
The Viral Susceptibility of the Haloferax Species

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
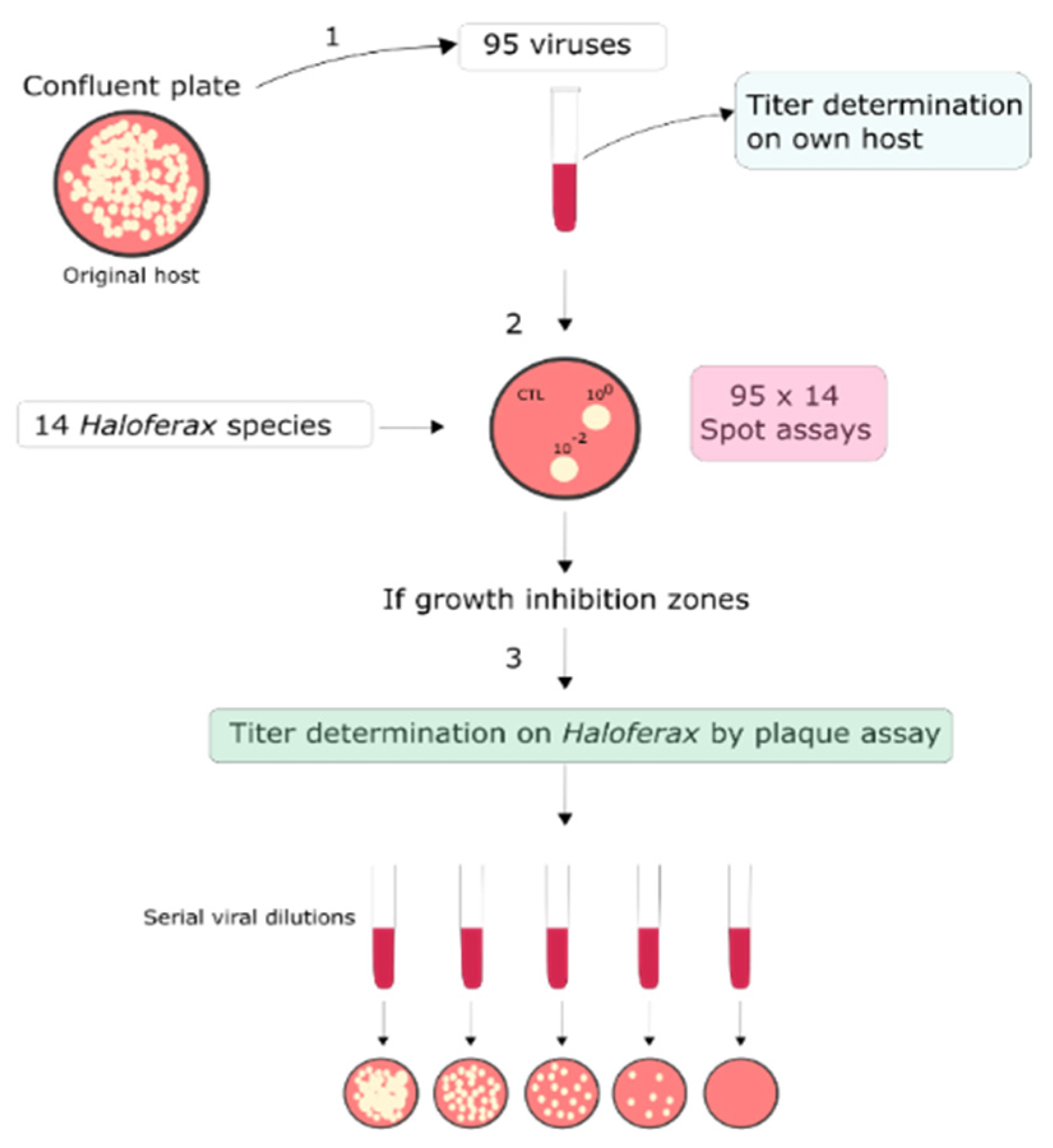
2.1. Archaeal Viruses and Strains and Growth Conditions
2.2. Sensitivity of Haloferax Strains to Euryarchaeal Viruses
2.3. Viral Comparative Genomics
3. Results and Discussion
3.1. Detection of Viruses Infecting Haloferax Strains
3.2. Characteristics of Haloferax Infecting Viruses
3.3. Restriction–Modification Systems
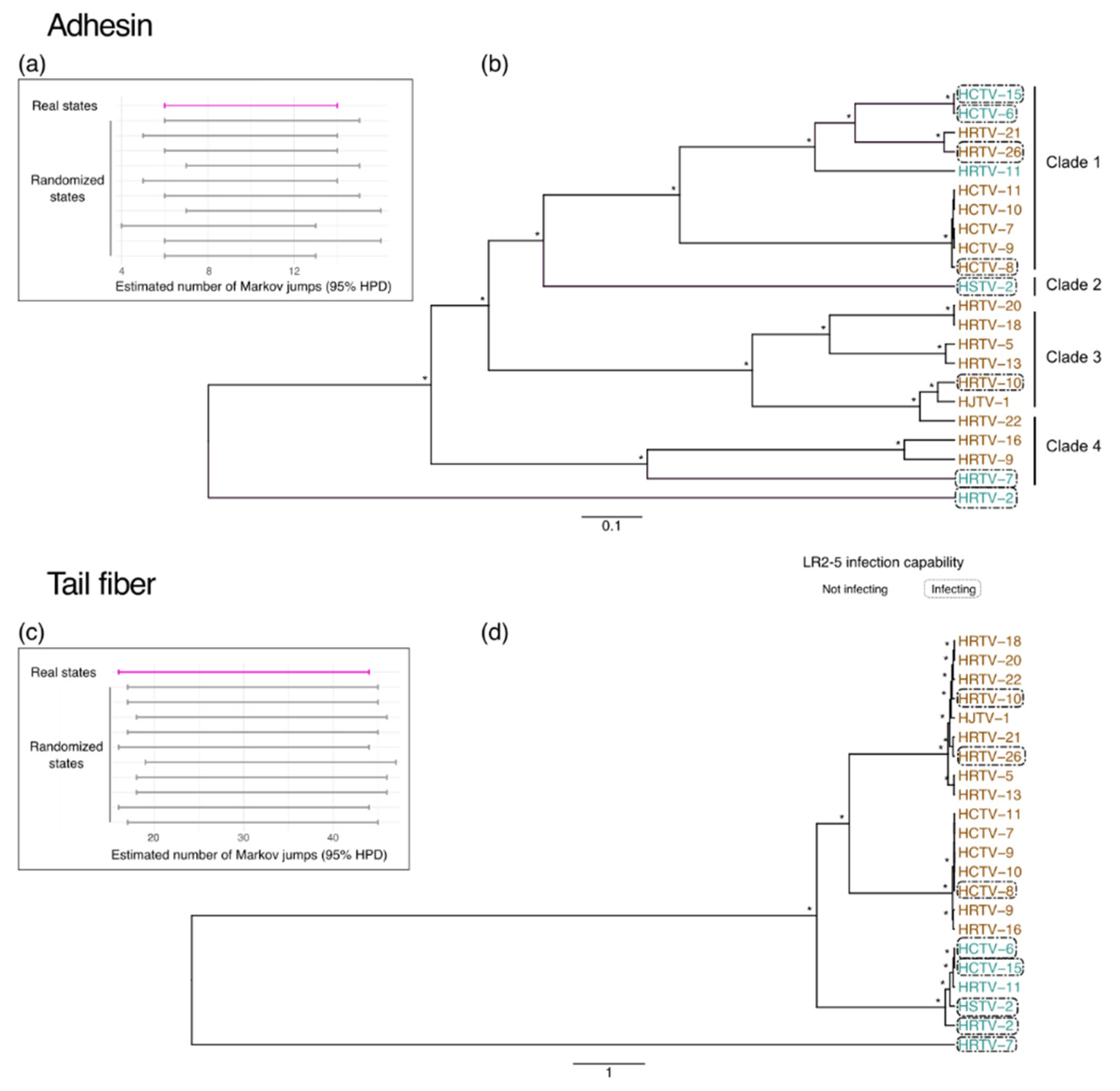
3.4. Adhesins and Tail Fiber Proteins
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Borrel, G.; Brugère, J.-F.; Gribaldo, S.; Schmitz, R.A.; Moissl-Eichinger, C. The Host-Associated Archaeome. Nat. Rev. Microbiol. 2020, 18, 622–636. [Google Scholar] [CrossRef]
- Lloyd, K.G.; May, M.K.; Kevorkian, R.T.; Steen, A.D. Meta-Analysis of Quantification Methods Shows That Archaea and Bacteria Have Similar Abundances in the Subseafloor. Appl. Environ. Microbiol. 2013, 79, 7790–7799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forterre, P.; Prangishvili, D. The Origin of Viruses. Res. Microbiol. 2009, 160, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Hartman, R.; Munson-McGee, J.; Young, M.J.; Lawrence, C.M. Survey of High-Resolution Archaeal Virus Structures. Curr. Opin. Virol. 2019, 36, 74–83. [Google Scholar] [CrossRef]
- Baquero, D.P.; Liu, Y.; Wang, F.; Egelman, E.H.; Prangishvili, D.; Krupovic, M. Structure and Assembly of Archaeal Viruses, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; Volume 108, ISBN 9780128207611. [Google Scholar]
- Prangishvili, D.; Bamford, D.H.; Forterre, P.; Iranzo, J.; Koonin, E.V.; Krupovic, M. The Enigmatic Archaeal Virosphere. Nat. Rev. Microbiol. 2017, 15, 724–739. [Google Scholar] [CrossRef]
- Pietilä, M.K.; Demina, T.A.; Atanasova, N.S.; Oksanen, H.M.; Bamford, D.H. Archaeal Viruses and Bacteriophages: Comparisons and Contrasts. Trends Microbiol. 2014, 22, 334–344. [Google Scholar] [CrossRef]
- Abrescia, N.G.A.; Bamford, D.H.; Grimes, J.M.; Stuart, D.I. Structure Unifies the Viral Universe. Annu. Rev. Biochem. 2012, 81, 795–822. [Google Scholar] [CrossRef]
- Koonin, E.v.; Dolja, V.v.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global Organization and Proposed Megataxonomy of the Virus World. Microbiol. Mol. Biol. Rev. 2020, 84, e00061-19. [Google Scholar] [CrossRef]
