Viral Proteins in SARS-CoV-2 Replication. Cell Biosci. 2021, 11, 140. [Google Scholar] [CrossRef] [PubMed]
- Springstein, B.L.; Deighan, P.; Grabe, G.J.; Hochschild, A. A Bacterial Cell-Based Assay to Study SARS-CoV-2 Protein-Protein Interactions. mBio 2021, 12, e02936-21. [Google Scholar] [CrossRef] [PubMed]
- Krafcikova, P.; Silhan, J.; Nencka, R.; Boura, E. Structural Analysis of the SARS-CoV-2 Methyltransferase Complex Involved in RNA Cap Creation Bound to Sinefungin. Nat. Commun. 2020, 11, 3717. [Google Scholar] [CrossRef] [PubMed]
- Vithani, N.; Ward, M.D.; Zimmerman, M.I.; Novak, B.; Borowsky, J.H.; Singh, S.; Bowman, G.R. SARS-CoV-2 Nsp16 Activation Mechanism and a Cryptic Pocket with Pan-Coronavirus Antiviral Potential. Biophys. J. 2021, 120, 2880–2889. [Google Scholar] [CrossRef] [PubMed]
- Decroly, E.; Debarnot, C.; Ferron, F.; Bouvet, M.; Coutard, B.; Imbert, I.; Gluais, L.; Papageorgiou, N.; Sharff, A.; Bricogne, G.; et al. Crystal Structure and Functional Analysis of the SARS-Coronavirus RNA Cap 2′-O-Methyltransferase Nsp10/Nsp16 Complex. PLoS Pathog. 2011, 7, e1002059. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Zhou, Z.-J.; Wang, Q.; He, Q.-N.; Zhao, M.-Y.; Qiu, Y.; Ge, X.-Y. Innate Immunity Evasion Strategies of Highly Pathogenic Coronaviruses: SARS-CoV, MERS-CoV, and SARS-CoV-2. Front. Microbiol. 2021, 12, 770656. [Google Scholar] [CrossRef] [PubMed]
- Gorkhali, R.; Koirala, P.; Rijal, S.; Mainali, A.; Baral, A.; Bhattarai, H.K. Structure and Function of Major SARS-CoV-2 and SARS-CoV Proteins. Bioinform. Biol. Insights 2021, 15, 117793222110258. [Google Scholar] [CrossRef]
- Lin, S.; Chen, H.; Chen, Z.; Yang, F.; Ye, F.; Zheng, Y.; Yang, J.; Lin, X.; Sun, H.; Wang, L.; et al. Crystal Structure of SARS-CoV-2 Nsp10 Bound to Nsp14-ExoN Domain Reveals an Exoribonuclease with Both Structural and Functional Integrity. Nucleic Acids Res. 2021, 49, 5382–5392. [Google Scholar] [CrossRef]
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural Basis and Functional Analysis of the SARS Coronavirus Nsp14–Nsp10 Complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef]
- Choudhury, A.; Das, N.C.; Patra, R.; Mukherjee, S. In Silico Analyses on the Comparative Sensing of SARS-CoV-2 MRNA by the Intracellular TLRs of Humans. J. Med. Virol. 2021, 93, 2476–2486. [Google Scholar] [CrossRef]
- Rosas-Lemus, M.; Minasov, G.; Shuvalova, L.; Inniss, N.L.; Kiryukhina, O.; Brunzelle, J.; Satchell, K.J.F. High-Resolution Structures of the SARS-CoV-2 2′-O-Methyltransferase Reveal Strategies for Structure-Based Inhibitor Design. Sci. Signal. 2020, 13, eabe1202. [Google Scholar] [CrossRef] [PubMed]
- Encinar, J.A.; Menendez, J.A. Potential Drugs Targeting Early Innate Immune Evasion of SARS-Coronavirus 2 via 2′-O-Methylation of Viral RNA. Viruses 2020, 12, 525. [Google Scholar] [CrossRef] [PubMed]
- Rona, G.; Zeke, A.; Miwatani-Minter, B.; de Vries, M.; Kaur, R.; Schinlever, A.; Garcia, S.F.; Goldberg, H.V.; Wang, H.; Hinds, T.R.; et al. The NSP14/NSP10 RNA Repair Complex as a Pan-Coronavirus Therapeutic Target. Cell Death Differ. 2022, 29, 285–292. [Google Scholar] [CrossRef]
