
Origins and Evolution of Seasonal Human Coronaviruses

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Datasets and Subsampling
2.2. Maximum Likelihood (ML) Trees and Temporal Signal
2.3. Comparative Genomics
2.4. Recombination Analysis
2.5. Phylodynamics and Host-Jump Analysis
2.6. Selection Analysis
2.7. Amino Acid Substitution Analysis
3. Results
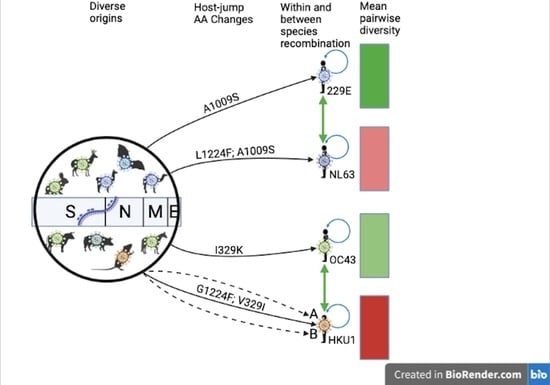
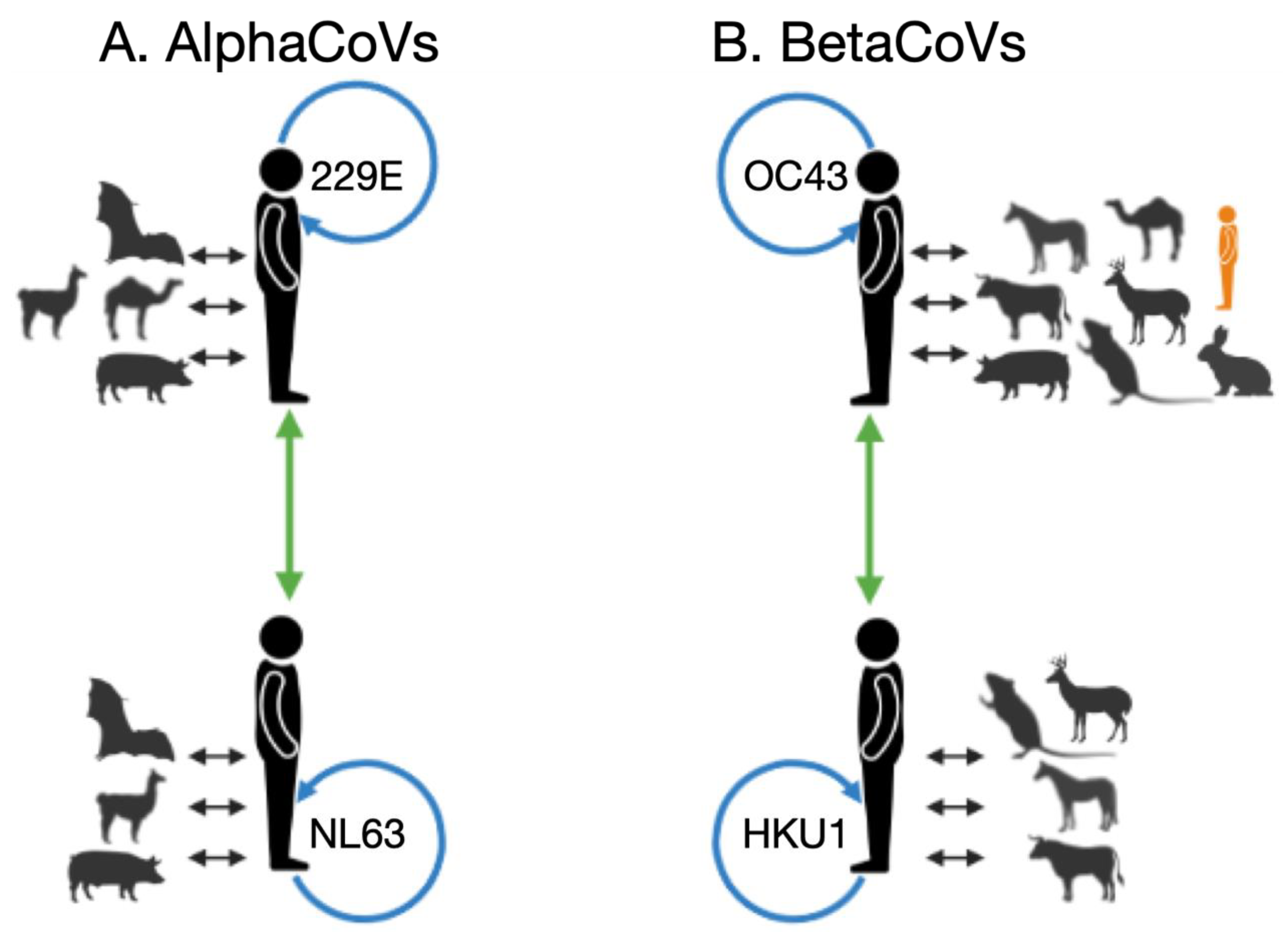
3.1. Zoonotic Origins of the Seasonal Human Coronaviruses
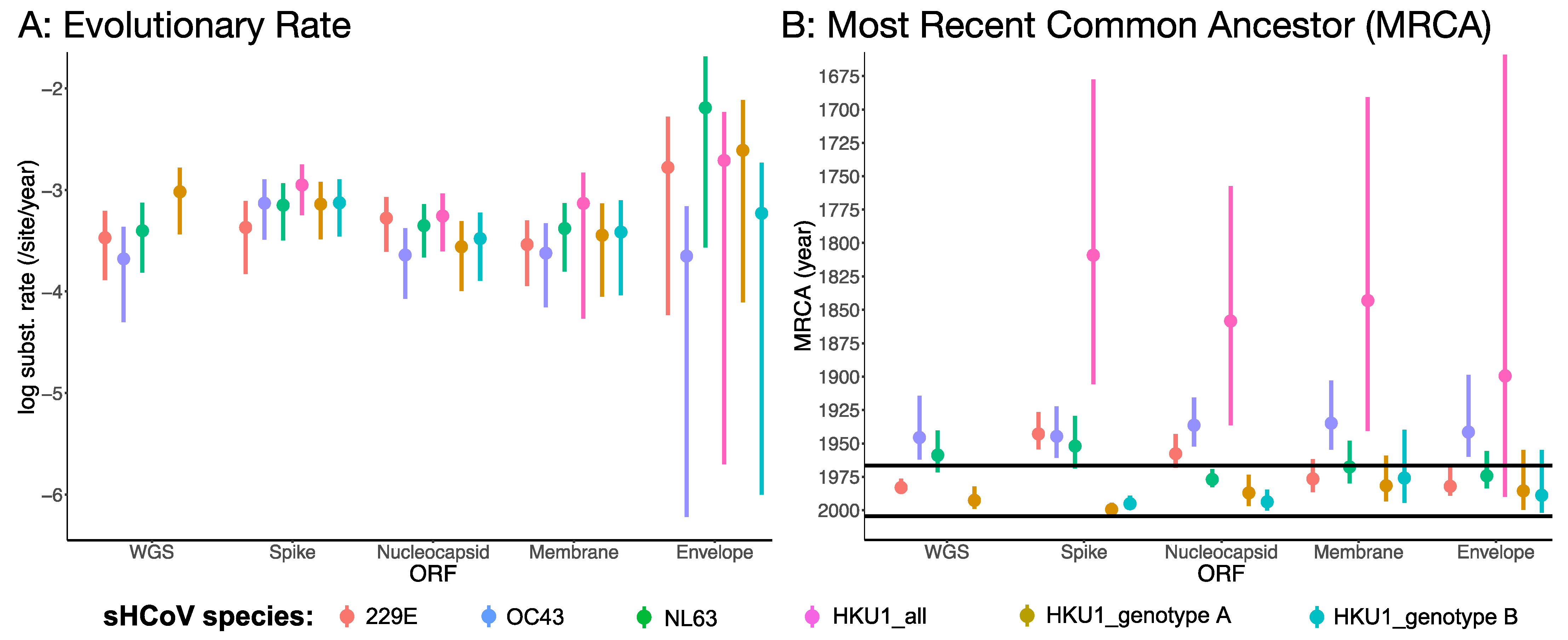
3.2. Rates of Evolution and Emergence Dates
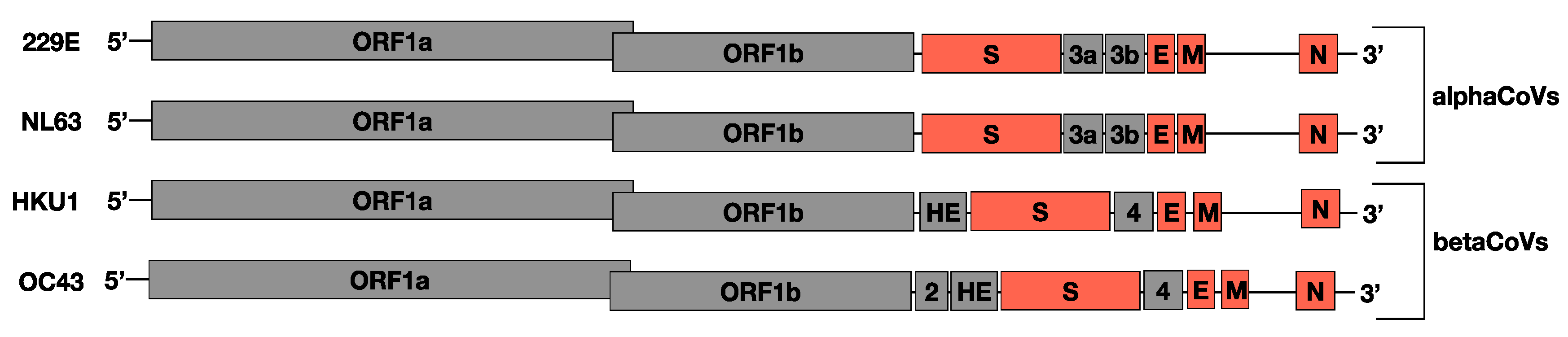
3.3. Recombination Patterns
3.4. Recombination Rates
3.5. Pairwise Diversity
3.6. Selection Analysis
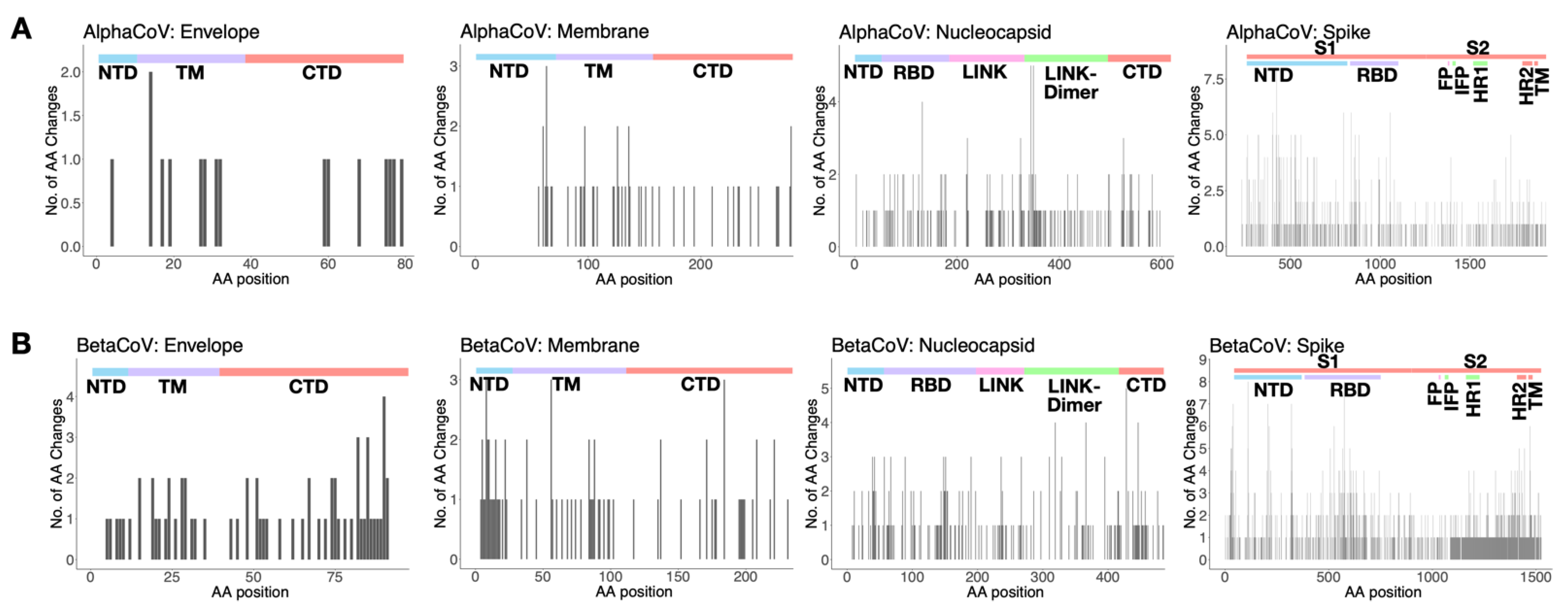
3.7. Amino Acid Substitutions
4. Discussion
Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuypers, J.; Martin, E.T.; Heugel, J.; Wright, N.; Morrow, R.; Englund, J.A. Clinical Disease in Children Associated With Newly Described Coronavirus Subtypes. Pediatrics 2007, 119, e70–e76. [Google Scholar] [CrossRef] [PubMed]
- Otieno, G.P.; Murunga, N.; Agoti, C.N.; Gallagher, K.E.; Awori, J.O.; Nokes, D.J. Surveillance of Endemic Human Coronaviruses (HCoV-NL63, OC43 and 229E) Associated with Childhood Pneumonia in Kilifi, Kenya. Wellcome Open Res. 2020, 5, 150. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Walsh, E.E.; Hayden, F.G. Rhinovirus and Coronavirus Infection—Associated Hospitalizations among Older Adults. J. Infect. Dis. 2002, 185, 1338–1341. [Google Scholar] [CrossRef] [Green Version]
- Esper, F.; Weibel, C.; Ferguson, D.; Landry, M.L.; Kahn, J.S. Coronavirus HKU1 Infection in the United States. Emerg. Infect. Dis. 2006, 12, 775–779. [Google Scholar] [CrossRef]
- Koetz, A.; Nilsson, P.; Lindén, M.; van der Hoek, L.; Ripa, T. Detection of Human Coronavirus NL63, Human Metapneumovirus and Respiratory Syncytial Virus in Children with Respiratory Tract Infections in South-West Sweden. Clin. Microbiol. Infect. 2006, 12, 1089–1096. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; Okamoto, M.; Ohmi, A.; Watanabe, O.; Miyabayashi, S.; Nishimura, H. Detection of Human Coronavirus-NL63 in Children in Japan. Pediatr. Infect. Dis. J. 2005, 24, 645–646. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Woo, P.C.Y.; Yip, C.C.Y.; Tse, H.; Tsoi, H.-W.; Cheng, V.C.C.; Lee, P.; Tang, B.S.F.; Cheung, C.H.Y.; Lee, R.A.; et al. Coronavirus HKU1 and Other Coronavirus Infections in Hong Kong. J. Clin. Microbiol. 2006, 44, 2063–2071. [Google Scholar] [CrossRef] [Green Version]
- Sipulwa, L.A.; Ongus, J.R.; Coldren, R.L.; Bulimo, W.D. Molecular Characterization of Human Coronaviruses and Their Circulation Dynamics in Kenya, 2009–2012. Virol. J. 2016, 13, 18. [Google Scholar] [CrossRef] [Green Version]
