1. Introduction
TRIM proteins comprise the largest family of E3 ligases in mammals. They are characterized by a tripartite motif of RING, B-Box and coiled-coil and may encode additional domains, of which the PRYSPRY domain is the most common. Many TRIM proteins have proposed roles in infection and immunity (for recent review, see [
1]) where they typically inhibit viral replication, although some are reported to promote replication [
2,
3]. A commonly reported mechanism is one in which the TRIM binds a viral protein and causes it to be degraded via ubiquitination. Targeting is usually mediated by the C-terminal PRYSPRY domain, whilst ubiquitination is catalyzed by the N-terminal RING domain, though TRIMs have been reported to degrade their target even when they lack a RING [
4]. Typical targets for degradation include the viral capsid or nucleocapsid: TRIM5 [
5], TRIM11 [
6], and TRIM34 [
7] are reported to target the capsid of HIV and TRIM14 [
4], TRIM22 [
8] and TRIM41 [
9] the nucleocapsid of Influenza.
Antiviral TRIMs have also been shown to stimulate immune signaling, providing a second mechanism of viral inhibition. This can be indirect, by promoting the activity of pattern-recognition receptors (PRRs). For instance, TRIM65 is reported to K63-polyubiquitinate MDA5 and promotes its signaling [
10]. RIPLET, technically not a TRIM but containing RING, coiled-coil and PRYSPRY domains, modifies RIG-I with K63-chains and stimulates its activity [
11,
12], while TRIM56 is implicated in DNA sensing by promoting the activity of cGAS [
13]. Other TRIMs promote signaling directly as primary PRRs. TRIM5 [
14] and TRIM21 [
15] both stimulate innate immune signaling upon detection of incoming virions. This activity occurs in parallel with their direct-acting restriction mechanisms, in which targeted viral capsids are degraded. The restriction and signaling activities of both TRIM5 and TRIM21 are inextricably linked—synthesis of K63-polyubiquitination leads to proteasomal recruitment, followed by concomitant liberation of free K63-chains and protein degradation [
16,
17].
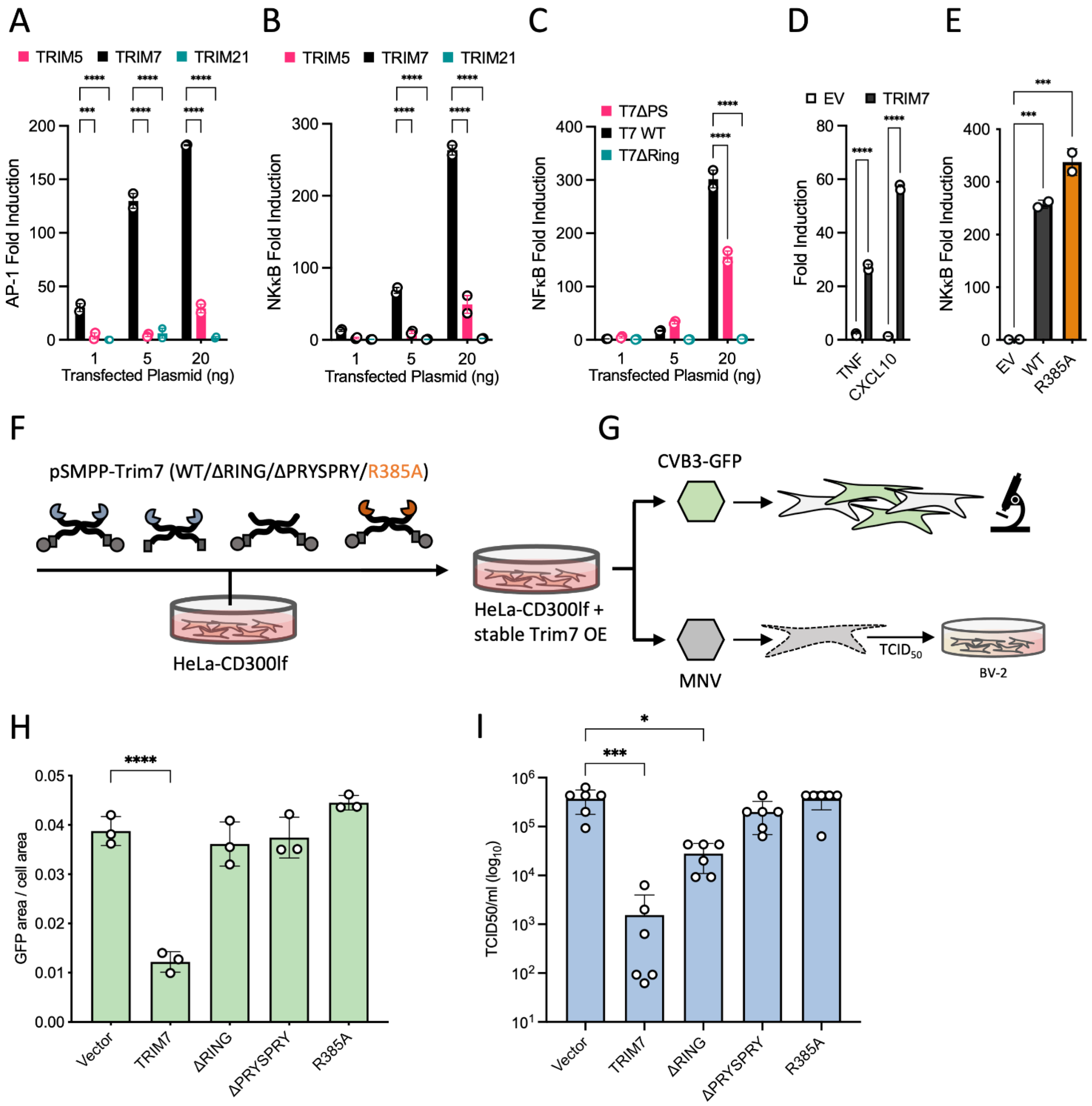
Exactly how many members of the TRIM family are involved in antiviral immunity, what proportion directly restrict or promote immune signaling and which are capable of both is unknown. In a transient over-expression screen of 36 human and 19 mouse TRIMs, over one-third were found to possess antiretroviral activity [
18]. In a follow-up study, 16 out of 43 over-expressed human TRIMs were shown to induce NF-κB or AP-1 signaling [
19]. Tellingly, there was a close correspondence between restriction and signaling; of 17 TRIMs that restricted MLV, 12 were also immune stimulatory. A more recent over-expression screen of 118 RING E3 ligases (including over 50 TRIMs) identified TRIM7 as a potent inhibitor of picornavirus Coxsackievirus B3 (CVB3) [
20]. Restriction required PRYSPRY binding to viral protein 2BC, followed by its RING-dependent ubiquitination and degradation. TRIM7 was also previously identified in a genome-wide overexpression screen as an inhibitor of the picorna-like virus calicivirus murine norovirus (MNV), an activity similarly found to be PRYSPRY-dependent [
21]. However, these reports of antiviral activity are at odds with other TRIM7 studies. TRIM7 has been proposed as a pro-viral factor in Zika virus infection, where it was shown to K63-polyubiquitinate the viral envelope protein [
3]. TRIM7 has also been reported as an agonist of TLR4-mediated signaling in macrophages [
22], a tumour suppressor through degradation of Src [
23], an oncogene through the K63-ubiquitination and stabilization of AP-1 co-activator RACO-1 [
24] and an interactor and regulator of glycogenin—the latter activity giving rise to TRIM7s original designation as ‘glycogenin-interacting protein’ (GNIP) [
25]. There is little that connects these disparate cellular roles.