- Liu, Y.; Demina, T.A.; Roux, S.; Aiewsakun, P.; Kazlauskas, D.; Simmonds, P.; Prangishvili, D.; Oksanen, H.M.; Krupovic, M. Diversity, Taxonomy, and Evolution of Archaeal Viruses of the Class Caudoviricetes. PLoS Biol. 2021, 19, e3001442. [Google Scholar] [CrossRef] [PubMed]
- Oren, A. Taxonomy of Halophilic Archaea: Current Status and Future Challenges. Extremophiles 2014, 18, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, N.S.; Bamford, D.H.; Oksanen, H.M. Haloarchaeal Virus Morphotypes. Biochimie 2015, 118, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Demina, T.A.; Pietilä, M.K.; Svirskaitė, J.; Ravantti, J.J.; Atanasova, N.S.; Bamford, D.H.; Oksanen, H.M. HCIV-1 and Other Tailless Icosahedral Internal Membrane-Containing Viruses of the Family Sphaerolipoviridae. Viruses. MDPI AG 2017, 9, 32. [Google Scholar] [CrossRef]
- Demina, T.A.; Oksanen, H.M. Pleomorphic Archaeal Viruses: The Family Pleolipoviridae Is Expanding by Seven New Species. Arch. Virol. 2020, 165, 2723–2731. [Google Scholar] [CrossRef]
- Bath, C.; Dyall-Smith, M.L. His1, an Archaeal Virus of the Fuselloviridae Family That Infects Haloarcula Hispanica. J. Virol. 1998, 72, 9392–9395. [Google Scholar] [CrossRef]
- Senčilo, A.; Jacobs-Sera, D.; Russell, D.A.; Ko, C.C.; Bowman, C.A.; Atanasova, N.S.; Osterlund, E.; Oksanen, H.M.; Bamford, D.H.; Hatfull, G.F.; et al. Snapshot of Haloarchaeal Tailed Virus Genomes. RNA Biol. 2013, 10, 803. [Google Scholar] [CrossRef] [PubMed]
- Trojet, S.N.; Caumont-Sarcos, A.; Perrody, E.; Comeau, A.M.; Krisch, H.M. The Gp38 Adhesins of the T4 Superfamily: A Complex Modular Determinant of the Phage’s Host Specificity. Genome Biol. Evol. 2011, 3, 674–686. [Google Scholar] [CrossRef] [Green Version]
- Dunne, M.; Denyes, J.M.; Arndt, H.; Loessner, M.J.; Leiman, P.G.; Correspondence, J.K.; Klumpp, J. Salmonella Phage S16 Tail Fiber Adhesin Features a Rare Polyglycine Rich Domain for Host Recognition. Structure 2018, 26, 1573–1582. [Google Scholar] [CrossRef] [Green Version]
- Atanasova, N.S.; Demina, T.A.; Buivydas, A.; Bamford, D.H.; Oksanen, H.M. Archaeal Viruses Multiply: Temporal Screening in a Solar Saltern. Viruses 2015, 7, 1902–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Pan, S.; Zhang, Y.; Ren, M.; Feng, M.; Peng, N.; Chen, L.; Liang, Y.X.; She, Q. Harnessing Type i and Type III CRISPR-Cas Systems for Genome Editing. Nucleic Acids Res. 2015, 44, e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albers, S.V.; Driessen, A.J.M. Conditions for Gene Disruption by Homologous Recombination of Exogenous DNA into the Sulfolobus Solfataricus Genome. Archaea 2008, 2, 145–149. [Google Scholar] [CrossRef] [Green Version]