- Desimmie, B.A.; Demeulemeester, J.; Suchaud, V.; Taltynov, O.; Billamboz, M.; Lion, C.; Bailly, F.; Strelkov, S.V.; Debyser, Z.; Cotelle, P.; et al. 2-Hydroxyisoquinoline-1,3(2H,4H)-Diones (HIDs), Novel Inhibitors of HIV Integrase with a High Barrier to Resistance. ACS Chem. Biol. 2013, 8, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Amor, S.; Fernández Blanco, L.; Baker, D. Innate Immunity during SARS-CoV-2: Evasion Strategies and Activation Trigger Hypoxia and Vascular Damage. Clin. Exp. Immunol. 2020, 202, 193–209. [Google Scholar] [CrossRef]
- Suryawanshi, R.K.; Koganti, R.; Agelidis, A.; Patil, C.D.; Shukla, D. Dysregulation of Cell Signaling by SARS-CoV-2. Trends Microbiol. 2021, 29, 224–237. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-Dependent RNA Polymerase from COVID-19 Virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef]
- Hartenian, E.; Nandakumar, D.; Lari, A.; Ly, M.; Tucker, J.M.; Glaunsinger, B.A. The Molecular Virology of Coronaviruses. J. Biol. Chem. 2020, 295, 12910–12934. [Google Scholar] [CrossRef]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of Replicating SARS-CoV-2 Polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Shannon, A.; Le, N.T.-T.; Selisko, B.; Eydoux, C.; Alvarez, K.; Guillemot, J.-C.; Decroly, E.; Peersen, O.; Ferron, F.; Canard, B. Remdesivir and SARS-CoV-2: Structural Requirements at Both Nsp12 RdRp and Nsp14 Exonuclease Active-Sites. Antivir. Res. 2020, 178, 104793. [Google Scholar] [CrossRef]
- Peng, Q.; Peng, R.; Yuan, B.; Zhao, J.; Wang, M.; Wang, X.; Wang, Q.; Sun, Y.; Fan, Z.; Qi, J.; et al. Structural and Biochemical Characterization of the Nsp12-Nsp7-Nsp8 Core Polymerase Complex from SARS-CoV-2. Cell Rep. 2020, 31, 107774. [Google Scholar] [CrossRef]
- Li, J.-Y.; Liao, C.-H.; Wang, Q.; Tan, Y.-J.; Luo, R.; Qiu, Y.; Ge, X.-Y. The ORF6, ORF8 and Nucleocapsid Proteins of SARS-CoV-2 Inhibit Type I Interferon Signaling Pathway. Virus Res. 2020, 286, 198074. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, Z.; Xiao, X.; Tian, Z.; Dong, X.; Wang, C.; Li, L.; Ren, L.; Lei, X.; Xiang, Z.; et al. SARS-CoV-2 Nsp12 Attenuates Type I Interferon Production by Inhibiting IRF3 Nuclear Translocation. Cell. Mol. Immunol. 2021, 18, 945–953. [Google Scholar] [CrossRef]
- Yuen, C.-K.; Lam, J.-Y.; Wong, W.-M.; Mak, L.-F.; Wang, X.; Chu, H.; Cai, J.-P.; Jin, D.-Y.; To, K.K.-W.; Chan, J.F.-W.; et al. SARS-CoV-2 Nsp13, Nsp14, Nsp15 and Orf6 Function as Potent Interferon Antagonists. Emerg. Microbes Infect. 2020, 9, 1418–1428. [Google Scholar] [CrossRef]
- Li, A.; Zhao, K.; Zhang, B.; Hua, R.; Fang, Y.; Jiang, W.; Zhang, J.; Hui, L.; Zheng, Y.; Li, Y.; et al. SARS-CoV-2 Nsp12 Protein Is Not an Interferon-β Antagonist. J. Virol. 2021, 95, e00747-21. [Google Scholar] [CrossRef]