In this study, we sought to determine how TRIM7 is capable of interacting with such diverse substrates and the mechanism by which it targets viral proteins for degradation. We show that the PRYSPRY domain of TRIM7 possesses a unique binding site that promiscuously interacts with proteins containing a C-terminus ending in a helix-ΦQ motif. The reason why TRIM7 has been reported to bind glycogenin-1 (GYG1), RACO-1 and CVB3 2BC is because they all possess this structural signature. We also show that it is this promiscuous helix-ΦQ motif that allows TRIM7 to restrict infection by both norovirus and Coxsackievirus. These viruses possess a viral 3C-protease that cleaves their polyproteins to generate viral proteins ending in glutamine. A TRIM7 point mutant that no longer binds this glutamine motif loses all viral restriction. 3C-like proteases are common to positive-strand RNA viruses from the Picornaviridae, Caliciviridae and Coronaviridae families, and all generate viral proteins with C-terminal glutamines that are potential substrates for TRIM7. However, we show that despite possessing multiple proteins ending in glutamine, SARS-CoV-2 is not restricted by TRIM7. Moreover, the tissue expression of TRIM7 and its gene regulation is not consistent with an antiviral TRIM, such as TRIM5 or TRIM21, suggesting that its described function as a restriction factor may be an artefact of overexpression.
2. Material and Methods
2.1. Proteins
Trim7 PRYSPRY constructs and all GYG constructs with an N terminal hexahistidine tag were expressed in E. coli (C41) and purified using Nickel affinity chromatography and Size Exclusion Chromatography (SEC). Briefly, cells were grown in 2XTY (supplemented with 0.5% glucose, 2 mM MgSO4 and appropriate antibiotics) at 37 °C for 2–3 h (OD600 around 0.6–1), after which they were induced with 1 mM IPTG and incubated at 18 °C overnight. Cells were pelleted with a Sorvall SLC-6000 compatible centrifuge at 4500× g for 25 min and the pellet frozen until processed. The pellet was resuspended in lysis buffer (50 mM Tris pH 8, 1 M NaCl, 10% v/v BugBuster (Merck, Gillingham, UK), 10 mM imidazole, 2 mM DTT and 1 × complete protease inhibitors (Roche, Basel, Switzerland) and sonicated for 15 min total time (10 s on/20 s off) at 70% amplitude. The soluble fraction was recovered by centrifugation at 40,000× g in a JLA25.50 rotor and put through a gravity flow column with 5 mL of NiNTA Agarose (Qiagen). The bound fraction was washed in Buffer B (300 mM NaCl, 50 mM Tris pH 8, 10 mM imidazole and 1 mM DTT) and eluted with Buffer E (300 mM NaCl, 50 mM Tris pH 8, 400 mM imidazole and 1 mM DTT). Fractions containing the protein were pooled, filtered and separated by SEC using HiLoad 26/600 Superdex 75 pg column (Cytiva, Marlborough, MA, USA) in 150 mM NaCl, 50 mM Tris pH 8 and 1 mM DTT. Full-length GYG proteins were separated using an equivalent 200 pg column. The appropriate fractions were pooled and concentrated to 10–15 mg/mL.
MBP-Trim7-CCPS was expressed and purified as described above but with the following adjustments; after elution from NiNTA agarose, the fractions were pooled and loaded onto an MBP-Trap (5 mL) column (300 mM NaCl, 50 mM Tris pH 8, 1 mM DTT) washed with the same buffer and then eluted with buffer supplemented with 10 mM Maltose. The peak fractions were then separated by SEC using HiLoad 26/600 Superdex 200 pg column in 300 mM NaCl, 50 mM Tris pH 8, 10% w/v glycerol and 1 mM DTT. The appropriate fractions were pooled and concentrated to 20 mg/mL.
TRIM7-RING and TRIM7-RING-Box were expressed in
E. coli C41 cells as GST–TEV fusion protein. Cleared cell lysates were prepared by sonication in 50 mM Tris at pH 8 (pH 9 for RING-Box), 150 mM NaCl, 2 mM DTT with the addition of 20% (
v/
v) BugBuster and complete protease inhibitors, followed by centrifugation 16,000×
g for 30 min. Lysates were loaded onto GST beads and washed with lysis buffer, then cleaved with TEV protease overnight at 4 °C. Cleaved proteins were concentrated and separated using a HiLoad 26/600 Superdex 75 size exclusion column. The peak fractions were pooled, concentrated and frozen in aliquots at −80 °C. Ube1, Ube2N, Ube2V2, Ube2W were produced as previously described [
26].
2.2. Constructs and Cloning
TRIM7-PRYSPRY constructs (both mouse and human) were ordered as synthetic genes encoding the residues 342–511 of TRIM7 and cloned into a double-digested pOPTH (introduces uncleavable MAHHHHHHM sequence at the N terminus) expression vector (KpnI, NdeI). MBP-hTRIM7-CC-PS was cloned by amplifying the hTrim7-PRYSPRY sequence and Gibson assembly with a synthetic gene fragment containing the CC sequence (residues 165–341) into a double-digested pOPTM (introduces hisMBP-TEVsite at the N terminus) expression vector (KpnI, NdeI). TRIM7-RING (residues 1–123) and TRIM7-RING-BOX (residues 1–168) constructs were cloned by PCR amplification from codon optimized (E. coli) synthetic DNA and Gibson assembly into double-digested (NdeI, HindIII) pOPTG expression vector (introduces GST-TEVsite at the N terminus).
