
Diversity of SIRV-like Viruses from a North American Population


Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Growth Conditions
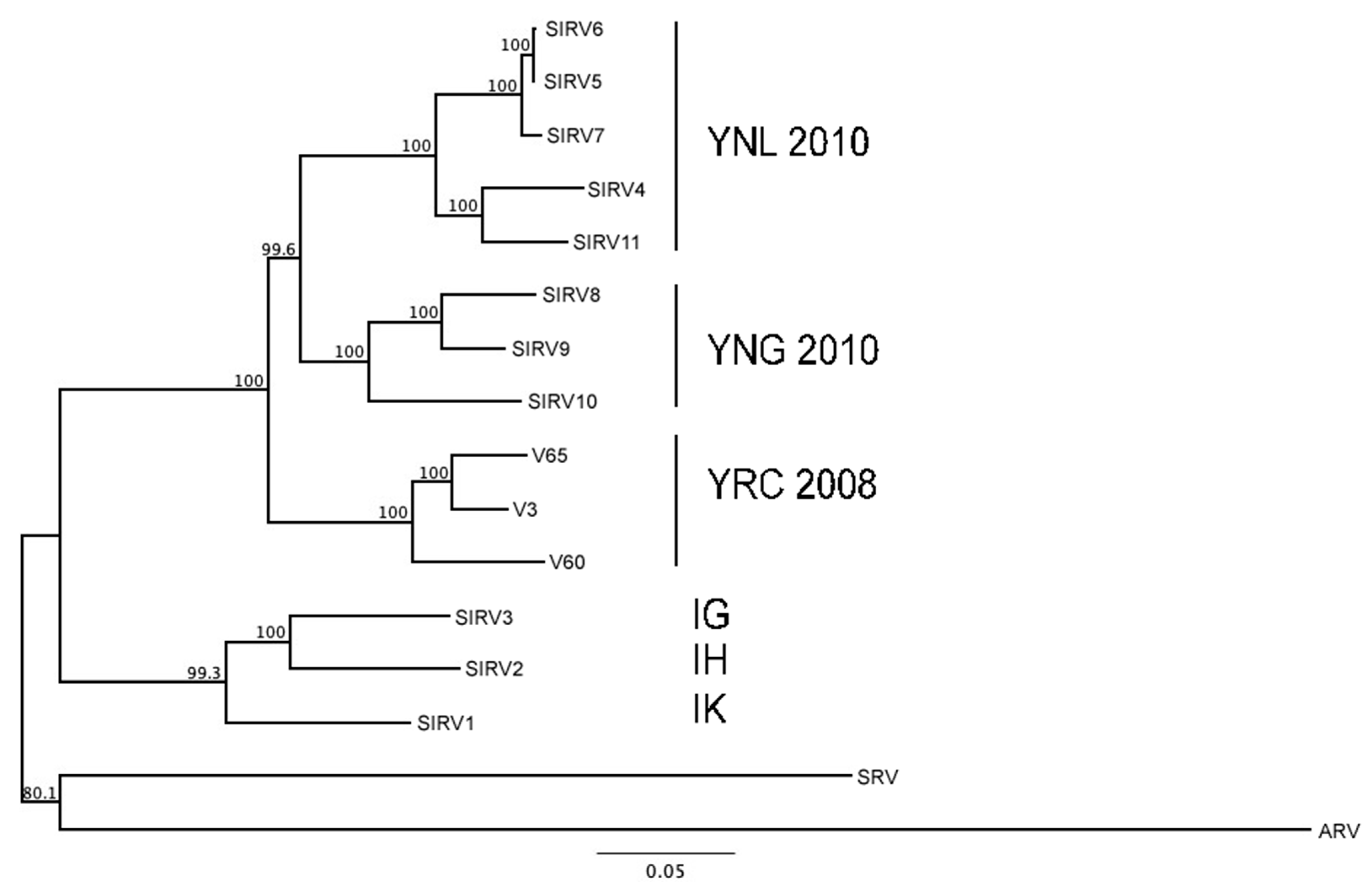
2.2. DNA Analyses
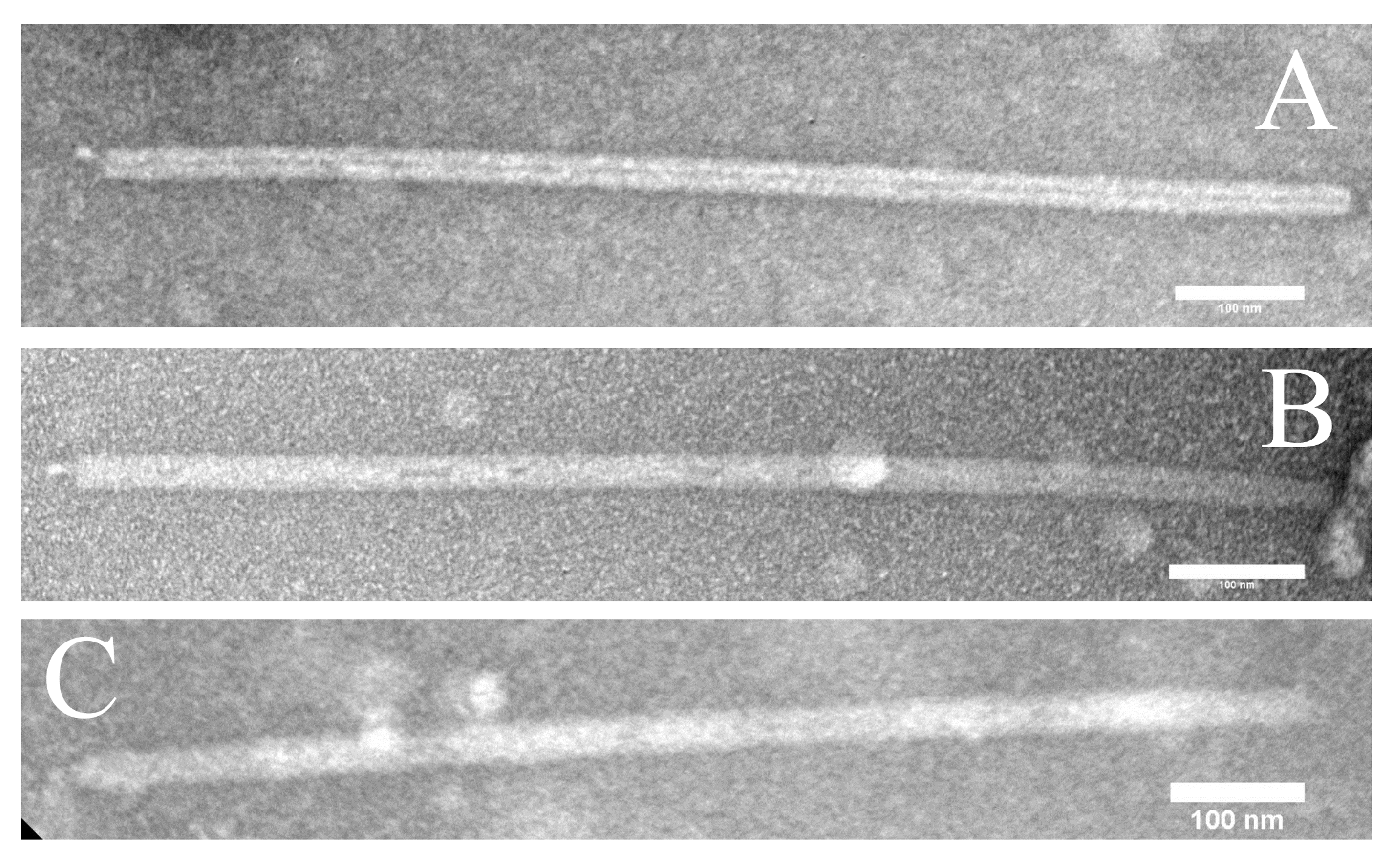
2.3. Electron Microscopy
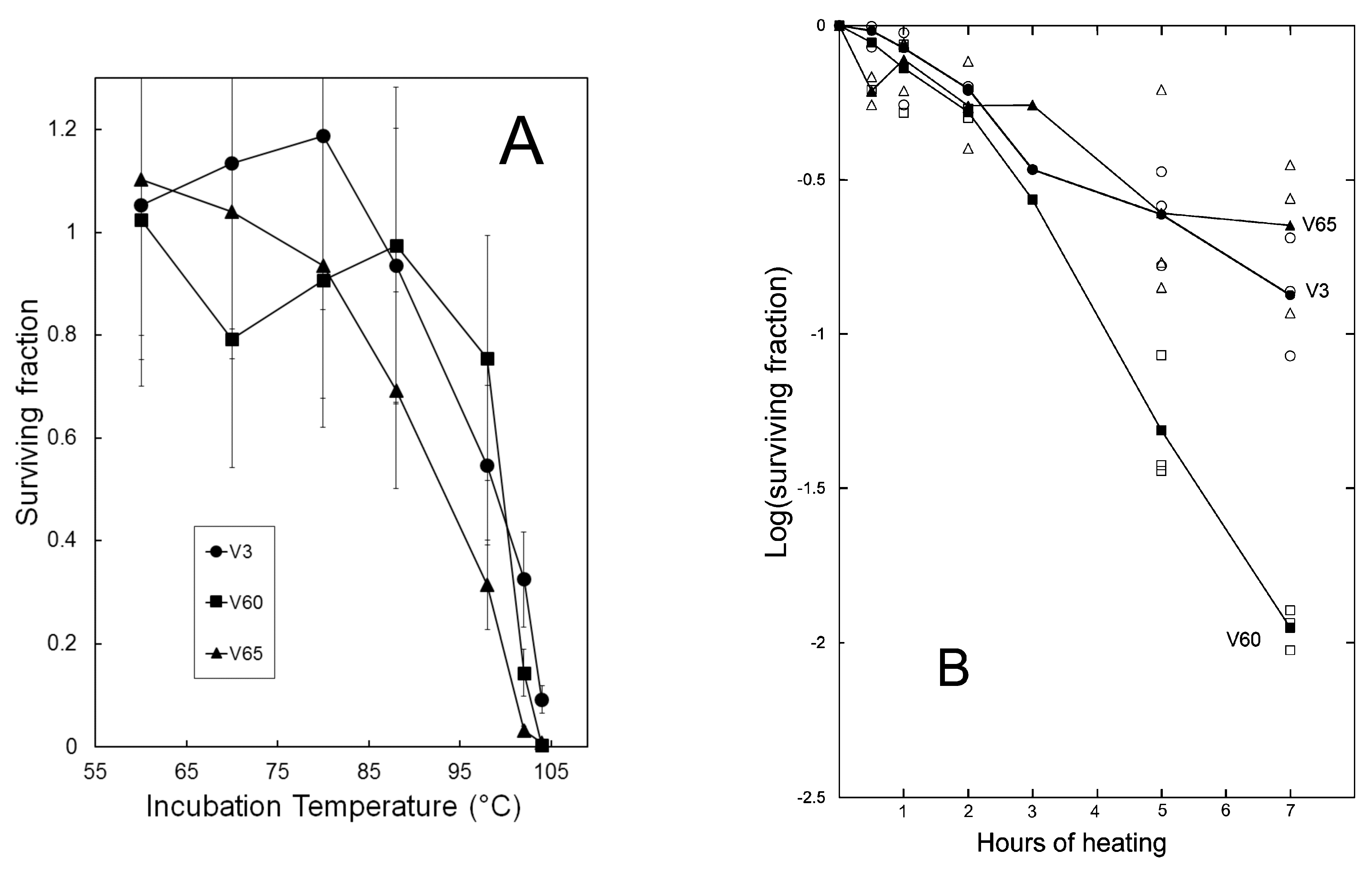
2.4. Inactivation Assays
2.5. Measurements of Acquired Resistance
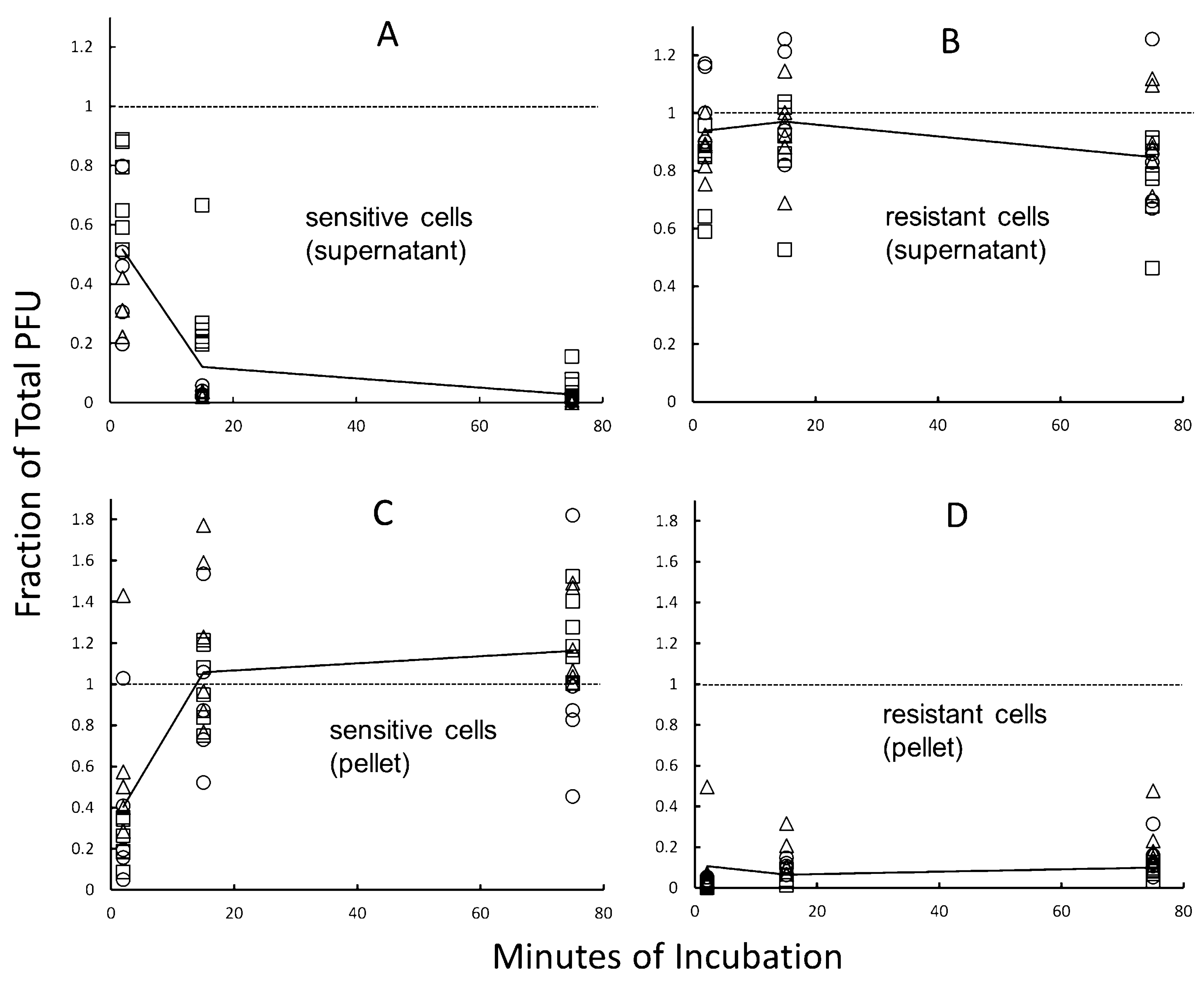
2.6. Virion Binding Assays
3. Results
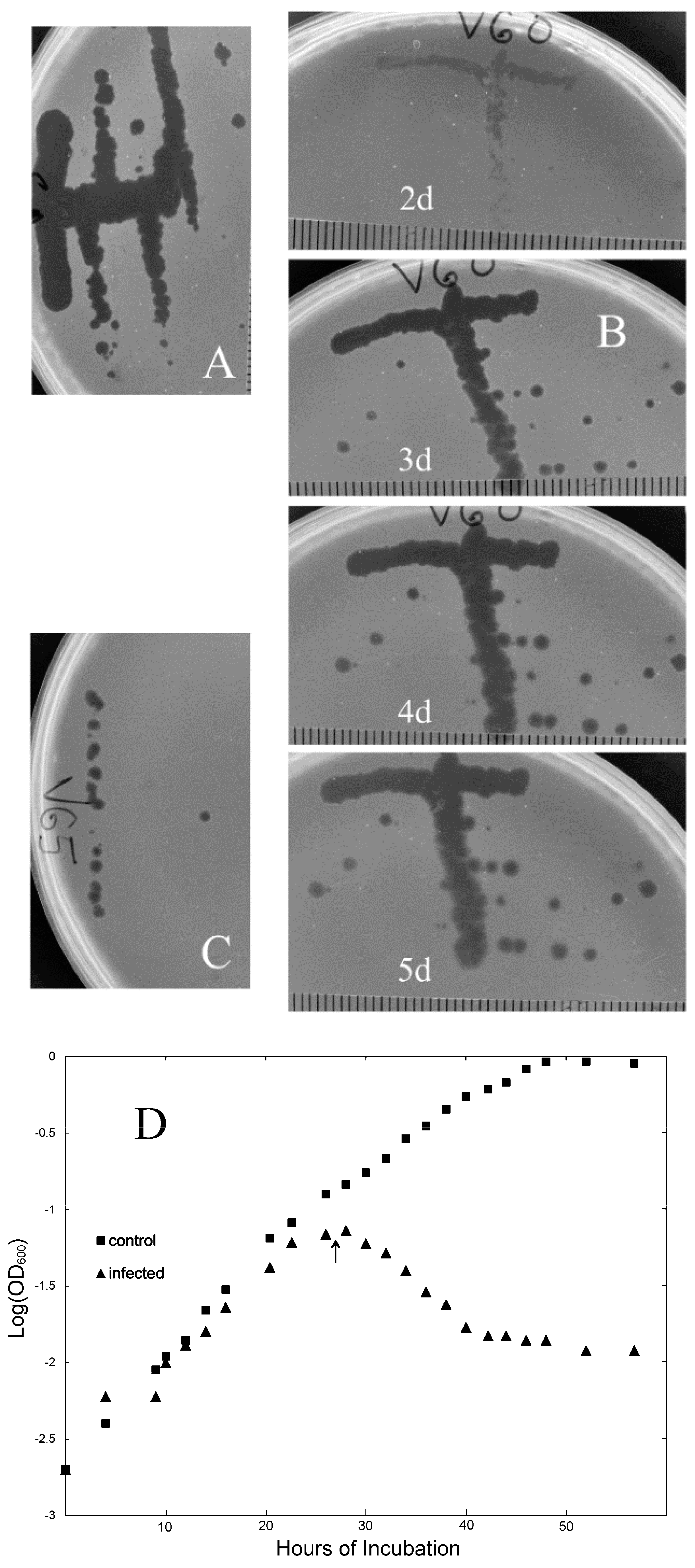
3.1. New Isolates of Lytic Archaeal Virus
3.2. Virion Stability
3.3. Evaluating Population-Level Variation
3.4. Acquired Resistance to Viral Infection
3.5. Cellular Basis of High-Frequency Resistance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, R.; Rossler, N.; Iro, M.; Scholz, H.; Witte, A. Haloarchaeal myovirus phiCh1 harbours a phase variation system for the production of protein variants with distinct cell surface adhesion specificities. Mol. Microbiol. 2012, 83, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.; Fischer, M.A.; Kupczok, A.; Reetz, J.; Kern, T.; Schmitz, R.A.; Rother, M. Characterization of the lytic archaeal virus Drs3 infecting Methanobacterium formicicum. Arch. Virol. 2019, 164, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.; Stedman, K.; Snyder, J.; Wiedenheft, B.; Willits, D.; Brumfield, S.; McDermott, T.; Young, M.J. Viruses from extreme thermal environments. Proc. Natl. Acad. Sci. USA 2001, 98, 13341–13345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietila, M.K.; Demina, T.A.; Atanasova, N.S.; Oksanen, H.M.; Bamford, D.H. Archaeal viruses and bacteriophages: Comparisons and contrasts. Trends Microbiol. 2014, 22, 334–344. [Google Scholar] [CrossRef]