- Lewis, A.M.; Recalde, A.; Bräsen, C.; Counts, J.A.; Nussbaum, P.; Bost, J.; Schocke, L.; Shen, L.; Willard, D.J.; Quax, T.E.F.F.; et al. The Biology of Thermoacidophilic Archaea from the Order Sulfolobales. FEMS Microbiol. Rev. 2021, 45, fuaa063. [Google Scholar] [CrossRef]
- Leigh, J.A.; Albers, S.V.; Atomi, H.; Allers, T. Model Organisms for Genetics in the Domain Archaea: Methanogens, Halophiles, Thermococcales and Sulfolobales. FEMS Microbiol. Rev. 2011, 35, 577–608. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; van Wolferen, M.; Wagner, A.; Lassak, K.; Meyer, B.H.; Reimann, J.; Albers, S.V. Versatile Genetic Tool Box for the Crenarchaeote Sulfolobus Acidocaldarius. Front. Microbiol. 2012, 3, 214. [Google Scholar] [CrossRef] [Green Version]
- Iverson, E.; Stedman, K. A Genetic Study of SSV1, the Prototypical Fusellovirus. Front. Microbiol. 2012, 3, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedenheft, B.; Stedman, K.; Roberto, F.; Willits, D.; Gleske, A.-K.; Zoeller, L.; Snyder, J.; Douglas, T.; Young, M. Comparative Genomic Analysis of Hyperthermophilic Archaeal Fuselloviridae Viruses. J. Virol. 2004, 78, 1954. [Google Scholar] [CrossRef] [Green Version]
- Fulton, J.; Bothner, B.; Lawrence, M.; Johnson, J.E.; Douglas, T.; Young, M. Genetics, Biochemistry and Structure of the Archaeal Virus STIV. Biochem. Soc. Trans. 2009, 37, 114–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, X.; Chen, L.; Huang, X.; Luo, Y.; She, Q.; Huang, L. Sulfolobus Tengchongensis Spindle-Shaped Virus STSV1: Virus-Host Interactions and Genomic Features. J. Virol. 2005, 79, 8677–8686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasova, N.S.; Roine, E.; Oren, A.; Bamford, D.H.; Oksanen, H.M. Global Network of Specific Virus-Host Interactions in Hypersaline Environments. Env. Microbiol. 2012, 14, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Svirskaitė, J.; Oksanen, H.; Daugelavičius, R.; Bamford, D. Monitoring Physiological Changes in Haloarchaeal Cell during Virus Release. Viruses 2016, 8, 59. [Google Scholar] [CrossRef]
- Santos-Pérez, I.; Charro, D.; Gil-Carton, D.; Azkargorta, M.; Elortza, F.; Bamford, D.H.; Oksanen, H.M.; Abrescia, N.G.A. Structural Basis for Assembly of Vertical Single β-Barrel Viruses. Nat. Commun. 2019, 10, 1184. [Google Scholar] [CrossRef] [Green Version]