The pEXN-2XHA plasmid backbone was used in comparison of signaling of different TRIM proteins. pEXN-2XHA-TRIM7 was generated by inserting a synthetic gene encoding full-length codon optimized TRIM7 sequence (GeneArt) into a double-digested pEXN-2XHA vector (EcoRI, XhoI). pEXN-2XHA-TRIM5 was used as previously described [
27]. pEXN-2XHA-TRIM21 was cloned from constructs published previously [
27]—sequence encoding full-length human TRIM21 was PCR amplified and inserted into double-digested pEXN-2XHA vector (EcoRI, XhoI).
pSMPP-2xHA-TRIM7 constructs were cloned from the pEXN-2XHA-TRIM7 plasmid. Relevant fragments (FL, ΔPRYSPRY(1–341), ΔRING(128–511)) were PCR amplified and cloned into BamHI linearized pSMPPv1 using Gibson assembly. Untagged pSMPP-TRIM7 constructs (FL, ΔPRYSPRY(1–341), ΔRING(128–511)) were ordered as synthetic gene fragments (GeneArt) designed for Gibson assembly into double-digested (NotI, BamHI) pSMPP vector. N terminally mCherry tagged Trim7 constructs were generated from pSMPP-2XHA-Trim7 by PCR amplification and Gibson assembly into double-digested (XhoI, KpnI) pmCherry-C1 plasmid vector.
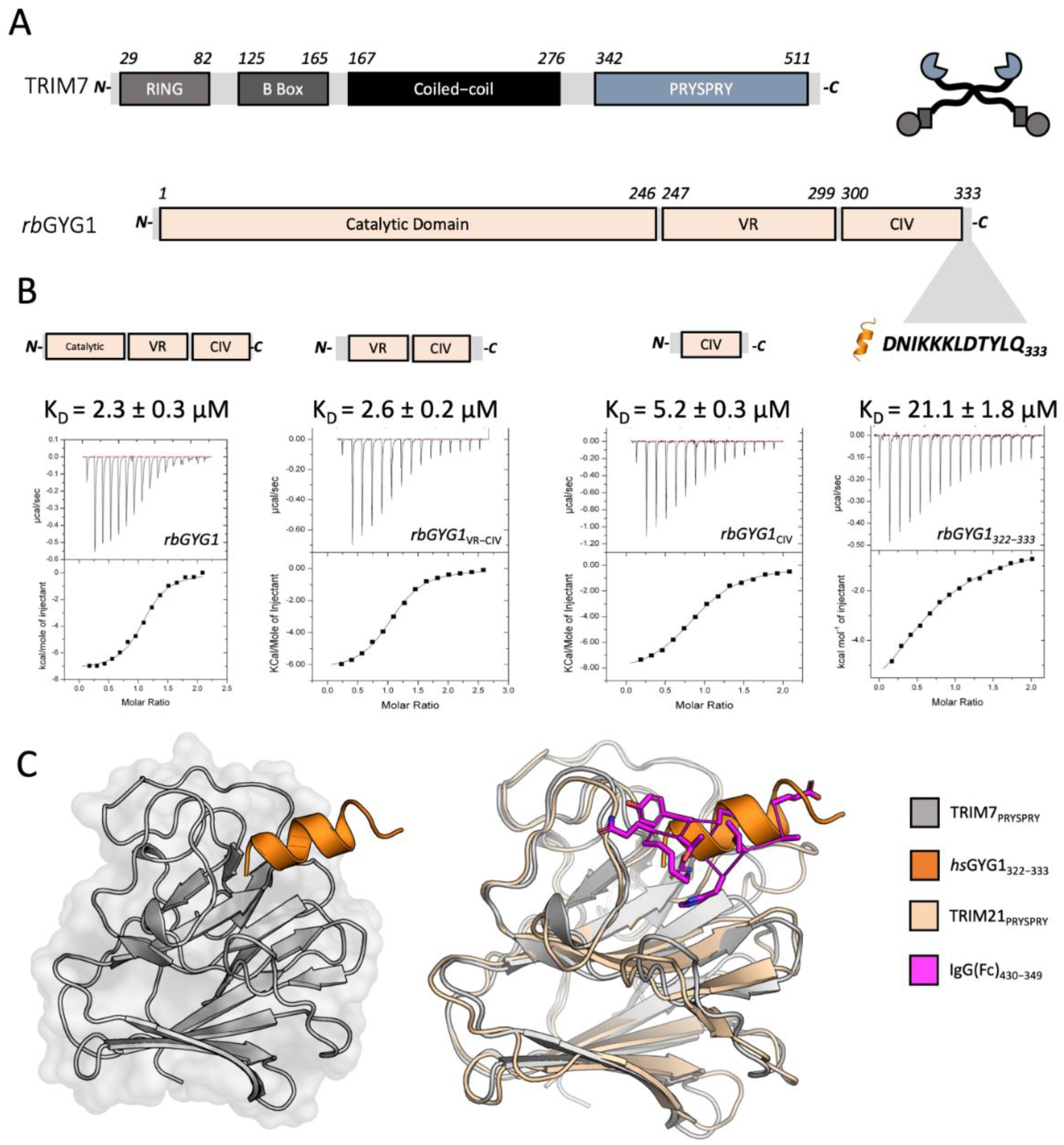
The pmEGFP-GYG1 plasmid was generated by restriction cloning of a synthesized gene fragment encoding the hsGYG1-GN1 sequence into the pmEGFP-C1 plasmid vector (BsrG1, EcoR1). For bacterial expression of hGYG1-GN1, the plasmid pOPTH-hGYG1-GN1 was generated by Q5 PCR amplification of the sequence from pmEGFP-GYG1 and Gibson assembled into a double-digested pOPTH (introduces uncleavable MAHHHHHH sequence at the N terminus) plasmid vector (KpnI, NdeI). The rest of the GYG1 protein constructs were ordered as synthetic genes and cloned into double-digested pOPTH (KpnI, HindIII). Plasmids encoding rbGYG1 and mGYG1 were ordered as synthetic genes encoding the full-length proteins and cloned into the pOPTH plasmid vector using restriction cloning. Truncated constructs rbGYG1VR-CIV, rbGYG1CIV, rbGYG11-246, rbGYG11-299 were generated from pOPTH-rbGYG1 by PCR amplification and restriction cloning into the same pOPTH vector.
All site-directed mutagenesis was performed with a Q5 PCR kit apart from mutagenesis of pSMPP-TRIM7-R385A, which was cloned by Gibson mutagenesis following manufacturer’s instructions (NEB).