- Pina, M.; Bize, A.; Forterre, P.; Prangishvili, D. The archeoviruses. FEMS Microbiol. Rev. 2011, 35, 1035–1054. [Google Scholar] [CrossRef]
- Haring, M.; Vestergaard, G.; Rachel, R.; Chen, L.; Garrett, R.A.; Prangishvili, D. Virology: Independent virus development outside a host. Nature 2005, 436, 1101–1102. [Google Scholar] [CrossRef]
- Brumfield, S.K.; Ortmann, A.C.; Ruigrok, V.; Suci, P.; Douglas, T.; Young, M.J. Particle assembly and ultrastructural features associated with replication of the lytic archaeal virus sulfolobus turreted icosahedral virus. J. Virol. 2009, 83, 5964–5970. [Google Scholar] [CrossRef] [Green Version]
- Bize, A.; Karlsson, E.A.; Ekefjard, K.; Quax, T.E.; Pina, M.; Prevost, M.C.; Forterre, P.; Tenaillon, O.; Bernander, R.; Prangishvili, D. A unique virus release mechanism in the Archaea. Proc. Natl. Acad. Sci. USA 2009, 106, 11306–11311. [Google Scholar] [CrossRef] [Green Version]
- DiMaio, F.; Yu, X.; Rensen, E.; Krupovic, M.; Prangishvili, D.; Egelman, E.H. Virology. A virus that infects a hyperthermophile encapsidates A-form DNA. Science 2015, 348, 914–917. [Google Scholar] [CrossRef] [Green Version]
- Sakai, H.D.; Kurosawa, N. Saccharolobus caldissimus gen. nov., sp. nov., a facultatively anaerobic iron-reducing hyperthermophilic archaeon isolated from an acidic terrestrial hot spring, and reclassification of Sulfolobus solfataricus as Saccharolobus solfataricus comb. nov. and Sulfolobus shibatae as Saccharolobus shibatae comb. nov. Int. J. Syst. Evol. Microbiol. 2018, 68, 1271–1278. [Google Scholar] [CrossRef]
- Zillig, W.; Kletzin, A.; Schleper, C.; Holz, I.; Janekovic, D.; Hain, J.; Lanzendorfer, M.; Kristjansson, J. Screening for Sulfolobales, their plasmids, and their viruses in Icelandic solfataras. Syst. Appl. Microbiol. 1994, 16, 609–628. [Google Scholar] [CrossRef]