- Pietilä, M.K.; Atanasova, N.S.; Manole, V.; Liljeroos, L.; Butcher, S.J.; Oksanen, H.M.; Bamford, D.H. Virion Architecture Unifies Globally Distributed Pleolipoviruses Infecting Halophilic Archaea. J. Virol. 2012, 86, 5067–5079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Omari, K.; Li, S.; Kotecha, A.; Walter, T.S.; Bignon, E.A.; Harlos, K.; Somerharju, P.; De Haas, F.; Clare, D.K.; Molin, M.; et al. The Structure of a Prokaryotic Viral Envelope Protein Expands the Landscape of Membrane Fusion Proteins. Nat. Commun. 2019, 10, 846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietilä, M.K.; Atanasova, N.S.; Oksanen, H.M.; Bamford, D.H. Modified Coat Protein Forms the Flexible Spindle-Shaped Virion of Haloarchaeal Virus His1. Environ. Microbiol. 2013, 15, 1674–1686. [Google Scholar] [CrossRef] [PubMed]
- Pohlschroder, M.; Schulze, S. Haloferax Volcanii. Trends Microbiol. 2019, 27, 86–87. [Google Scholar] [CrossRef]
- Hartman, A.L.; Norais, C.; Badger, J.H.; Delmas, S.; Haldenby, S.; Madupu, R.; Robinson, J.; Khouri, H.; Ren, Q.; Lowe, T.M.; et al. The Complete Genome Sequence of Haloferax Volcanii DS2, a Model Archaeon. PLoS ONE 2010, 5, e9605. [Google Scholar] [CrossRef] [Green Version]
- Allers, T.; Ngo, H.P.; Mevarech, M.; Lloyd, R.G. Development of Additional Selectable Markers for the Halophilic Archaeon Haloferax Volcanii Based on the LeuB and TrpA Genes. Appl. Environ. Microbiol. 2004, 70, 943–953. [Google Scholar] [CrossRef] [Green Version]
- Strillinger, E.; Grötzinger, S.W.; Allers, T.; Eppinger, J.; Weuster-Botz, D. Production of Halophilic Proteins Using Haloferax Volcanii H1895 in a Stirred-Tank Bioreactor. Appl. Microbiol. Biotechnol. 2016, 100, 1183–1195. [Google Scholar] [CrossRef] [Green Version]
- Stachler, A.E.; Schwarz, T.S.; Schreiber, S.; Marchfelder, A. CRISPRi as an Efficient Tool for Gene Repression in Archaea. Methods 2020, 172, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Bisson-Filho, A.W.; Zheng, J.; Garner, E. Archaeal Imaging: Leading the Hunt for New Discoveries. Mol. Biol. Cell 2018, 29, 1675–1681. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.C.; Angstmann, C.N.; Bisson-Filho, A.W.; Garner, E.C.; Duggin, I.G.; Curmi, P.M.G. Division Plane Placement in Pleomorphic Archaea Is Dynamically Coupled to Cell Shape. Mol. Microbiol. 2019, 112, 785–799. [Google Scholar] [CrossRef]