2.3. Cell Culture
HEK293T, BV-2 and HeLa-CD300lf cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 2 mM L-glutamine, 100 U/mL penicillin and 100 mg/mL streptomycin (GIBCO) at 37 °C with 5% CO2. U2OS cells were grown in McCoy-5a medium supplemented with 10% FBS, 1XGlutaMAX, 100 U/mL penicillin and 100 mg/mL streptomycin.
2.4. NF-κB and AP-1 Signalling Experiments
HEK293T cells were seeded at 1 × 105 cells per well in 24 well plates 24 h before transfection. Each well was transfected with the indicated amounts of pEXN-2XHA-TRIM5/7/21 plasmids, together with 10 ng pGL4.32[luc2P/NF-κB-RE/Hygro] (NF-κB response element-dependent firefly luciferase, Cat. no 9PIE849) or pGL4.44 [luc2P/AP-1-RE/Hygro] (AP-1 response element-dependent firefly luciferase, Cat. no 9PIE411) and 5 ng pRL-TK (Cat. no E2241) using the Dual Luciferase Reporter Assay (Promega, Madison, WI, USA). Twenty-four hours post-transfection, cells were lysed in Passive Lysis Buffer, and sequential firefly and Renilla luminescence measured (BMG PHERAstar plate reader), according to manufacturer’s instructions (Promega). Firefly luciferase luminescence was normalized to Renilla luciferase luminescence, and these values normalized to those of empty vector-transfected cells.
2.5. qPCR Signalling Experiments
HEK293T cells were seeded at 2.5 × 105 cells per well in a 6-well plate. The next day the cells were transfected with 250 ng of either pEXN-2XHA-TRIM7 or empty pEXN-HA vector, and the cells harvested after 8 h. RNA was extracted using the RNeasy mini kit (Qiagen, Hilden, Germany) and cDNA produced from 1 µg RNA using oligo dT and Superscript RT. Gene expression was monitored by TaqMan Gene Expression Assays (Applied Biosystems, Waltham, MA, USA) on a StepOnePlus Real-Time PCR System (Life Technologies, Carlsbad, CA, USA). Gene expression assays were from Applied Biosystems: ACTB (Hs01060665_g1), CXCL10 (Hs01124251_g1). Relative expression was quantified using the 2−ΔΔCt method.
2.6. Stable Cell Line Generation
Stable cell lines were generated by stable lentiviral transduction of recipient cell lines. To generate the relevant lentiviruses, the untagged pSMPP-TRIM7 constructs were co-transfected into HEK293T cells with pMDG2 (a gift from D. Trono [EPFL; Addgene, plasmid 12259]) and pCRV-GagPol (a gift from S. Neil, Kings College London) plasmids using FuGENE6 (Promega) reagent and incubated for 3 days. Supernatants were harvested by filtration and used for transduction of HeLa cells expressing the mouse CD300lf [
28] as previously described [
20]. Briefly, 2.5 × 10
5 cells were infected with the appropriate lentivirus in a 6-well plate in DMEM supplemented with 3% FBS, 20 mM HEPES and 4 μg/mL Polybrene, and spinoculated at 1000×
g and 25 °C for 45 min. The cells were then incubated at 37 °C for 48 h. Antibiotic selection was carried out with 2 μg puromycin until all untransduced control cells were dead. For SARS-CoV-2 infection experiments, HeLa cells expressing TRIM7 constructs and mouse CD300lf were further transduced with lentiviruses encoding the ACE2 receptor [
29] and selected with 10 µg/mL of Blasticidin.
2.7. CVB3 Experiments
For production of Coxsackievirus, plasmid encoding for strain pH3 with an eGFP (eGFP-CVB3) (kind gift from Professor Charles Rice) was transfected into HEK293T cells using PEI. After 48 h, stock was harvested by 3 cycles of freeze-thawing and centrifugation to remove the debris. Collected stock (P0) was used to generate working stock (P1) by infecting fresh plate for HEK293T cells and harvesting as above. Virus titer was assessed by TCID50 using HeLa cells. For infection experiments, Hela-CD300lf stably expressing different TRIM7 constructs were seeded onto 24-well plates 24 h prior to infection. Cells were infected at MOI of 0.2, and infection levels determined by measuring eGFP fluorescence after 12 h on the incuCyte.
2.8. MNV1 Experiments
HeLa-CD300lf-TRIM7 cells suspended in DMEM supplemented with 10% FBS were incubated with the virus on an end-to-end rotor at 37 °C for 1 h. The cells were then washed twice in DMEM to remove the unbound virus and then incubated at 37 °C for the duration of the infection. The plate (both cells and supernatants) was frozen at −80 °C for 24 h post-infection, and TCID50 assays were subsequently carried out in BV2 cells as previously described [
30].
2.9. SARS-CoV-2 Experiments
HeLa cells expressing versions of TRIM7, CD300lf and ACE2 were seeded into 96-well plates at a density of 1.5 × 10
4 day prior to infection. For infections, we used isolate of omicron variant BA.1 (EPI_ISL_7886688), which was a gift from Ravi Gupta (University of Cambridge). Media containing 2% FBS and virus at MOI = 0.2 PFU/cell was added to wells and incubated for 18 h. Cells were freeze-thawed and then lysed with buffer containing 0.25% Triton X-100, 50 mM KCl, 100 mM Tris–HCl pH 7.4, glycerol 40% and RNAsecure (1/100) for 5 min. Infectious virus was then inactivated at 95 °C for 5 min, and samples processed for RT-qPCR as described previously [
31]. We used Luna Universal Probe One-Step kit following manufacturer’s recommendations. Primer/probe for genomic viral RNA were CDC-N1. Primer probes for 18S control were described previously [
32]. SARS-CoV-2_N_Positive control RNA was used as standard for the viral genomic N reactions. For 18S standard, DNA was synthesized and kindly gifted by Jordan Clarks and James Stewart (University of Liver-pool). Final concentrations of 500 nM for each primer and 125 nM for the probe were used. RT–qPCRs were run on ABI StepOnePlus PCR System with the following program: 55 °C for 10 min, 95 °C for 1 min and then 40 cycles of 95 °C denaturation for 10 s and 60 °C extension for 30 s. RNA copy numbers were obtained from standards, and then genomic copies of N normalized to 1010 copies of 18S.