- Prangishvili, D.; Arnold, H.P.; Gotz, D.; Ziese, U.; Holz, I.; Kristjansson, J.K.; Zillig, W. A novel virus family, the Rudiviridae: Structure, virus-host interactions and genome variability of the Sulfolobus viruses SIRV1 and SIRV2. Genetics 1999, 152, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, S.; Le Moine Bauer, S.; Garrett, R.A. Inter-viral conflicts that exploit host CRISPR immune systems of Sulfolobus. Mol. Microbiol. 2014, 91, 900–917. [Google Scholar] [CrossRef] [Green Version]
- Bautista, M.A.; Black, J.A.; Youngblut, N.D.; Whitaker, R.J. Differentiation and Structure in Sulfolobus islandicus Rod-Shaped Virus Populations. Viruses 2017, 9, 120. [Google Scholar] [CrossRef] [Green Version]
- Steinmetz, N.F.; Bize, A.; Findlay, K.C.; Lomonossoff, G.P.; Manchester, M.; Evans, D.J.; Prangishvili, D. Site-specific and Spatially Controlled Addressability of a New Viral Nanobuilding Block: Sulfolobus islandicus Rod-shaped Virus 2. Adv. Funct. Mater. 2008, 18, 3478–3486. [Google Scholar] [CrossRef]
- Grogan, D.W.; Ozarzak, M.A.; Bernander, R. Variation in gene content among geographically diverse Sulfolobus isolates. Environ. Microbiol. 2008, 10, 137–146. [Google Scholar] [CrossRef]
- Blount, Z.D.; Grogan, D.W. New insertion sequences of Sulfolobus: Functional properties and implications for genome evolution in hyperthermophilic archaea. Mol. Microbiol. 2005, 55, 312–325. [Google Scholar] [CrossRef]
- Whitaker, R.J.; Grogan, D.W.; Taylor, J.W. Geographic barriers isolate endemic populations of hyperthermophilic archaea. Science 2003, 301, 976–978. [Google Scholar] [CrossRef]
- Grogan, D.W.; Gunsalus, R.P. Sulfolobus acidocaldarius synthesizes UMP via a standard de novo pathway: Results of biochemical-genetic study. J. Bacteriol. 1993, 175, 1500–1507. [Google Scholar] [CrossRef] [Green Version]