- Nyaguthii, D.M.; Otieno, G.P.; Kombe, I.K.; Koech, D.; Mutunga, M.; Medley, G.F.; Nokes, D.J.; Munywoki, P.K. Infection Patterns of Endemic Human Coronaviruses in Rural Households in Coastal Kenya. Wellcome Open Res. 2021, 6, 27. [Google Scholar] [CrossRef]
- Gaunt, E.R.; Hardie, A.; Claas, E.C.J.; Simmonds, P.; Templeton, K.E. Epidemiology and Clinical Presentations of the Four Human Coronaviruses 229E, HKU1, NL63, and OC43 Detected over 3 Years Using a Novel Multiplex Real-Time PCR Method. J. Clin. Microbiol. 2010, 48, 2940–2947. [Google Scholar] [CrossRef] [Green Version]
- Nickbakhsh, S.; Ho, A.; Marques, D.F.P.; McMenamin, J.; Gunson, R.N.; Murcia, P.R. Epidemiology of Seasonal Coronaviruses: Establishing the Context for the Emergence of Coronavirus Disease 2019. J. Infect. Dis. 2020, 222, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabeça, T.K.; Granato, C.; Bellei, N. Epidemiological and Clinical Features of Human Coronavirus Infections among Different Subsets of Patients. Influ. Other Respir. Viruses 2013, 7, 1040–1047. [Google Scholar] [CrossRef]
- Kiyuka, P.K.; Agoti, C.N.; Munywoki, P.K.; Njeru, R.; Bett, A.; Otieno, J.R.; Otieno, G.P.; Kamau, E.; Clark, T.G.; Van Der Hoek, L.; et al. Human Coronavirus NL63 Molecular Epidemiology and Evolutionary Patterns in Rural Coastal Kenya. J. Infect. Dis. 2018, 217, 1728–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munywoki, P.K.; Koech, D.C.; Agoti, C.N.; Lewa, C.; Cane, P.A.; Medley, G.F.; Nokes, D.J. The Source of Respiratory Syncytial Virus Infection In Infants: A Household Cohort Study In Rural Kenya. J. Infect. Dis. 2013, 209, 1685–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Dempsey, D.M.; Dutilh, B.E.; Harrach, B.; Harrison, R.L.; Hendrickson, R.C.; et al. Changes to Virus Taxonomy and the Statutes Ratified by the International Committee on Taxonomy of Viruses. Arch. Virol. 2020, 165, 2737–2748. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Huang, Y.; Lau, S.K.P.; Yuen, K.-Y. Coronavirus Genomics and Bioinformatics Analysis. Viruses 2010, 2, 1804–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, M.M. RNA Recombination in Animal and Plant Viruses. Microbiol. Rev. 1992, 56, 61–79. [Google Scholar] [CrossRef]
- Pasternak, A.; Spaan, W.J.M.; Snijder, E. Nidovirus Transcription: How to make sense…? J. Gen. Virol. 2006, 87, 1403–1421. [Google Scholar] [CrossRef]
- Li, X.; Luk, H.K.H.; Lau, S.K.P.; Woo, P.C.Y. Human Coronaviruses: General Features. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Hulswit, R.J.G.; De Haan, C.A.M.; Bosch, B.J. Coronavirus Spike Protein and Tropism Changes. Adv. Virus Res. 2016, 96, 29–57. [Google Scholar] [CrossRef]