- Duggin, I.G.; Aylett, C.H.S.; Walsh, J.C.; Michie, K.A.; Wang, Q.; Turnbull, L.; Dawson, E.M.; Harry, E.J.; Whitchurch, C.B.; Amos, L.A.; et al. CetZ Tubulin-like Proteins Control Archaeal Cell Shape. Nature 2015, 519, 362–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Silva, R.T.; Abdul-Halim, M.F.; Pittrich, D.A.; Brown, H.J.; Pohlschroder, M.; Duggin, I.G. Improved Growth and Morphological Plasticity of Haloferax Volcanii. Microbiology 2021, 167, 001012. [Google Scholar] [CrossRef] [PubMed]
- Kinosita, Y.; Mikami, N.; Li, Z.; Braun, F.; Quax, T.E.F.; van der Does, C.; Ishmukhametov, R.; Albers, S.V.; Berry, R.M. Motile Ghosts of the Halophilic Archaeon, Haloferax Volcanii. Proc. Natl. Acad. Sci. USA 2020, 117, 26766–26772. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rodriguez-Franco, M.; Albers, S.-V.V.; Quax, T.E.F.F. The Switch Complex ArlCDE Connects the Chemotaxis System and the Archaellum. Mol. Microbiol. 2020, 114, 468–479. [Google Scholar] [CrossRef]
- Quax, T.E.F.; Altegoer, F.; Rossi, F.; Li, Z.; Rodriguez-Franco, M.; Kraus, F.; Bange, G.; Albers, S.V. Structure and Function of the Archaeal Response Regulator CheY. Proc. Natl. Acad. Sci. USA 2018, 115, E1259–E1268. [Google Scholar] [CrossRef] [Green Version]
- Esquivel, R.N.; Schulze, S.; Xu, R.; Hippler, M.; Pohlschroder, M. Identification of Haloferax Volcanii Pilin N-Glycans with Diverse Roles in Pilus Biosynthesis, Adhesion, and Microcolony Formation. J. Biol. Chem. 2016, 291, 10602–10614. [Google Scholar] [CrossRef] [Green Version]
- Esquivel, R.N.; Xu, R.; Pohlschroder, M. Novel Archaeal Adhesion Pilins with a Conserved N Terminus. J. Bacteriol. 2013, 195, 3808. [Google Scholar] [CrossRef] [Green Version]
- Maier, L.K.; Marchfelder, A. It’s All about the T: Transcription Termination in Archaea. Biochem. Soc. Trans. 2019, 47, 461–468. [Google Scholar] [CrossRef]
- Humbard, M.A.; Miranda, H.v.; Lim, J.M.; Krause, D.J.; Pritz, J.R.; Zhou, G.; Chen, S.; Wells, L.; Maupin-Furlow, J.A. Ubiquitin-like Small Archaeal Modifier Proteins (SAMPs) in Haloferax Volcanii. Nature 2010, 463, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, M.; Malla, S.; Blythe, M.J.; Nieduszynski, C.A.; Allers, T. Accelerated Growth in the Absence of DNA Replication Origins. Nature 2013, 503, 544–547. [Google Scholar] [CrossRef]