- Boom, R.; Sol, C.J.; Salimans, M.M.; Jansen, C.L.; Wertheim-van Dillen, P.M.; van der Noordaa, J. Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 1990, 28, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grogan, D.W. Isolation of Sulfolobus acidocaldarius mutants. In Archaea: A Laboratory Manual; Robb, F.T., DasSarma, S., Fleischmann, E.M., Place, A.R., Schreier, H.J., Sowers, K.R., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1995; pp. 125–132. [Google Scholar]
- Hall, B.M.; Ma, C.X.; Liang, P.; Singh, K.K. Fluctuation analysis CalculatOR: A web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics 2009, 25, 1564–1565. [Google Scholar] [CrossRef] [Green Version]
- Flynn, K.M.; Vohr, S.H.; Hatcher, P.J.; Cooper, V.S. Evolutionary rates and gene dispensability associate with replication timing in the archaeon Sulfolobus islandicus. Genome Biol. Evol. 2010, 2, 859–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quemin, E.R.; Lucas, S.; Daum, B.; Quax, T.E.; Kuhlbrandt, W.; Forterre, P.; Albers, S.V.; Prangishvili, D.; Krupovic, M. First insights into the entry process of hyperthermophilic archaeal viruses. J. Virol. 2013, 87, 13379–13385. [Google Scholar] [CrossRef] [Green Version]
- Meyer, J.R.; Dobias, D.T.; Weitz, J.S.; Barrick, J.E.; Quick, R.T.; Lenski, R.E. Repeatability and contingency in the evolution of a key innovation in phage lambda. Science 2012, 335, 428–432. [Google Scholar] [CrossRef] [Green Version]
- Setlow, P. I will survive: DNA protection in bacterial spores. Trends Microbiol. 2007, 15, 172–180. [Google Scholar] [CrossRef]