- De Haan, C.A.M.; Rottier, P.J.M. Molecular Interactions in the Assembly of Coronaviruses. Adv. Virus Res. 2005, 64, 165–230. [Google Scholar]
- Xu, R.H.; He, J.F.; Evans, M.R.; Peng, G.W.; Field, H.E.; Yu, D.W.; Lee, C.K.; Luo, H.M.; Lin, W.S.; Lin, P.; et al. Epidemiologic Clues to SARS Origin in China. Emerg. Infect. Dis. 2004, 10, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.; Yuen, K.-Y. Infectious Diseases Emerging from Chinese Wet-Markets: Zoonotic Origins of Severe Respiratory Viral Infections. Curr. Opin. Infect. Dis. 2006, 19, 401–407. [Google Scholar] [CrossRef]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and Sources of Endemic Human Coronaviruses. Adv. Virus Res. 2018, 100, 163–188. [Google Scholar] [CrossRef]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Günther, S.; Preiser, W.; Van Der Werf, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.M.; Guan, Y.; Yuen, K.-Y. Severe Acute Respiratory Syndrome. Nat. Med. 2004, 10, S88–S97. [Google Scholar] [CrossRef]
- McIntosh, K.; Becker, W.B.; Chanock, R.M. Growth in Suckling-Mouse Brain of “IBV-like” Viruses from Patients with Upper Respiratory Tract Disease. Proc. Natl. Acad. Sci. USA 1967, 58, 2268–2273. [Google Scholar] [CrossRef] [Green Version]
- Bradburne, A.F.; Bynoe, M.L.; Tyrrell, D.A. Effects of a “New” Human Respiratory Virus in Volunteers. BMJ 1967, 3, 767–769. [Google Scholar] [CrossRef] [Green Version]
- Hamre, D.; Procknow, J.J. A New Virus Isolated from the Human Respiratory Tract. Proc. Soc. Exp. Biol. Med. 1966, 121, 190–193. [Google Scholar] [CrossRef]
- Van Der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.; Wolthers, K.C.; Wertheim-van Dillen, P.M.E.; Kaandorp, J.; Spaargaren, J.; Berkhout, B. Identification of a New Human Coronavirus. Nat. Med. 2004, 10, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.P.; Chu, C.; Chan, K.; Tsoi, H.; Huang, Y.; Wong, B.H.L.; Poon, R.W.S.; Cai, J.J.; Luk, W.; et al. Characterization and Complete Genome Sequence of a Novel Coronavirus, Coronavirus HKU1, from Patients with Pneumonia. J. Virol. 2005, 79, 884–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.; Lau, S.; Woo, P.; Yuen, K.-Y. Bats as a Continuing Source of Emerging Infections in Humans. Rev. Med. Virol. 2007, 17, 67–91. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Lau, C.C.Y.; Tsang, A.K.L.; Lau, J.H.N.; Bai, R.; Teng, J.L.L.; Tsang, C.C.C.; Wang, M.; et al. Discovery of Seven Novel Mammalian and Avian Coronaviruses in the Genus Deltacoronavirus Supports Bat Coronaviruses as the Gene Source of Alphacoronavirus and Betacoronavirus and Avian Coronaviruses as the Gene Source of Gammacoronavirus and Deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.F.-W.; To, K.K.-W.; Tse, H.; Jin, D.Y.; Yuen, K.Y. Interspecies Transmission and Emergence of Novel Viruses: Lessons from Bats and Birds. Trends Microbiol. 2013, 21, 544–555. [Google Scholar] [CrossRef]