2.10. Co-Localisation and Degradation Experiment
U2OS cells were seeded into 8-well μ-slide (ibidi, polymer) for co-localization or into standard tissue culture 24-well plates for bulk fluorescence quantification and protein quantification. For the former, 2–3 × 104 cells were seeded, and 5 × 105 cells were seeded in the latter. The next day, cells were co-transfected by a 1:1 mixture of the relevant plasmids using FuGENE6 and placed in the incuCyte (Sartorius, Göttingen, Germany) for live imaging. After 18–20 h of transfections, the cells were harvested, and the cell pellets processed as described below. To quantify the degradation of the EGFP-GYG1 using bulk fluorescence by incuCyte, the total integrated intensity in each condition was divided by the green fluorescence area to account for different transfection efficiencies. Co-localization experiment was imaged using live-cell imagining with the LUMASCOPE LS720 (Etaluma, Carlsbad, CA, USA). Cells expressing low levels of each plasmid were selected for analysis to avoid overexpression artefacts and aggregated proteins. Images were processed in ImageJ, and data plotted in PRISM (GraphPad Software, San Diego, CA, USA).
2.11. Protein Quantification Using Capillary Based Western Blot (Simple Western)
Cells were lysed in 40 µL RIPA buffer supplemented with 1 × complete protease inhibitors (Roche, Basel, Switzerland), and the soluble protein recovered by centrifugation (40 min at 21 k × g); 30 µL of the lysate was diluted with 90 µL 0.1 × Sample Buffer 2 and further processed according to manufacturer’s instructions. Jess instrument was used to analyze the samples (Protein Simple). Actin was used as the loading control (MAB8929, 1:1000) and detected with anti-mouse HRP. mCh-Trim7 was detected with rabbit polyclonal anti-mCherry (ab167453, 1:200), and EGFP-GYG1 was detected with rabbit polyclonal anti-GFP (NB600-303, 1:1000), both detected with anti-rabbit NIR secondary antibody. Protein levels were normalized to actin and to the fraction of cells transfected (determined with the incuCyte as the fraction of cells with detectable green fluorescence).
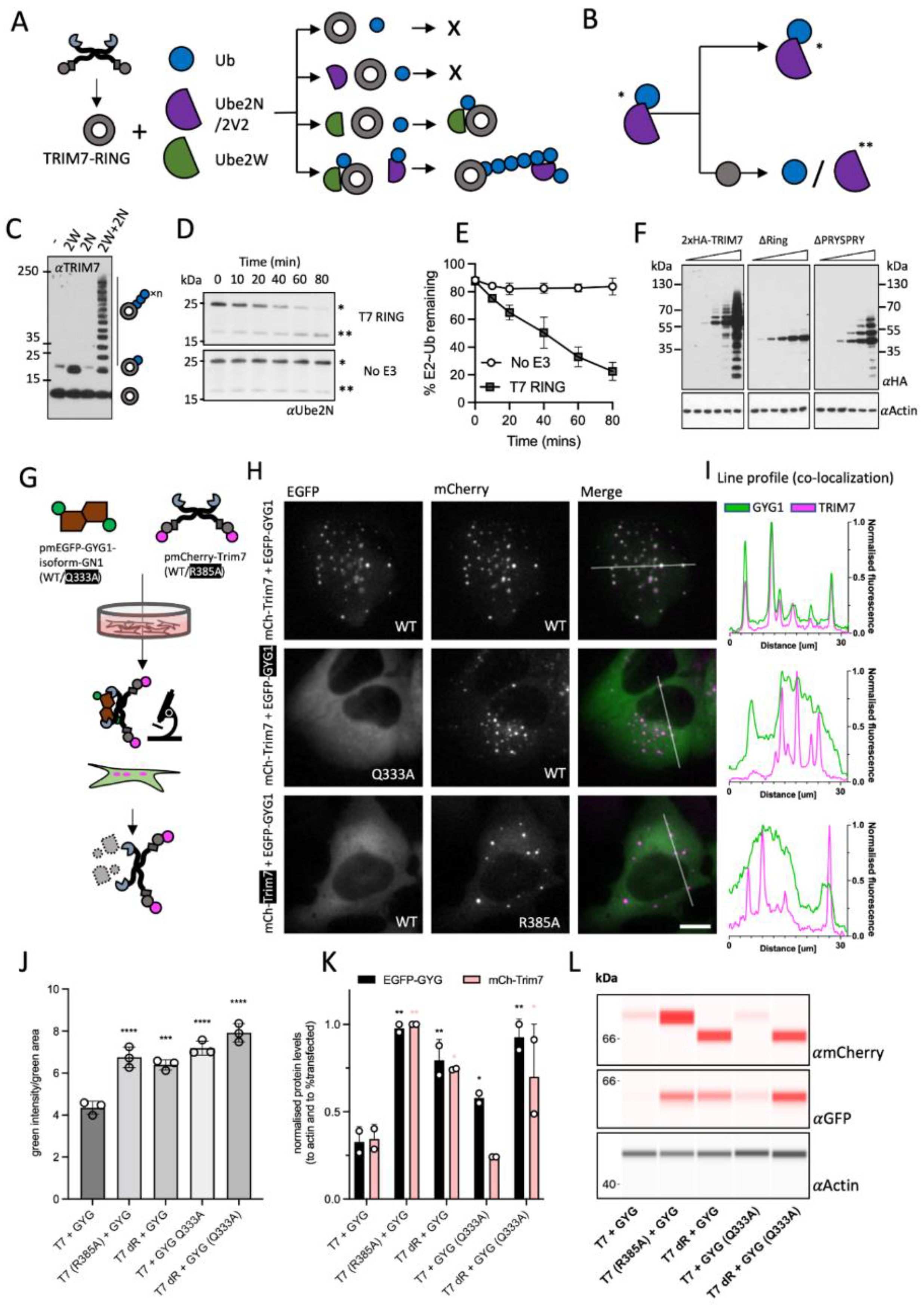
2.12. In Vitro Ubiquitination Experiment
In vitro ubiquitination reactions were carried out in 1× ubiquitination buffer (50 mM Tris⋅HCl, pH 7.4, 2.5 mM MgCl2, 0.5 mM DTT) with the addition of 2 mM ATP, 0.5 µM His-E1, 1 µM Ube2W, Ube2N/Ube2V2, 8 µg ubiquitin (Ub) and 400 ng TRIM7-RING. Reaction mixtures were incubated at 37 °C for 1–4 h, quenched by the addition of LDS sample buffer and boiling at 95 °C for 5 min. Samples were resolved by LDS-PAGE and detected by immunoblot with TRIM7 or anti-Ub-HRP (Santa Cruz, sc8017-HRP P4D1, 1:1000) antibodies.