- Xu, G.; Li, Y.; Zhang, S.; Peng, H.; Wang, Y.; Li, D.; Jin, T.; He, Z.; Tong, Y.; Qi, C.; et al. SARS-CoV-2 Promotes RIPK1 Activation to Facilitate Viral Propagation. Cell Res. 2021, 31, 1230–1243. [Google Scholar] [CrossRef]
- Cheng, L.; Zhang, X.; Chen, Y.; Wang, D.; Zhang, D.; Yan, S.; Wang, H.; Xiao, M.; Liang, T.; Li, H.; et al. Dynamic Landscape Mapping of Humoral Immunity to SARS-CoV-2 Identifies Non-Structural Protein Antibodies Associated with the Survival of Critical COVID-19 Patients. Signal Transduct. Target. Ther. 2021, 6, 304. [Google Scholar] [CrossRef]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Correa Marrero, M.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182, 685–712.e19. [Google Scholar] [CrossRef]
- Te Velthuis, A.J.W.; Fodor, E. Influenza Virus RNA Polymerase: Insights into the Mechanisms of Viral RNA Synthesis. Nat. Rev. Microbiol. 2016, 14, 479–493. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, J.; Wang, H.; Gao, Y.; Liu, Q.; Mu, A.; Ji, W.; Yan, L.; Zhu, Y.; Zhu, C.; et al. Structural Basis for RNA Replication by the SARS-CoV-2 Polymerase. Cell 2020, 182, 417–428.e13. [Google Scholar] [CrossRef]
- Sahin, E.; Bozdayi, G.; Yigit, S.; Muftah, H.; Dizbay, M.; Tunccan, O.G.; Fidan, I.; Caglar, K. Genomic Characterization of SARS-CoV-2 Isolates from Patients in Turkey Reveals the Presence of Novel Mutations in Spike and Nsp12 Proteins. J. Med. Virol. 2021, 93, 6016–6026. [Google Scholar] [CrossRef]
- Narayanan, N.; Nair, D.T. Vitamin B12 May Inhibit RNA-Dependent-RNA Polymerase Activity of Nsp12 from the SARS-CoV-2 Virus. IUBMB Life 2020, 72, 2112–2120. [Google Scholar] [CrossRef]
- Mirza, M.U.; Froeyen, M. Structural Elucidation of SARS-CoV-2 Vital Proteins: Computational Methods Reveal Potential Drug Candidates against Main Protease, Nsp12 Polymerase and Nsp13 Helicase. J. Pharm. Anal. 2020, 10, 320–328. [Google Scholar] [CrossRef]
- Newman, J.A.; Douangamath, A.; Yadzani, S.; Yosaatmadja, Y.; Aimon, A.; Brandão-Neto, J.; Dunnett, L.; Gorrie-stone, T.; Skyner, R.; Fearon, D.; et al. Structure, Mechanism and Crystallographic Fragment Screening of the SARS-CoV-2 Nsp13 Helicase. Nat. Commun. 2021, 12, 4848. [Google Scholar] [CrossRef]
- White, M.A.; Lin, W.; Cheng, X. Discovery of COVID-19 Inhibitors Targeting the SARS-CoV-2 Nsp13 Helicase. J. Phys. Chem. Lett. 2020, 11, 9144–9151. [Google Scholar] [CrossRef]
- Jia, Z.; Yan, L.; Ren, Z.; Wu, L.; Wang, J.; Guo, J.; Zheng, L.; Ming, Z.; Zhang, L.; Lou, Z.; et al. Delicate Structural Coordination of the Severe Acute Respiratory Syndrome Coronavirus Nsp13 upon ATP Hydrolysis. Nucleic Acids Res. 2019, 47, 6538–6550. [Google Scholar] [CrossRef]
- Habtemariam, S.; Nabavi, S.F.; Banach, M.; Berindan-Neagoe, I.; Sarkar, K.; Sil, P.C.; Nabavi, S.M. Should We Try SARS-CoV-2 Helicase Inhibitors for COVID-19 Therapy? Arch. Med. Res. 2020, 51, 733–735. [Google Scholar] [CrossRef]
- Vazquez, C.; Swanson, S.E.; Negatu, S.G.; Dittmar, M.; Miller, J.; Ramage, H.R.; Cherry, S.; Jurado, K.A. SARS-CoV-2 Viral Proteins NSP1 and NSP13 Inhibit Interferon Activation through Distinct Mechanisms. PLoS ONE 2021, 16, e0253089. [Google Scholar] [CrossRef]