- Vijgen, L.; Keyaerts, E.; Moës, E.; Thoelen, I.; Wollants, E.; Lemey, P.; Vandamme, A.-M.; Van Ranst, M. Complete Genomic Sequence of Human Coronavirus OC43: Molecular Clock Analysis Suggests a Relatively Recent Zoonotic Coronavirus Transmission Event. J. Virol. 2005, 79, 1595–1604. [Google Scholar] [CrossRef] [Green Version]
- Vijgen, L.; Keyaerts, E.; Lemey, P.; Maes, P.; Van Reeth, K.; Nauwynck, H.; Pensaert, M.; Van Ranst, M. Evolutionary History of the Closely Related Group 2 Coronaviruses: Porcine Hemagglutinating Encephalomyelitis Virus, Bovine Coronavirus, and Human Coronavirus OC43. J. Virol. 2006, 80, 7270–7274. [Google Scholar] [CrossRef] [Green Version]
- Corman, V.M.; Eckerle, I.; Memish, Z.A.; Liljander, A.M.; Dijkman, R.; Jonsdottir, H.R.; Ngeiywa, K.J.Z.J.; Kamau, E.; Younan, M.; Al Masri, M.; et al. Link of a ubiquitous human coronavirus to dromedary camels. Proc. Natl. Acad. Sci. USA 2016, 113, 9864–9869. [Google Scholar] [CrossRef] [Green Version]
- Corman, V.M.; Baldwin, H.J.; Tateno, A.F.; Zerbinati, R.M.; Annan, A.; Owusu, M.; Nkrumah, E.E.; Maganga, G.D.; Oppong, S.; Adu-Sarkodie, Y.; et al. Evidence for an Ancestral Association of Human Coronavirus 229E with Bats. J. Virol. 2015, 89, 11858–11870. [Google Scholar] [CrossRef] [Green Version]
- Pfefferle, S.; Oppong, S.; Drexler, J.F.; Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Müller, M.A.; Annan, A.; Vallo, P.; Adu-Sarkodie, Y.; et al. Distant Relatives of Severe Acute Respiratory Syndrome Coronavirus and Close Relatives of Human Coronavirus 229E in Bats, Ghana. Emerg. Infect. Dis. 2009, 15, 1377–1384. [Google Scholar] [CrossRef]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2016, 25, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakulenko, Y.; Deviatkin, A.; Lukashev, A. The Effect of Sample Bias and Experimental Artefacts on the Statistical Phylogenetic Analysis of Picornaviruses. Viruses 2019, 11, 1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, M.D.; Woolhouse, M.; Rambaut, A. The Effects of Sampling Strategy on the Quality of Reconstruction of Viral Population Dynamics Using Bayesian Skyline Family Coalescent Methods: A simulation study. Virus Evol. 2016, 2, vew003. [Google Scholar] [CrossRef] [Green Version]