- Wood, E.R.; Ghane, F.; Grogan, D.W. Genetic responses of the thermophilic archaeon Sulfolobus acidocaldarius to short-wavelength UV light. J. Bacteriol. 1997, 179, 5693–5698. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; Volume 3, p. A1.21. [Google Scholar]
- Pollard, E.C.; Solosko, W. The thermal inactivation of T4 and lambda bacteriophage. Biophys. J. 1971, 11, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.X.; Slater, M.R.; Ackermann, H.-W. Isolation and characterization of Thermus bacteriophages. Arch. Virol. 2006, 151, 663–679. [Google Scholar] [CrossRef]
- Halsall, T.G.; Hirst, E.L.; Jones, J.K. Oxidation of carbohydrates by the periodate ion. J. Chem. Soc. 1947, 172, 1427–1432. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.L.; Grogan, D.W. Rates of spontaneous mutation in an archaeon from geothermal environments. J. Bacteriol. 1997, 179, 3298–3303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martusewitsch, E.; Sensen, C.W.; Schleper, C. High spontaneous mutation rate in the hyperthermophilic archaeon Sulfolobus solfataricus is mediated by transposable elements. J. Bacteriol. 2000, 182, 2574–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkner, S.; Lipps, G. Mutation and reversion frequencies of different Sulfolobus species and strains. Extremophiles 2008, 12, 263–270. [Google Scholar] [CrossRef]
- Drake, J.W. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. USA 1991, 88, 7160–7164. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; He, F.; Bhoobalan-Chitty, Y.; Martinez-Alvarez, L.; Guo, Y.; Peng, X. Unveiling cell surface and type IV secretion proteins responsible for archaeal rudivirus entry. J. Virol. 2014, 88, 10264–10268. [Google Scholar] [CrossRef] [Green Version]