- Gordon, D.E.; Hiatt, J.; Bouhaddou, M.; Rezelj, V.V.; Ulferts, S.; Braberg, H.; Jureka, A.S.; Obernier, K.; Guo, J.Z.; Batra, J.; et al. Comparative Host-Coronavirus Protein Interaction Networks Reveal Pan-Viral Disease Mechanisms. Science 2020, 370, eabe9403. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Y.; Liu, X.; Lu, J.; Qian, C.; Wan, Z.; Yan, X.; Zheng, H.; Zhang, M.; Xiong, S.; et al. Antiviral Activity of Cepharanthine against Severe Acute Respiratory Syndrome Coronavirus In Vitro. Chin. Med. J. 2005, 118, 493–496. [Google Scholar] [CrossRef]
- Fan, H.-H.; Wang, L.-Q.; Liu, W.-L.; An, X.-P.; Liu, Z.-D.; He, X.-Q.; Song, L.-H.; Tong, Y.-G. Repurposing of Clinically Approved Drugs for Treatment of Coronavirus Disease 2019 in a 2019-Novel Coronavirus-Related Coronavirus Model. Chin. Med. J. 2020, 133, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Briguglio, I.; Piras, S.; Corona, P.; Carta, A. Inhibition of RNA Helicases of SsRNA+ Virus Belonging to Flaviviridae, Coronaviridae and Picornaviridae Families. Int. J. Med. Chem. 2011, 2011, 1–22. [Google Scholar] [CrossRef]
- Hsu, J.C.-C.; Laurent-Rolle, M.; Pawlak, J.B.; Wilen, C.B.; Cresswell, P. Translational Shutdown and Evasion of the Innate Immune Response by SARS-CoV-2 NSP14 Protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2101161118. [Google Scholar] [CrossRef]
- Minskaia, E.; Hertzig, T.; Gorbalenya, A.E.; Campanacci, V.; Cambillau, C.; Canard, B.; Ziebuhr, J. Discovery of an RNA Virus 3′→5′ Exoribonuclease That Is Critically Involved in Coronavirus RNA Synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5108–5113. [Google Scholar] [CrossRef]
- Bouvet, M.; Imbert, I.; Subissi, L.; Gluais, L.; Canard, B.; Decroly, E. RNA 3′-End Mismatch Excision by the Severe Acute Respiratory Syndrome Coronavirus Nonstructural Protein Nsp10/Nsp14 Exoribonuclease Complex. Proc. Natl. Acad. Sci. USA 2012, 109, 9372–9377. [Google Scholar] [CrossRef]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.-Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2′-O Methylation of the Viral MRNA Cap Evades Host Restriction by IFIT Family Members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef]
- Hayn, M.; Hirschenberger, M.; Koepke, L.; Nchioua, R.; Straub, J.H.; Klute, S.; Hunszinger, V.; Zech, F.; Prelli Bozzo, C.; Aftab, W.; et al. Systematic Functional Analysis of SARS-CoV-2 Proteins Uncovers Viral Innate Immune Antagonists and Remaining Vulnerabilities. Cell Rep. 2021, 35, 109126. [Google Scholar] [CrossRef]
- Narayanan, N.; Nair, D.T. Ritonavir May Inhibit Exoribonuclease Activity of Nsp14 from the SARS-CoV-2 Virus and Potentiate the Activity of Chain Terminating Drugs. Int. J. Biol. Macromol. 2021, 168, 272–278. [Google Scholar] [CrossRef]
- European Medicines Agency (EMA). Norvir. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/norvir (accessed on 10 August 2022).