- Chernomor, O.; Minh, B.Q.; Forest, F.; Klaere, S.; Ingram, T.; Henzinger, M.; Von Haeseler, A. Split Diversity in Constrained Conservation Prioritization Using Integer Linear Programming. Methods Ecol. Evol. 2014, 6, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.P.; Yip, C.C.Y.; Huang, Y.; Tsoi, H.-W.; Chan, K.-H.; Yuen, K.-Y. Comparative Analysis of 22 Coronavirus HKU1 Genomes Reveals a Novel Genotype and Evidence of Natural Recombination in Coronavirus HKU1. J. Virol. 2006, 80, 7136–7145. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for Inferring Very Large Phylogenies by Using the Neighbor-Joining Method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, M.M.; Cavanagh, D. The Molecular Biology of Coronaviruses. Adv. Virus Res. 1997, 48, 1–100. [Google Scholar] [CrossRef]
- Graham, R.L.; Baric, R.S. Recombination, Reservoirs, and the Modular Spike: Mechanisms of Coronavirus Cross-Species Transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef] [Green Version]
- Vakulenko, Y.; Deviatkin, A.; Drexler, J.; Lukashev, A. Modular Evolution of Coronavirus Genomes. Viruses 2021, 13, 1270. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. The Effect of Recombination on the Accuracy of Phylogeny Estimation. J. Mol. Evol. 2002, 54, 396–402. [Google Scholar] [CrossRef]
- Didelot, X.; Maiden, M.C. Impact of Recombination on Bacterial Evolution. Trends Microbiol. 2010, 18, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Jo, W.K.; Drosten, C.; Drexler, J.F. The Evolutionary Dynamics of Endemic Human Coronaviruses. Virus Evol. 2021, 7, veab020. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayres, D.L.; Darling, A.; Zwickl, D.J.; Beerli, P.; Holder, M.; Lewis, P.O.; Huelsenbeck, J.P.; Ronquist, F.; Swofford, D.L.; Cummings, M.P.; et al. BEAGLE: An Application Programming Interface and High-Performance Computing Library for Statistical Phylogenetics. Syst. Biol. 2011, 61, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, B.; Rambaut, A.; Drummond, A. Choosing Appropriate Substitution Models for the Phylogenetic Analysis of Protein-Coding Sequences. Mol. Biol. Evol. 2005, 23, 7–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian Coalescent Inference of Past Population Dynamics from Molecular Sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. Ggtree: An R Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Minin, V.N.; Suchard, M.A. Counting Labeled Transitions in Continuous-Time Markov Models of Evolution. J. Math. Biol. 2007, 56, 391–412. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Pond, S.L.K. Less Is More: An Adaptive Branch-Site Random Effects Model for Efficient Detection of Episodic Diversifying Selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Weaver, S.; Smith, M.D.; Wertheim, J.O.; Murrell, S.; Aylward, A.; Eren, K.; Pollner, T.; Martin, D.P.; Smith, D.M.; et al. Gene-Wide Identification of Episodic Selection. Mol. Biol. Evol. 2015, 32, 1365–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trovão, N.S.; Khan, S.M.; Lemey, P.; Nelson, M.I.; Cherry, J.L. Evolution of Influenza A Virus Hemagglutinin H1 and H3 across Host Species. bioRxiv 2022, 488870. [Google Scholar] [CrossRef]