2.13. Ubiquitin Discharge Assay
To prevent auto-ubiquitination, lysine 92 of Ube2N was replaced with arginine [
33]. Ube2NK92R was loaded with ubiquitin by mixing 40 µM of the E2 with 1 µM Ube1, 0.37 mM Ub and 3 mM ATP in 50 mM HEPES pH 7.5, 150 mM NaCl, 20 mM MgCl
2 and incubating the reaction at 37 °C for 30 min. The reaction was transferred to ice and used immediately. To observe E3 mediated discharge of ubiquitin, 2 µM ubiquitin-loaded E2 was mixed with 1.5 µM TRIM7-RING in 50 mM HEPES pH 7.5, 150 mM NaCl, 20 mM MgCl
2, 50 mM L-lysine, 2.5 µM Ube2V2. Samples were taken at the time points indicated in the text and mixed immediately with LDS sample buffer at 4 °C. The samples were boiled for exactly 20 s, resolved by LDS-PAGE and observed by immunoblot using anti-Ube2N (Bio-Rad, Hercules, CA, USA, AHP974, 1:1000) and detected by near-infrared detection (Odyssey, LI-COR, Lincoln, NE, USA).
2.14. Western Blotting
Stable cell lines were confirmed for overexpression of TRIM7 by Western blotting. Cells were lysed in RIPA buffer supplemented with 1 × complete protease inhibitors (Roche). Lysates were separated on a precast SDS-PAGE gel (NuPAGE, 4–12%), transferred onto nitrocellulose membranes and blotted for GAPDH (Ambion, AM4300; 1:20,000) and TRIM7 (generated in house, αTrim7; 1:250)
2.15. Antibody Generation and Animal Handling
C57BL/6 wild-type mice were obtained from Jackson Laboratories and bred in LMB ARES facility under SPF conditions. Thirteen-week-old mice were used for the immunization experiment, which was conducted in accordance with the moderate severity limit protocol 7 and Home Office Animals (Scientific Procedures) Act (1986). All animal work was licensed under the UK Animals (Scientific Procedures) Act, 1986 and approved by the Medical Research Council Animal Welfare and Ethical Review Body. Mice (n = 12) were immunized subcutaneously (s.c) with 100 μg TRIM7-Ring-Box protein in PBS mixed with complete Freund’s adjuvant, 200 μl of emulsion per animal. Mice received 3 rounds of s.c boosting with 50 μg protein mixed with incomplete Freund’s adjuvant. Boosting was done on days 21, 42 and 79. Tail bleeds for ELISA analyses were collected on days 31, 58. Mice were sacrificed on day 93, and intracardiac blood was collected.
2.16. ELISA
A 96-well plate (Nunc) was coated overnight with 1 μg/mL of TRIM7-RING-BOX. The plate was blocked with 2% Marvell in PBS, 0.05% Tween 20 (MPBST) and incubated with diluted mouse sera. Bound antibodies were detected with goat anti-mouse IgG-GRP (Jackson Immunoresearch, 115-035-071).
2.17. Peptides
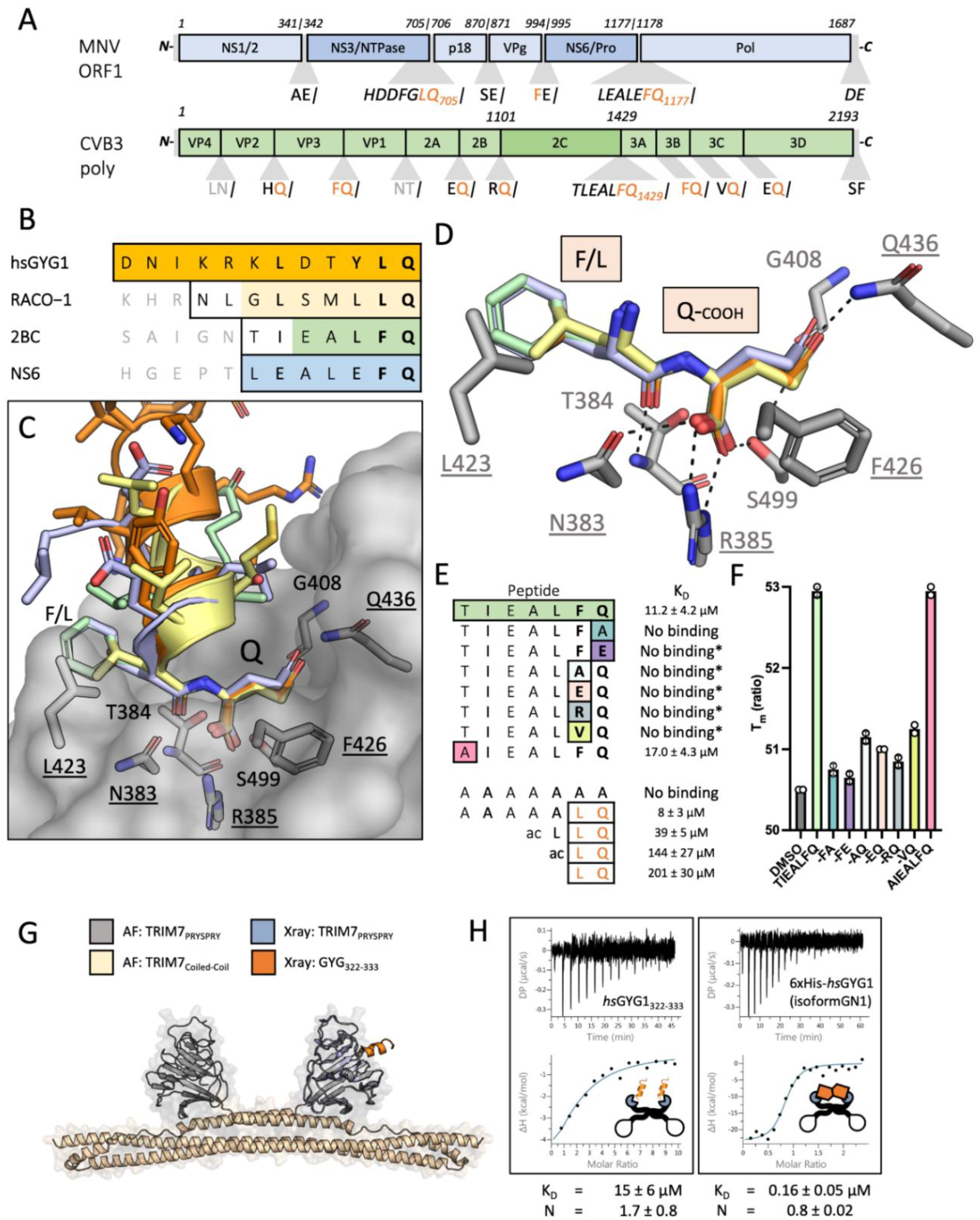
The peptides DNIKKKLDTYLQ and NLGLSMLLQ were ordered from St John’s Innovation Centre, Cambridge, UK. Peptides DNIKRKLDTYLQ, TIEALFQ, AIEALFQ, TIEALFA, TIEALAQ, TIEALRQ, TIEALVQ, TIEALFE, TIEALEQ, LQ, LLQ, Ac-LLQ, AAAAAAA, AAAAALQ were synthesized by DesignerBioscience (Cambridge, UK), dissolved in DMSO to 50–200 mM and stored at −80 °C. Peptides HDDFGLQ, LEALEFQ were synthesized by GenScript (Oxford, UK) and dissolved in DMSO to 50 mM and stored at −80 °C.