- Devkota, K.; Schapira, M.; Perveen, S.; Khalili Yazdi, A.; Li, F.; Chau, I.; Ghiabi, P.; Hajian, T.; Loppnau, P.; Bolotokova, A.; et al. Probing the SAM Binding Site of SARS-CoV-2 Nsp14 In Vitro Using SAM Competitive Inhibitors Guides Developing Selective Bisubstrate Inhibitors. SLAS Discov. Adv. Sci. Drug Discov. 2021, 26, 1200–1211. [Google Scholar] [CrossRef]
- Nencka, R.; Silhan, J.; Klima, M.; Otava, T.; Kocek, H.; Krafcikova, P.; Boura, E. Coronaviral RNA-Methyltransferases: Function, Structure, and Inhibition. Nucleic Acids Res. 2022, 50, 635–650. [Google Scholar] [CrossRef]
- Gurung, A.B.; Ali, M.A.; Lee, J.; Farah, M.A.; Al-Anazi, K.M. An Updated Review of Computer-Aided Drug Design and Its Application to COVID-19. BioMed Res. Int. 2021, 2021, 8853056. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, F.K. A Biochemical Perspective of the Nonstructural Proteins (Nsps) and the Spike Protein of SARS-CoV-2. Protein J. 2021, 40, 260–295. [Google Scholar] [CrossRef] [PubMed]
- Ulferts, R.; Ziebuhr, J. Nidovirus Ribonucleases: Structures and Functions in Viral Replication. RNA Biol. 2011, 8, 295–304. [Google Scholar] [CrossRef]
- Al-Rashedi, N.A.M.; Munahi, M.G.; Ah ALObaidi, L. Prediction of Potential Inhibitors against SARS-CoV-2 Endoribonuclease: RNA Immunity Sensing. J. Biomol. Struct. Dyn. 2020, 40, 4879–4892. [Google Scholar] [CrossRef]
- Kim, Y.; Jedrzejczak, R.; Maltseva, N.I.; Wilamowski, M.; Endres, M.; Godzik, A.; Michalska, K.; Joachimiak, A. Crystal Structure of Nsp15 Endoribonuclease NendoU from SARS-CoV-2. Protein Sci. 2020, 29, 1596–1605. [Google Scholar] [CrossRef]
- Bhardwaj, K.; Guarino, L.; Kao, C.C. The Severe Acute Respiratory Syndrome Coronavirus Nsp15 Protein Is an Endoribonuclease That Prefers Manganese as a Cofactor. J. Virol. 2004, 78, 12218–12224. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Kirsebom, F.C.M.; Kausar, F.; Nuriev, R.; Makris, S.; Johansson, C. Neutrophil Recruitment and Activation Are Differentially Dependent on MyD88/TRIF and MAVS Signaling during RSV Infection. Mucosal Immunol. 2019, 12, 1244–1255. [Google Scholar] [CrossRef]
- Huang, S.; Liu, K.; Cheng, A.; Wang, M.; Cui, M.; Huang, J.; Zhu, D.; Chen, S.; Liu, M.; Zhao, X.; et al. SOCS Proteins Participate in the Regulation of Innate Immune Response Caused by Viruses. Front. Immunol. 2020, 11, 558341. [Google Scholar] [CrossRef]
- Krähling, V.; Stein, D.A.; Spiegel, M.; Weber, F.; Mühlberger, E. Severe Acute Respiratory Syndrome Coronavirus Triggers Apoptosis via Protein Kinase R but Is Resistant to Its Antiviral Activity. J. Virol. 2009, 83, 2298–2309. [Google Scholar] [CrossRef]