- Tagliamonte, M.S.; Abid, N.; Borocci, S.; SanGiovanni, E.; Ostrov, D.A.; Pond, S.L.K.; Salemi, M.; Chillemi, G.; Mavian, C. Multiple Recombination Events and Strong Purifying Selection at the Origin of SARS-CoV-2 Spike Glycoprotein Increased Correlated Dynamic Movements. Int. J. Mol. Sci. 2020, 22, 80. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Lee, P.; Tsang, A.K.L.; Yip, C.C.Y.; Tse, H.; Lee, R.A.; So, L.-Y.; Lau, Y.-L.; Chan, K.-H.; Woo, P.C.Y.; et al. Molecular Epidemiology of Human Coronavirus OC43 Reveals Evolution of Different Genotypes over Time and Recent Emergence of a Novel Genotype due to Natural Recombination. J. Virol. 2011, 85, 11325–11337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyrc, K.; Dijkman, R.; Deng, L.; Jebbink, M.F.; Ross, H.A.; Berkhout, B.; van der Hoek, L. Mosaic Structure of Human Coronavirus NL63, One Thousand Years of Evolution. J. Mol. Biol. 2006, 364, 964–973. [Google Scholar] [CrossRef]
- Klinakis, A.; Cournia, Z.; Rampias, T. N-Terminal Domain Mutations of the Spike Protein Are Structurally Implicated in Epitope Recognition in Emerging SARS-CoV-2 Strains. Comput. Struct. Biotechnol. J. 2021, 19, 5556–5567. [Google Scholar] [CrossRef]
- Cui, Z.; Liu, P.; Wang, N.; Wang, L.; Fan, K.; Zhu, Q.; Wang, K.; Chen, R.; Feng, R.; Jia, Z.; et al. Structural and Functional Characterizations of Infectivity and Immune Evasion of SARS-CoV-2 Omicron. Cell 2022, 185, 860–871. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron Escapes the Majority of Existing SARS-CoV-2 Neutralizing Antibodies. Nature 2021, 602, 657–663. [Google Scholar] [CrossRef]
- Kumar, S.; Thambiraja, T.S.; Karuppanan, K.; Subramaniam, G. Omicron and Delta Variant of SARS-CoV-2: A Comparative Computational Study of Spike Protein. J. Med. Virol. 2021, 94, 1641–1649. [Google Scholar] [CrossRef]
- Maaroufi, H. The N764K and N856K Mutations in SARS-CoV-2 Omicron S Protein Generate Potential Cleavage Sites for SKI-1/S1P Protease. bioRxiv 2022, 477298. [Google Scholar] [CrossRef]
- Dudas, G.; Carvalho, L.M.; Rambaut, A.; Bedford, T. MERS-CoV Spillover at the Camel-Human Interface. eLife 2018, 7, e31257. [Google Scholar] [CrossRef] [PubMed]
- VanInsberghe, D.; Neish, A.S.; Lowen, A.C.; Koelle, K. Recombinant SARS-CoV-2 Genomes Are Currently Circulating at Low Levels. bioRxiv Prepr. Serv. Biol. 2021. [Google Scholar] [CrossRef]