2.18. Isothermal Titration Calorimetry (ITC)
Titrations were performed using the MicroCal iTC200 and MicroCal Auto-iTC200 instruments (Malvern Panalytical, Malvern, UK). Binding assays were carried out in 150 mM NaCl, 50 mM Tris pH 8, 1 mM DTT and 0–1%
v/
v DMSO. Generally, 13–20 injections were carried out with a DP of 6 μcal/s, 750 RPM, 20–25 °C, 150–180 s spacing and 5–6 s injection times of 2.5–3 μL. Titrations were performed with protein and peptide in cell and syringe interchangeably. Generally, TRIM7 in the cell was kept at 20 μM with peptides diluted to 400 μM and up to 2 mM for dipeptides. See
Table S2 for details. Control titrations of titrant into buffer were carried out where appropriate. Data were analyzed using MicroCal PEAQ-ITC analysis software, using one site model to fit the data. For low c-value experiments (such as the LQ titration), the N was fixed to 1. For experiments with MBP-T7CCPS, the buffer was 165 mM NaCl, 50 mM Tris pH 8, 1 mM DTT, 0.5%
w/
v glycerol and 0–0.8%
v/
v DMSO. For experiments with rbGYG, mGYG and mTrim7-PRYPSRY, the protein samples were dialysed against 50 mM phosphate buffer, pH 6.5 and 1 mM DTT. ITC experiments were conducted on MicroCal ITC 200 at 15 °C or an AutoITC and analysed using a standard one-state model within MicroCal instrument software.
2.19. Differential Scanning Fluorimetry
Thermal stabilization assays were performed using the NanoTemper Prometheus NT48 instrument (Nanotemper Technologies, München, Germany). All experiments were performed in 150 mM NaCl, 50 mM Tris pH 8, 1 mM DTT and 1% v/v DMSO unless otherwise noted. Trim7 PRYSPRY was used at 10 μM and peptides at 90 μM. Samples were heated at 2 °C/min from 15 to 95 °C. Data were analyzed using the NanoTemper Prometheus Control Software, and the first derivative of the melt curves used to define the Tm for the apo and complexed samples. Independent experiments used the same peptide and protein stocks.
2.20. Crystallography
TRIM7 PRYSPRY protein was dialyzed against 20 mM HEPES, 50 mM NaCl, 1 mM DTT, pH 8.0) and concentrated to 15 mg/mL; 4 mM peptide was added and the complex crystallized in 0.2 M KCSN, 15% PEG 4000, 0.1 M NaOAc, pH 5.5 for GYG1 (NH2-DNIKRKLDTYLQ-COOH, 100 mM in DMSO), for RACO-1 (NH2-NLGLSMLLQ-COOH, 50 mM in DMSO) in 1.5 M NaPO
4H
2 and 1 M Sodium Citrate pH 5.5. For 2BC, Trim7 PRYSPRY at 13.15 mg/mL in 150 mM NaCl, 50 mM Tris pH 8 and 1 mM DTT was mixed 100:1 with the 2BC peptide (NH2-TIEALFQ-COOH, 100 mM in DMSO) and crystallized in 1 M Sodium Potassium Tartrate, 0.1 M HEPES pH 6.8. For the NS6 peptide (NH2-LEALEFQ-COOH, 50 mM in DMSO), the protein was mixed 50:1 and co-crystallized in 1 M Na/K Phosphate pH 5. Crystals were cryoprotected with reservoir solution containing 20–30%
v/
v glycerol. Data for GYG1 was collected on an in-house FR-E SuperBright high-brilliance rotating anode linked to an automated crystal mounting system (ACTOR; Rigaku, Tokyo, Japan) and processed with CCP4i [
34] package as follows: data were indexed in iMOSFILM, point group was determined in POINTLESS and scaled in SCALA. Molecular replacement was performed with PHASER [
35] using the TRIM21 PRYSPRY model 2IWG. After the first iteration, further structures were solved using the Trim7 model from the GYG structure. The MR model was refined with COOT [
36] and REF-MAC5 [
37]. Data for RACO-1 was collected at the ESRF on beamline ID23 and processed as for GYG1. Data for 2BC and NS6 were collected at the DLS (Didcot, UK) on beamline i24 and i04, respectively, processed using the FASTDP [
38] and Xia2dials [
39] pipeline and processed as described.
2.21. Phylogenetic Analysis
Coding sequences for TRIM7 and GYG1 were obtained from Ensembl v. 106 [
40]. Multiple sequence alignments were constructed with Muscle v. 3.8.31 with default arguments [
41]. Phylogenetic trees were reconstructed using raxml v. 8.2.12 (with “-f a -m PROTCATLGX -# 100” arguments) [
42].
4. Discussion
The data presented here show that TRIM7 targets diverse substrates through a highly unusual binding mechanism in which it recognizes a C-terminal helix-ΦQ motif. This mechanism explains the promiscuous binding reported for TRIM7 against unrelated proteins GYG1, RACO-1, 2C, NS3 and NS6. During preparation of our manuscript similar data were deposited in bioRxiv [
49], showing that TRIM7 binds to a ΦQ motif. The helix-ΦQ motif recognized by TRIM7 is reminiscent of the N-end degron system. In the ‘N-end rule’ system [
50], N-recognin proteins hydrogen bond with free α-amino groups at protein N-termini [
51]. TRIM7 recognizes the other end of proteins, using an arginine residue (R385) to form a bidentate salt bridge with the carboxylic acid at C-termini. Both systems also share similarities in how substrates can be generated. In the N-end system, proteolytic cleavage of N-terminal methionine exposes amino acids at the termini that differ in their suitability as substrates for degradation. Likewise, proteolysis can reveal novel substrates for TRIM7 by exposing C-terminal glutamines. We propose that it is the action of 3C proteases that generates viral substrates for TRIM7 and results in the restriction of CVB3 and MNV1 infection. In support of our hypothesis, we show here that when C-terminal glutamine binding is abolished by mutating TRIM7 residue R385, CVB3 and MNV1 are no longer restricted.