- Rowland, E.F.; Bautista, M.A.; Zhang, C.; Whitaker, R.J. Surface resistance to SSVs and SIRVs in pilin deletions of Sulfolobus islandicus. Mol. Microbiol. 2020, 113, 718–727. [Google Scholar] [CrossRef] [Green Version]
- Grogan, D.W. Isolation and fractionation of cell envelope from the extreme thermo-acidophile Sulfolobus acidocaldarius. J. Microbiol. Methods 1996, 26, 35–43. [Google Scholar] [CrossRef]
- Albers, S.V.; Meyer, B.H. The archaeal cell envelope. Nat. Rev. Microbiol. 2011, 9, 414–426. [Google Scholar] [CrossRef]
- Papathanasiou, P.; Erdmann, S.; Leon-Sobrino, C.; Sharma, K.; Urlaub, H.; Garrett, R.A.; Peng, X. Stable maintenance of the rudivirus SIRV3 in a carrier state in Sulfolobus islandicus despite activation of the CRISPR-Cas immune response by a second virus SMV1. RNA Biol. 2019, 16, 557–565. [Google Scholar] [CrossRef] [Green Version]
- Casadesus, J.; Low, D.A. Programmed heterogeneity: Epigenetic mechanisms in bacteria. J. Biol. Chem. 2013, 288, 13929–13935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaumont, H.J.; Gallie, J.; Kost, C.; Ferguson, G.C.; Rainey, P.B. Experimental evolution of bet hedging. Nature 2009, 462, 90–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppenheim, A.B.; Kobiler, O.; Stavans, J.; Court, D.L.; Adhya, S. Switches in bacteriophage lambda development. Annu. Rev. Genet 2005, 39, 409–429. [Google Scholar] [CrossRef] [PubMed]
- Sandmeier, H. Acquisition and rearrangement of sequence motifs in the evolution of bacteriophage tail fibres. Mol. Microbiol. 1994, 12, 343–350. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Designation | Origin | Reference |
|---|---|---|
| SIRV1 | Iceland, Kverkjoll solfatara | [12] |
| SIRV2 | Iceland, Hveragerdi solfatara | [13] |
| SIRV3 | Iceland, Gunnuhver solfatara | [14] |
| SIRV4–7, 11 | Wyoming (USA), Nymph Lake | [15] |