- Simon-Loriere, E.; Holmes, E.C. Why Do RNA Viruses Recombine? Nat. Rev. Genet. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S. Why Are RNA Virus Mutation Rates so Damn High? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kistler, K.E.; Bedford, T. Evidence for Adaptive Evolution in the Receptor-Binding Domain of Seasonal Coronaviruses OC43 and 229e. eLife 2021, 10, e64509. [Google Scholar] [CrossRef]
- Müller, N.F.; Kistler, K.E.; Bedford, T. Recombination Patterns in Coronaviruses. bioRxiv Prepr. Serv. Biol. 2021, 441806. [Google Scholar] [CrossRef]
- Al-Khannaq, M.N.; Ng, K.T.; Oong, X.Y.; Pang, Y.K.; Takebe, Y.; Chook, J.B.; Hanafi, N.S.; Kamarulzaman, A.; Tee, K.K. Molecular Epidemiology and Evolutionary Histories of Human Coronavirus OC43 and HKU1 among Patients with Upper Respiratory Tract Infections in Kuala Lumpur, Malaysia. Virol. J. 2016, 13, 33. [Google Scholar] [CrossRef] [Green Version]
- Marin, J.; Hedges, S.B. Undersampling Genomes Has Biased Time and Rate Estimates Throughout the Tree of Life. Mol. Biol. Evol. 2018, 35, 2077–2084. [Google Scholar] [CrossRef] [Green Version]
- Flint, J.; Racaniello, V.R.; Skalka, A.M.; Rall, G. Principles of Virology, Bundle. Princ. Virol. Bundle 2015. [Google Scholar] [CrossRef]
- Banner, L.R.; Mc Lai, M. Random Nature of Coronavirus RNA Recombination in the Absence of Selection Pressure. Virology 1991, 185, 441–445. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Species | WGS/ORF | Zoonotic Origins of the Human Coronavirus {% Markov Jumps} |
|---|---|---|---|
| alphaCoVs | 229E | WGS | Bat {65} Camelid {34} |
| Spike | Bat {73} Camelid {27} | ||
| Nucleocapsid | Bat {99} | ||
| Membrane | Bat {98} | ||
| Envelope | Bat {99} | ||
| NL63 | WGS | Bat {97} | |
| Spike | Bat {99} | ||
| Nucleocapsid | Bat {95} | ||
| Membrane | Bat {97} | ||
| Envelope | Bat {98} | ||
| betaCoVs | HKU1 | WGS | Murine {83} |
| Spike | Murine {92} | ||
| Nucleocapsid | Murine {72} | ||
| Membrane | Murine {78} | ||
| Envelope | Murine {71} | ||
| OC43 | WGS | Bovine {36} Murine {34} Rabbit {10} Canine {9} | |
| Spike | Bovine {35} Murine {19} Camel {14} Rabbit {14} Canine {8} Equine {5} | ||
| Nucleocapsid | Porcine {25} Bovine {23} Murine {18} Rabbit {15} Equine {10} | ||
| Membrane | Camel {75} Murine {6} Rabbit {3} Porcine {3} Equine {2} Bovine {2} Bat {2} | ||
| Envelope | Murine {30} Porcine {24} Bovine {20} Equine {8} Rabbit {6} Camel {3} |
| Genome Segment | Coronavirus Genus/Species | R/Theta | r/m | δν | δ |
|---|---|---|---|---|---|
| (a) Spike | alphaCoVs | 1.103 | 41.590 | 37.718 | 335.413 |
| betaCoVs | 2.193 | 28.494 | 12.994 | 208.896 | |
| (b) WGS | alphaCoVs | 0.014 | 0.840 | 58.043 | 1032.937 |
| betaCoVs | 0.025 | 0.867 | 35.369 | 780.250 | |