As many positive-stranded RNA viruses utilize a 3C or 3C-like mechanism to liberate their proteins from an initial single polypeptide, there is the possibility that TRIM7 could function as a more broadly-acting restriction factor than antiretroviral ligase TRIM5. However, the ends generated by 3C proteases are not strictly conserved. For example, human norovirus proteins do not have identical C-termini as MNV1 proteins (e.g., in Norwalk virus, NS6 ends in EGETALE rather than LEALEFQ, but NS3 maintains a terminal LQ). Moreover, not all viral proteins with a helix-ΦQ motif will be accessible in the full-length form or if the protein is sequestered during viral replication in the cells. Nevertheless, it is surprising that we observed no restriction of live SARS-CoV-2, as the virus possesses 10 proteins ending in ‘LQ’ or ‘FQ’. We purposefully performed our experiments in the same cells where we had observed restriction of both MNV1 and CVB3 so we could be sure there was functional TRIM7 present. However, it is possible that restriction requires a cell line that is more physiologically relevant for SARS-CoV-2 or that expresses even higher levels of TRIM7. During preparation of this manuscript, a pre-print report was submitted that claims TRIM7 can bind to and degrade multiple SARS-CoV-2 proteins, including NSP5 and NSP8 [
49]. If similar conditions can be recreated during an infection, then perhaps restriction would be observed. Of course, it may also be that binding does not happen during live virus infection, or there is insufficient degradation to impact viral replication. Alternatively, SARS-CoV-2 may antagonize TRIM7 function.
An important question raised by this study is what the principle physiological function of TRIM7 actually is. Just because TRIM7 possesses antiviral activity does not mean this is its natural cellular function, particularly as we show such activity is dependent upon a highly promiscuous binding mechanism that makes ~1% of cellular proteins potential substrates. Both CVB3 and MNV viral screens were based on TRIM7 over-expression, as were many of the other reports identifying TRIM7 ligands. Ectopic TRIM7 over-expression may have created an opportunity for binding and ubiquitination that does not exist under normal physiological conditions. The link between TRIM7s helix-ΦQ binding mechanism and 3C-protease processing, while highly attractive, may be coincidental. A decreased CVB3 viral yield was reported in mouse myoblasts and cardiomyoctes treated with TRIM7 siRNAs [
20], however, this may have been an indirect effect caused by reduced cell growth and metabolism in the depleted cells. TRIM7s link to glycogenin makes this a particularly valid possibility. We strongly suggest that future studies of TRIM7 restriction phenotypes, such as with SARS-CoV-2, avoid overexpression and use a knockout approach to avoid possible artefacts. Even so, it will be important to rule out confounding factors such as altered cell growth, and ultimately viral challenge experiments in TRIM7 knockout animals may be required for a definitive answer.
We have shown here that in addition to its reported degradation activity, over-expressed TRIM7 stimulates signaling pathways, including AP-1 and NF-κB, by synthesizing K63-polyubiquitin. Dual signaling/effector function is a signature of bona fide antiviral TRIMs, TRIM5 and TRIM21. Indeed, TRIM-based overexpression screens have previously identified multiple TRIMs with antiviral restriction and NF-κB activity [
19]. However, it is possible that overexpression creates artefactual conditions for signaling. Over-expression can push TRIM proteins into forming cytoplasmic bodies that would not normally assemble under resting conditions [
46], and these bodies are a source of ubiquitination and downstream signal transduction.
Phylogeny and tissue expression offer a useful perspective into TRIM7 function. TRIMs have undergone a rapid expansion in mammals: there are over 70 TRIMs in humans, whereas only 20 in worms and 10 in flies [
52]. Recently expanded TRIMs comprise a sub-group that evolves at a faster rate and contains a PRYSPRY domain [
53]. Positive selection has been identified in many of these proteins, and while this includes TRIMs that have no known antiviral role, such a signature can be indicative of pathogen evolutionary pressure [
54]. TRIM7 contains a single positively selected site in its coiled-coil, in contrast to the 21 sites found in antiretroviral TRIM5 [
54]. TRIM7 is also older than antiviral TRIMs such as TRIM5 and TRIM21, which are only found in mammals. The evolutionary age of TRIM7 is a reasonable match to the appearance of the helix-ΦQ motif in glycogenin. Glycogenin arose in early eukaryotes, but the final ~30 residues did not become fixed until jawed vertebrates and the helix-ΦQ motif only in amniotes (~319 Mya;
Figure S8 and [
55]). TRIM7 is present in birds and reptiles, suggesting it is slightly younger (~261 Mya;
Figure S8 and [
55]). Tissue expression is another useful predictor of function, and in this respect TRIM7 and glycogenin are also a good match. TRIM7 is expressed principally in skeletal muscle and in the brain, both regions where glycogenin is also highly expressed (
Figure S9). TRIM7 expression is highly tissue-specific, unlike antivirals TRIM5 and TRIM21, which are expressed almost everywhere. Moreover, TRIM7 tissue expression does not match that of the MNV1 receptor CD300lf, the CVB3 receptor CXADR or the SARS-CoV-2 receptor ACE2 (
Figure S9), which would be strange if it had evolved to restrict these viruses. Finally, in contrast to Trim5 and Trim21, the Trim7 gene does not possess either an interferon-stimulated response element (ISRE) or gamma interferon-activated site (GAS), and its expression is not upregulated by interferon [
56]. Taken together, neither phylogeny, tissue expression nor gene regulation are particularly supportive of TRIM7 being a broad-acting antiviral restriction factor. It is possible that none of the currently identified TRIM7 interacting proteins are the natural substrate—indeed, the real TRIM7 substrate may not be a protein at all but a metabolite. The malonate observed to bind apo TRIM7 makes the same core salt bridge as the glutamate side chain and carboxylic acid in the protein ligands. However, this moiety is not sufficient for full binding affinity, meaning that any natural metabolite would likely need to make additional interactions. Typically, an enzyme’s function is revealed with its substrate, but TRIM7s unusual binding mechanism obviates such an approach. Future work will need to focus on TRIM7 in the tissues where it is naturally found, using knockout-out approaches and whole organism models.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}