- Haque, R.U.; Paradisi, F.; Allers, T. Haloferax Volcanii for Biotechnology Applications: Challenges, Current State and Perspectives. Appl. Microbiol. Biotechnol. 2020, 104, 1371–1382. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Halim, M.F.; Schulze, S.; DiLucido, A.; Pfeiffer, F.; Filho, A.W.B.; Pohlschroder, M. Lipid Anchoring of Archaeosortase Substrates and Midcell Growth in Haloarchaea. mBio 2020, 11, e00349-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Kinosita, Y.; Rodriguez-Franco, M.; Nußbaum, P.; Braun, F.; Delpech, F.; Quax, T.E.F.; Albers, S.V. Positioning of the Motility Machinery in Halophilic Archaea. mBio 2019, 10, e00377-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nußbaum, P.; Ithurbide, S.; Walsh, J.C.; Patro, M.; Delpech, F.; Rodriguez-Franco, M.; Curmi, P.M.G.; Duggin, I.G.; Quax, T.E.F.; Albers, S.V. An Oscillating MinD Protein Determines the Cellular Positioning of the Motility Machinery in Archaea. Curr. Biol. 2020, 30, 4956–4972.e4. [Google Scholar] [CrossRef]
- Jarrell, K.F.; Ding, Y.; Meyer, B.H.; Albers, S.-V.; Kaminski, L.; Eichler, J. N-Linked Glycosylation in Archaea: A Structural, Functional, and Genetic Analysis. Microbiol. Mol. Biol. Rev. 2014, 78, 304–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Liu, H.; Han, J.; Liu, J.; Wang, R.; Zhao, D.; Zhou, J.; Xiang, H. Characterization of CRISPR RNA Biogenesis and Cas6 Cleavage-Mediated Inhibition of a Provirus in the Haloarchaeon Haloferax Mediterranei. J. Bacteriol. 2013, 195, 867–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuttall, S.D.; Smith, M.L.D. HF1 and HF2: Novel Bacteriophages of Halophilic Archaea. Virology 1993, 197, 678–684. [Google Scholar] [CrossRef]
- Mizuno, C.M.; Prajapati, B.; Lucas-Staat, S.; Sime-Ngando, T.; Forterre, P.; Bamford, D.H.; Prangishvili, D.; Krupovic, M.; Oksanen, H.M. Novel Haloarchaeal Viruses from Lake Retba Infecting Haloferax and Halorubrum Species. Environ. Microbiol. 2019, 21, 2129–2147. [Google Scholar] [CrossRef] [Green Version]
- Tittes, C.; Schwarzer, S.; Pfeiffer, F.; Dyall-Smith, M.; Rodriguez-Franco, M.; Oksanen, H.M.; Quax, T.E.F. Cellular and Genomic Properties of Haloferax Gibbonsii LR2-5, the Host of Euryarchaeal Virus HFTV1. Front. Microbiol. 2021, 12, 625599. [Google Scholar] [CrossRef]
- Dyall-Smith, M. The Halohandbook: Protocols for haloarchaeal genetics. Halohandb. Protoc. haloarchaeal Genet. 2009. Available online: https://haloarchaea.com/wp-content/uploads/2018/10/Halohandbook_2009_v7.3mds.pdf (accessed on 10 January 2022).