| (44.7519, −110.7286) a | ||
| SIRV8–10 | Wyoming (USA), Norris Geyser Basin | [15] |
| (44.7278, −110.7139) a | ||
| V1–65 | Wyoming (USA), Midway Geyser Basin | this work |
| (44.5089, −110.8089) a | ||
| Y11 40.32 | Wyoming (USA), Midway Geyser Basin | this work |
| (44.5089, −110.8089) a | ||
| Y11 27.5 | Wyoming (USA), Norris Geyser Basin | this work |
| (44.7286, −110.7134) a |
| Surviving Fraction (Std. Dev.) | |||
|---|---|---|---|
| Treatment | V3 | V60 | V65 |
| NaIO4 | 0.0247 (0.0189) | 0.0016 (0.00074) | 0.00014 (3.8 × 10−5) |
| CHCl3 | 0.0210 (0.0189) | 0.0338 (0.0126) | 0.180 (0.248) |
| Urea | 0.0067 (0.0075) | 0.0071 (0.0092) | 0.0503 (0.0690) |
| Guanidine HCl | 0.219 (0.222) | 0.544 (0.564) | 0.0010 (0.0012) |
| Thermal Area | Sample No. | pH | Temp., °C | CFU/mL | PFU/mL |
|---|---|---|---|---|---|
| Ebro Springs | |||||
| 1 | 3.5 | 58 | <10 | <5 | |
| 2 | - | 57 | <10 | <5 | |
| 3 | - | 58 | <10 | <5 | |
| Washburn area | |||||
| 4 | 2.4 | 71 | <10 | <5 | |
| 5 | 4.2 | 69 | <10 | <5 | |
| 6 | 3.2 | 69 | 550 | <5 | |
| 7 | 3 | 76 | 160 | <5 | |
| 8 | 3.3 | 87 | 170 | <5 | |
| 9 | 3.1 | 76 | 200 | <5 | |
| 10 | 2.5 | 78 | <10 | <5 | |
| 11 | 2.6 | 82 | 20 | <5 | |
| 12 | 2.8 | 73 | 110 | <5 | |
| 13 | 3 | 61 | 30 | <5 | |
| Norris Geyser Basin | |||||
| 14 | 5 | 83 | 270 | <5 | |
| 15 | 3.5 | 86 | <10 | <5 | |
| 16 | 3.9 | 69 | 5000 | <5 | |
| 17 | 3.2 | 76 | 12,000 | <5 | |
| 18 | 3.2 | 83 | 140 | <5 | |
| 19 | 3 | 77 | 500 | <5 | |
| 20 | 3 | 83 | 25 | <5 | |
| 21 | 2.7 | 89 | <10 | <5 | |
| 22 | 3 | 59 | 5000 | <5 | |
| 23 | 3 | 82 | <10 | <5 | |
| 24 | 2.7 | 82 | <10 | <5 | |
| 25 | 3.1 | 77 | 7000 | <5 | |
| 26 | 3 | 82 | <10 | <5 | |
| 27 | 2.5 | 70 | 10,000 | 155 | |
| 28 | 3 | 75 | 2200 | 25 | |