- Pindel, A.; Sadler, A. The Role of Protein Kinase R in the Interferon Response. J. Interferon Cytokine Res. 2011, 31, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Hackbart, M.; Mettelman, R.C.; O’Brien, A.; Mielech, A.M.; Yi, G.; Kao, C.C.; Baker, S.C. Coronavirus Nonstructural Protein 15 Mediates Evasion of DsRNA Sensors and Limits Apoptosis in Macrophages. Proc. Natl. Acad. Sci. USA 2017, 114, E4251–E4260. [Google Scholar] [CrossRef]
- Khan, R.J.; Jha, R.K.; Singh, E.; Jain, M.; Amera, G.M.; Singh, R.P.; Muthukumaran, J.; Singh, A.K. Identification of Promising Antiviral Drug Candidates against Non-Structural Protein 15 (Nsp15) from SARS-CoV-2: An In Silico Assisted Drug-Repurposing Study. J. Biomol. Struct. Dyn. 2022, 40, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Wower, J.; Maltseva, N.; Chang, C.; Jedrzejczak, R.; Wilamowski, M.; Kang, S.; Nicolaescu, V.; Randall, G.; Michalska, K.; et al. Tipiracil Binds to Uridine Site and Inhibits Nsp15 Endoribonuclease NendoU from SARS-CoV-2. Commun. Biol. 2021, 4, 193. [Google Scholar] [CrossRef]
- Hong, S.; Seo, S.H.; Woo, S.-J.; Kwon, Y.; Song, M.; Ha, N.-C. Epigallocatechin Gallate Inhibits the Uridylate-Specific Endoribonuclease Nsp15 and Efficiently Neutralizes the SARS-CoV-2 Strain. J. Agric. Food Chem. 2021, 69, 5948–5954. [Google Scholar] [CrossRef]
- Kumar, S.; Kashyap, P.; Chowdhury, S.; Kumar, S.; Panwar, A.; Kumar, A. Identification of Phytochemicals as Potential Therapeutic Agents That Binds to Nsp15 Protein Target of Coronavirus (SARS-CoV-2) That Are Capable of Inhibiting Virus Replication. Phytomedicine 2021, 85, 153317. [Google Scholar] [CrossRef]
- Hoever, G.; Baltina, L.; Michaelis, M.; Kondratenko, R.; Baltina, L.; Tolstikov, G.A.; Doerr, H.W.; Cinatl, J. Antiviral Activity of Glycyrrhizic Acid Derivatives against SARS−coronavirus. J. Med. Chem. 2005, 48, 1256–1259. [Google Scholar] [CrossRef]
- Ghosh, R.; Chakraborty, A.; Biswas, A.; Chowdhuri, S. Evaluation of Green Tea Polyphenols as Novel Corona Virus (SARS-CoV-2) Main Protease (Mpro) Inhibitors—An In Silico Docking and Molecular Dynamics Simulation Study. J. Biomol. Struct. Dyn. 2021, 39, 4362–4374. [Google Scholar] [CrossRef]
- Sinha, S.K.; Prasad, S.K.; Islam, M.A.; Gurav, S.S.; Patil, R.B.; AlFaris, N.A.; Aldayel, T.S.; AlKehayez, N.M.; Wabaidur, S.M.; Shakya, A. Identification of Bioactive Compounds from Glycyrrhiza glabra as Possible Inhibitor of SARS-CoV-2 Spike Glycoprotein and Non-Structural Protein-15: A Pharmacoinformatics Study. J. Biomol. Struct. Dyn. 2021, 39, 4686–4700. [Google Scholar] [CrossRef]
- Azad, G.K. Identification of Novel Mutations in the Methyltransferase Complex (Nsp10-Nsp16) of SARS-CoV-2. Biochem. Biophys. Rep. 2020, 24, 100833. [Google Scholar] [CrossRef]
- Morales, P.; Curtis, N.; Zárate, S.; Bastida, A.; Bolanos-Garcia, V. Interfering with MRNA Methylation by the 2′O-Methyltransferase (Nsp16) from SARS-CoV-2 to Tackle the COVID-19 Disease. Catalysts 2020, 10, 1023. [Google Scholar] [CrossRef]