| (c) WGS: Interspecies analysis (between one sHCoV species and all other non-human CoVs) | 229E | 0.013 | 0.741 | 57.465 | 1146.815 |
| NL63 | 0.016 | 0.943 | 59.561 | 970.252 | |
| OC43 | 0.027 | 0.888 | 32.291 | 687.266 | |
| HKU1_all | 0.025 | 0.890 | 35.890 | 760.242 | |
| HKU1 Genotype A | 0.024 | 0.840 | 35.170 | 747.680 | |
| HKU1 Genotype B | 0.024 | 0.865 | 36.718 | 787.036 | |
| (d) WGS: Intraspecies analysis (within one sHCoV species) | 229E | 1.693 | 2.915 | 1.722 | 649.287 |
| NL63 | 0.138 | 0.885 | 6.415 | 106.019 | |
| OC43 | 0.329 | 1.636 | 4.967 | 675.283 | |
| HKU1_all | 0.029 | 1.912 | 66.927 | 743.307 | |
| HKU1 Genotype A | 0.216 | 0.537 | 2.493 | 176.197 | |
| HKU1 Genotype B | 0.056 | 2.123 | 37.595 | 811.754 |
| sHCoV Species | WGS | Spike | Envelope | Membrane | Nucleocapsid |
|---|---|---|---|---|---|
| 229E | 0.0048 | 0.0348 | 0.0040 | 0.0079 | 0.0121 |
| NL63 | 0.0081 | 0.0362 | 0.0077 | 0.0081 | 0.0076 |
| OC43 | 0.0076 | 0.0268 | 0.0085 | 0.0082 | 0.0077 |
| HKU1_all | 0.0222 | 0.1040 | 0.0795 | 0.0176 | 0.0231 |
| HKU1 Genotype A | 0.0028 | 0.0049 | 0.0010 | 0.0020 | 0.0062 |
| HKU1 Genotype B | 0.0122 | 0.0120 | 0.0072 | 0.0039 | 0.0049 |
| ORF | Positions Shared by Alpha sHCoVs 229E and NL63 | Positions Shared by Beta sHCoVs OC43 and HKU1 | Positions Shared between Alpha and Beta sHCoVs |
|---|---|---|---|
| Spike | 769, 1009, 1210, and 1253 | 8.21, 8.27, 8.31, 146, 154, 257, 329, 463.11, 489, 490.5, 490.7, 490.9, 497, 504.51, 1135, 1181, and 1221 | 60, 74, 152, 162, 208, 271, 295, 321, 367, 441, 476, 580, 624, 628, 629, 641, 680, 684, 751, 776, 795, 817, 932, 939, 957, 975, 1061, 1071, 1076, 1101, 1104, 1116, 1124, 1142, 1153, 1154, 1165, 1170, 1171, 1174, 1185, 1192, 1224, 1225, 1226, 1227, 1228, and 1272 |
| Nucleocapsid | 158, 160, 205, 216, 236, 252.7, 366, and 402 | 32 and 373.3 | 37, 62, 125, 127, 166, 182, 208, 210, 212, 240, 241, 249, 256, 297, 321, 323, 354, 364, 365, 384, 389, 390, 392, and 394 |
| Membrane | 8, 11, 72, and 82 | - | 10, 12, 15, 188, and 222 |
| Envelope | - | - | 3, 26, 27, 66, 72, and 73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otieno, J.R.; Cherry, J.L.; Spiro, D.J.; Nelson, M.I.; Trovão, N.S. Origins and Evolution of Seasonal Human Coronaviruses. Viruses 2022, 14, 1551. https://doi.org/10.3390/v14071551
Otieno JR, Cherry JL, Spiro DJ, Nelson MI, Trovão NS. Origins and Evolution of Seasonal Human Coronaviruses. Viruses. 2022; 14(7):1551. https://doi.org/10.3390/v14071551
Chicago/Turabian StyleOtieno, James R., Joshua L. Cherry, David J. Spiro, Martha I. Nelson, and Nídia S. Trovão. 2022. "Origins and Evolution of Seasonal Human Coronaviruses" Viruses 14, no. 7: 1551. https://doi.org/10.3390/v14071551