| 29 | 3 | 60 | <10 | <5 | |
| 30 | 3 | 70 | 900 | <5 | |
| 31 | 4.5 | 79 | 3800 | <5 | |
| 32 | 2.7 | 61 | <10 | <5 | |
| 33 | 2.7 | 73 | <10 | <5 | |
| 34 | 2.4 | 84 | <10 | <5 | |
| 35 | 2.4 | 84 | <10 | <5 | |
| 36 | 4.7 | 81 | <10 | <5 | |
| 37 | 4 | 72 | <10 | <5 | |
| 38 | 4 | 80 | 5500 | <5 | |
| Rabbit Creek area | |||||
| 39 | 4 | 81.5 | 30,000 | 50 | |
| 40 | 2.4 | 80 | 40,000 | 170 | |
| 41 | 3.9 | 82 | 30,000 | <5 | |
| 42 | 3.9 | 82 | 35,000 | <5 | |
| 43 | - | 81 | 14,000 | 5 | |
| 44 | 3.9 | 78 | 47,000 | <5 | |
| 45 | - | 81 | 30,000 | 5 | |
| 46 | 4 | 79 | 20,000 | <5 | |
| 47 | - | 74 | 30,000 | <5 | |
| 48 | 3.9 | 69 | 47,000 | <5 | |
| 49 | - | 66 | 97,000 | <5 | |
| 50 | 3.5 | 80 | 5000 | <5 | |
| 51 | - | 69 | 650 | <5 |
| Selected Resistance a | ||
| Challenge Virus | µ | 95% Confidence Limits |
| V3 | 4.47 × 10−4 | 2.87, 8.49 × 10−4 |
| V60 | 3.46 × 10−4 | 2.48, 5.28 × 10−4 |
| V65 | 6.65 × 10−4 | 4.17, 8.02 × 10−4 |
| Mean | 4.86 × 10−4 | |
| Non-Selected Resistance b | ||
| Resistant to | Number Observed | |
| V3 only | 1 | |
| V60 only | 3 | |
| V65 only | 0 | |
| All three | 2 | |
| (Total: 6) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fackler, J.R.; Dworjan, M.; Gazi, K.S.; Grogan, D.W. Diversity of SIRV-like Viruses from a North American Population. Viruses 2022, 14, 1439. https://doi.org/10.3390/v14071439
Fackler JR, Dworjan M, Gazi KS, Grogan DW. Diversity of SIRV-like Viruses from a North American Population. Viruses. 2022; 14(7):1439. https://doi.org/10.3390/v14071439
Chicago/Turabian StyleFackler, Joseph R., Michael Dworjan, Khaled S. Gazi, and Dennis W. Grogan. 2022. "Diversity of SIRV-like Viruses from a North American Population" Viruses 14, no. 7: 1439. https://doi.org/10.3390/v14071439