- Nakagawa, K.; Lokugamage, K.G.; Makino, S. Viral and Cellular MRNA Translation in Coronavirus-Infected Cells. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2016; Volume 96, pp. 165–192. ISBN 978-012-804-736-1. [Google Scholar]
- Sk, M.F.; Jonniya, N.A.; Roy, R.; Poddar, S.; Kar, P. Computational Investigation of Structural Dynamics of SARS-CoV-2 Methyltransferase-Stimulatory Factor Heterodimer Nsp16/Nsp10 Bound to the Cofactor SAM. Front. Mol. Biosci. 2020, 7, 590165. [Google Scholar] [CrossRef]
- Shuman, S. Structure, Mechanism, and Evolution of the MRNA Capping Apparatus. In Progress in Nucleic Acid Research and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2000; Volume 66, pp. 1–40. ISBN 978-012-540-066-4. [Google Scholar]
- Ghosh, A.; Lima, C.D. Enzymology of RNA Cap Synthesis. Wiley Interdiscip. Rev. RNA 2010, 1, 152–172. [Google Scholar] [CrossRef]
- Viswanathan, T.; Misra, A.; Chan, S.-H.; Qi, S.; Dai, N.; Arya, S.; Martinez-Sobrido, L.; Gupta, Y.K. A Metal Ion Orients SARS-CoV-2 MRNA to Ensure Accurate 2′-O Methylation of Its First Nucleotide. Nat. Commun. 2021, 12, 3287. [Google Scholar] [CrossRef]
- Züst, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2′-O-Methylation Provides a Molecular Signature for the Distinction of Self and Non-Self MRNA Dependent on the RNA Sensor MDA5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef]
- Alshiraihi, I.M.; Klein, G.L.; Brown, M.A. Targeting NSP16 Methyltransferase for the Broad-Spectrum Clinical Management of Coronaviruses: Managing the Next Pandemic. Diseases 2021, 9, 12. [Google Scholar] [CrossRef]
- Tazikeh-Lemeski, E.; Moradi, S.; Raoufi, R.; Shahlaei, M.; Janlou, M.A.M.; Zolghadri, S. Targeting SARS-CoV-2 Non-Structural Protein 16: A Virtual Drug Repurposing Study. J. Biomol. Struct. Dyn. 2021, 39, 4633–4646. [Google Scholar] [CrossRef]
- Patil, S.P.; Pacitti, M.F.; Gilroy, K.S.; Ruggiero, J.C.; Griffin, J.D.; Butera, J.J.; Notarfrancesco, J.M.; Tran, S.; Stoddart, J.W. Identification of Antipsychotic Drug Fluspirilene as a Potential P53-MDM2 Inhibitor: A Combined Computational and Experimental Study. J. Comput. Aided Mol. Des. 2015, 29, 155–163. [Google Scholar] [CrossRef]
- Kim, Y.; George, D.; Prior, A.M.; Prasain, K.; Hao, S.; Le, D.D.; Hua, D.H.; Chang, K.-O. Novel Triacsin C Analogs as Potential Antivirals against Rotavirus Infections. Eur. J. Med. Chem. 2012, 50, 311–318. [Google Scholar] [CrossRef]