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. Ggtree: An r Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A Genome Comparison Visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Whelan, S.; Goldman, N. A General Empirical Model of Protein Evolution Derived from Multiple Protein Families Using a Maximum-Likelihood Approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Henikoff, S.; Henikoff, J.G. Amino Acid Substitution Matrices from Protein Blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915. [Google Scholar] [CrossRef] [Green Version]
- Kingman, J.F.C. The Coalescent. Stoch. Proc. Appl. 1982, 13, 235–248. [Google Scholar] [CrossRef] [Green Version]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLOS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minin, V.N.; Suchard, M.A. Counting Labeled Transitions in Continuous-Time Markov Models of Evolution. J. Math Biol. 2008, 56, 391–412. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Valera, F.; Juez, G.; Kushner, D.J. Halobacterium Mediterranei Spec, Nov., a New 93 Carbohydrate-Utilizing Extreme Halophile. Syst. Appl. Microbiol. 1983, 4, 369–381. [Google Scholar] [CrossRef]
- Kumar, V.; Singh, B.; van Belkum, M.J.; Diep, D.B.; Chikindas, M.L.; Ermakov, A.M.; Tiwari, S.K. Halocins, Natural Antimicrobials of Archaea: Exotic or Special or Both? Biotechnol. Adv. 2021, 53, 107834. [Google Scholar] [CrossRef]
- Atanasova, N.S.; Pietilä, M.K.; Oksanen, H.M. Diverse Antimicrobial Interactions of Halophilic Archaea and Bacteria Extend over Geographical Distances and Cross the Domain Barrier. Microbiologyopen 2013, 2, 811–825. [Google Scholar] [CrossRef]
- Dyall-Smith, M.; Tang, S.L.; Russ, B.; Chiang, P.W.; Pfeiffer, F. Comparative Genomics of Two New HF1-like Haloviruses. Genes 2020, 11, 405. [Google Scholar] [CrossRef] [Green Version]
- Cerritelli, M.E.; Wall, J.S.; Simon, M.N.; Conway, J.F.; Steven, A.C. Stoichiometry and Domainal Organization of the Long Tail-Fiber of Bacteriophage T4: A Hinged Viral Adhesin. J. Mol. Biol. 1996, 260, 767–780. [Google Scholar] [CrossRef]
- Gambelli, L.; Meyer, B.H.; McLaren, M.; Sanders, K.; Quax, T.E.F.; Gold, V.A.M.; Albers, S.V.; Daum, B. Architecture and Modular Assembly of Sulfolobus S-Layers Revealed by Electron Cryotomography. Proc. Natl. Acad. Sci. USA 2019, 116, 25278–25286. [Google Scholar] [CrossRef] [Green Version]
- Zink, I.A.; Pfeifer, K.; Wimmer, E.; Sleytr, U.B.; Schuster, B.; Schleper, C. CRISPR-Mediated Gene Silencing Reveals Involvement of the Archaeal S-Layer in Cell Division and Virus Infection. Nat. Commun. 2019, 10, 4797. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Virus Morphology (5) | Host Strain to Grow the Virus | Titer on Own Host, pfu/mL | Titer on LR2-5, pfu/mL | EOP on LR2-5 (6) |