- Hebner, C.M.; Han, B.; Brendza, K.M.; Nash, M.; Sulfab, M.; Tian, Y.; Hung, M.; Fung, W.; Vivian, R.W.; Trenkle, J.; et al. The HCV Non-Nucleoside Inhibitor Tegobuvir Utilizes a Novel Mechanism of Action to Inhibit Ns5b Polymerase Function. PLoS ONE 2012, 7, e39163. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Yunaev, A.; Kreiserman, R.; Kaplan, A.; Stark, M.; Assaraf, Y.G. Lysosomotropic Drugs Activate TFEB via Lysosomal Membrane Fluidization and Consequent Inhibition of MTORC1 Activity. Cell Death Dis. 2018, 9, 1191. [Google Scholar] [CrossRef] [PubMed]
- Landolt, L.; Furriol, J.; Babickova, J.; Ahmed, L.; Eikrem, Ø.; Skogstrand, T.; Scherer, A.; Suliman, S.; Leh, S.; Lorens, J.B.; et al. AXL Targeting Reduces Fibrosis Development in Experimental Unilateral Ureteral Obstruction. Physiol. Rep. 2019, 7, e14091. [Google Scholar] [CrossRef] [PubMed]
- Tutusaus, A.; de Gregorio, E.; Cucarull, B.; Cristóbal, H.; Aresté, C.; Graupera, I.; Coll, M.; Colell, A.; Gausdal, G.; Lorens, J.B.; et al. A Functional Role of GAS6/TAM in Nonalcoholic Steatohepatitis Progression Implicates AXL as Therapeutic Target. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Smelkinson, M. The Hedgehog Signaling Pathway Emerges as a Pathogenic Target. J. Dev. Biol. 2017, 5, 14. [Google Scholar] [CrossRef]
- Roskoski, R. Janus Kinase (JAK) Inhibitors in the Treatment of Inflammatory and Neoplastic Diseases. Pharmacol. Res. 2016, 111, 784–803. [Google Scholar] [CrossRef]
- Liang, J.; Pitsillou, E.; Burbury, L.; Hung, A.; Karagiannis, T.C. In Silico Investigation of Potential Small Molecule Inhibitors of the SARS-CoV-2 Nsp10-Nsp16 Methyltransferase Complex. Chem. Phys. Lett. 2021, 774, 138618. [Google Scholar] [CrossRef]

| Structural Protein | Functions | Reference |
|---|---|---|
| S | Binds to host angiotensin-converting enzyme-2 (ACE2) receptor for fusion with the host cell membrane via endocytosis. | [11] |
| M | Promotes viral assembly by stabilizing the N protein-RNA complex inside the virion. | [12] |
| E | Forms the “viroporin” ion channel. Interacts with M protein to form viral particles. Acts on virion release and pathogenesis. | [13] |
| N | Packages viral genomic RNA into a helical ribonucleoprotein form. | [14] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Low, Z.Y.; Zabidi, N.Z.; Yip, A.J.W.; Puniyamurti, A.; Chow, V.T.K.; Lal, S.K. SARS-CoV-2 Non-Structural Proteins and Their Roles in Host Immune Evasion. Viruses 2022, 14, 1991. https://doi.org/10.3390/v14091991
Low ZY, Zabidi NZ, Yip AJW, Puniyamurti A, Chow VTK, Lal SK. SARS-CoV-2 Non-Structural Proteins and Their Roles in Host Immune Evasion. Viruses. 2022; 14(9):1991. https://doi.org/10.3390/v14091991
Chicago/Turabian StyleLow, Zheng Yao, Nur Zawanah Zabidi, Ashley Jia Wen Yip, Ashwini Puniyamurti, Vincent T. K. Chow, and Sunil K. Lal. 2022. "SARS-CoV-2 Non-Structural Proteins and Their Roles in Host Immune Evasion" Viruses 14, no. 9: 1991. https://doi.org/10.3390/v14091991