|---|---|---|---|---|---|
| Hafunaviridae (F) (1) | |||||
| Haloferacalesvirus (G) (2) | |||||
| HRTV-10 | M | Halorubrum sp. B2-2 | 1.2 × 109 | 6.8 × 103 | 6 × 10−6 |
| HRTV-18 | M | Halorubrum sp. SS10-3 | 7.5 × 109 | - | |
| HRTV-20 | M | Halorubrum sp. SS10-9 | 7.0 × 1010 | - | |
| HRTV-22 | M | Halorubrum sp. SS10-9 | 1.1 × 1010 | - | |
| HRTV-26 | M | Halorubrum sp. SS10-9 | 1.4 × 109 | 4.3 × 106 | 3 × 10−3 |
| HRTV-5 | M | Halorubrum sp. s5a-3 | 2.0 × 1010 | - | |
| HCTV-7 (3) | M | Haloarcula californiae | 4.8 × 1010 | - | |
| [HCTV-12] (3) | M | Haloarcula californiae | 1.3 × 1010 | - | |
| HCTV-9 | M | Haloarcula californiae | 2.8 × 1010 | - | |
| HCTV-11 | M | Haloarcula californiae | 4.0 × 1010 | - | |
| HRTV-9 | M | Halorubrum sp. B2-2 | 5.3 × 109 | - | |
| HRTV-16 | M | Haloterrigena sp. SS13-7 | nd | nd | |
| HCTV-8 | M | Haloarcula californiae | 2.3 × 1010 | 3.1 × 105 | 1 × 10−5 |
| HCTV-10 | M | Halorubrum sodomense | 2.3 × 109 | - | |
| HJTV-1 | M | Haloarcula japonica | 1.6 × 109 | - | |
| HRTV-13 | M | Halorubrum sp. SS8-2 | 5.1 × 109 | - | |
| HRTV-21 | M | Halorubrum sp. SS10-9 | 2.8 × 109 | - | |
| Mincapvirus (G) (2) | |||||
| HSTV-2 | M | Halorubrum sodomense | 8.2 × 109 | 2.3 × 1010 | 3 |
| HRTV-7 | M | Halorubrum sp. B2-2 | 2.1 × 109 | 7.9 × 105 | 4 × 10−4 |
| HRTV-2 | M | Halorubrum sp. s1-2 | 2.0 × 1010 | 4.6 × 1010 | 2 |
| HRTV-11 | M | Halorubrum sp. SL-5 | 4.7 × 1010 | - | |
| HCTV-6 (4) | M | Haloarcula californiae | 1.6 × 1010 | 3.9 × 109 | 2 × 10−1 |
| [HCTV-13] (4) | M | Haloarcula californiae | 2.5 × 109 | [1.3 × 107] | [5 × 10−3] |
| HCTV-15 | M | Halorubrum sp. SS6-2 | 1.9 × 1010 | 9.6 × 106 | 5 × 10−4 |
| Haloferuviridae (F) (1) | |||||
| Retbasiphovirus (G) (2) | |||||
| HFTV1 | S | Haloferax gibbonsii LR2-5 | 2.9 × 1012 | 2.9 × 1012 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguirre Sourrouille, Z.; Schwarzer, S.; Lequime, S.; Oksanen, H.M.; Quax, T.E.F. The Viral Susceptibility of the Haloferax Species. Viruses 2022, 14, 1344. https://doi.org/10.3390/v14061344
Aguirre Sourrouille Z, Schwarzer S, Lequime S, Oksanen HM, Quax TEF. The Viral Susceptibility of the Haloferax Species. Viruses. 2022; 14(6):1344. https://doi.org/10.3390/v14061344
Chicago/Turabian StyleAguirre Sourrouille, Zaloa, Sabine Schwarzer, Sebastian Lequime, Hanna M. Oksanen, and Tessa E. F. Quax. 2022. "The Viral Susceptibility of the Haloferax Species" Viruses 14, no. 6: 1344. https://doi.org/10.3390/v14061344