
Recombination in Coronaviruses, with a Focus on SARS-CoV-2


1
North-Western Tuscany Blood Bank, Pisa University Hospital, 56124 Pisa, Italy
2
Department of Medicine and Surgery, University of Insubria, 21100 Varese, Italy
*
Author to whom correspondence should be addressed.
Viruses 2022, 14(6), 1239; https://doi.org/10.3390/v14061239
Submission received: 30 April 2022
/
Revised: 6 June 2022
/
Accepted: 6 June 2022
/
Published: 7 June 2022
(This article belongs to the Special Issue SARS-CoV-2 inside and outside the Respiratory Tract: From Diagnostics to Therapies)
Abstract
:Recombination is a common evolutionary tool for RNA viruses, and coronaviruses are no exception. We review here the evidence for recombination in SARS-CoV-2 and reconcile nomenclature for recombinants, discuss their origin and fitness, and speculate how recombinants could make a difference in the future of the COVID-19 pandemics.
1. Recombination in Coronaviruses Other Than SARS-CoV-2
Recombination represents a major contributor to RNA virus evolution [1] together with re-assortment (which exclusively operates in RNA viruses with segmented genomes). Recombination can occur both in segmented [2,3] and non-segmented RNA viruses [4,5,6,7] and avoids an accumulation of irreversible deleterious mutations typical of asexual reproduction (so called “Muller’s ratchet” [8]). “Donor” and “acceptor” are conventional terms used to refer to the strain represented in a greater and lesser amount, respectively. Recombination within different sublineages of the same virus invariably requires co-circulation and co-infection of the same host.
Recombination can be difficult to detect whenever the sublineages have minimal differences and requires whole-genome sequencing (WGS). Unlabeled private mutations can help track the spread of the recombinant lineage more easily: they are defined as private mutations that are neither reversions nor labeled (i.e., they are not mutations to a genotype that is known to be common in a clade) [9]. Deletions are generally considered useful landmarks for recombination because they are unlikely to be reverted (except through recombination), but they can spontaneously occur across different sublineages (convergent evolution) independently from recombination (as seen, e.g., by the NSP6 SGF- reported in Alpha, Beta, and Gamma variants of concern of SARS-CoV-2 [10]).
We review here former evidence for recombination in betacoronaviruses and then focus on SARS-CoV-2 recombinants.
Coronaviridae undergo both homologous recombination (HR) and nonhomologous recombination (NHR). Only a minority of recombinants are likely detected in surveys since most of them are unlikely to be fitter than the currently dominant strain.
A high frequency of HR occurs across all three coronavirus groups, e.g., in murine hepatitis virus [11,12,13,14], transmissible gastroenteritis virus [15], bovine coronavirus [16], feline infectious peritonitis virus [17,18], and infectious bronchitis virus (IBV) [19,20,21,22]. RNA recombination is thought to be similar to poliovirus [23]: in this scheme, the viral RNA-dependent RNA polymerase (RdRp) detaches from one template with the nascent RNA strand intact and resumes elongation at the identical or similar position on another template. Recombination in MHV was reported at levels as high as 25% [24], a record for RNA viruses.
By virtue of an RdRp template switch likely occurring during synthesis of the (−)-strand templates for subgenomic mRNA (sgmRNA) synthesis, coronaviruses generate a 3′-coterminal nested set of sgmRNAs sharing a 65–90 nt long common leader. One form of NHR that occurs between genomic and sgmRNA has been hypothesized to result from the collapse of the transcription complex during (-)-strand discontinuous transcription [25]. Such a disruption would leave a partial copy of the leader sequence within the genome near the junction between two genes. Remnants of leader RNAs were found in the genomes of wild-type HCoV-OC43 [26], and the HCoV-HKU1 genome contains two very significant segments of embedded leader sequence (Woo et al., 2005) [27,28,29].
Among human coronaviruses, recombination was first reported in 2006 for both HCoV-HKU1 [30] and HCoV-NL63 [31,32], then in 2011 for HCoV-OC43 (genotype D since 2004) [33], in 2004 for SARS-CoV [34,35], and finally in 2014 for MERS-CoV [36,37] (leading to at least five strains with parts from both humans and camels [38]). Nevertheless, recombination rates across the genome of the human seasonal coronaviruses 229E, OC43, and NL63 vary with rates of adaptation [39]. To date, there is no well-documented example of recombination between extant coronaviruses of different groups.
2. Recombination in SARS-CoV-2
2.1. Recombinant Origin of SARS-CoV-2
Li et al. initially showed in March 2020 that SARS-CoV-2’s entire receptor-binding motif (RBM) was introduced through recombination with coronaviruses from pangolins, possibly a critical step in the evolution of SARS-CoV-2’s ability to infect humans [40]. This was later confirmed by Zhu et al. in December 2020 [41]. However, more recently, using sliding window bootstrap (SWB) to highlight the regions supporting phylogenetic relationships, SARS-CoV-2 was defined as a mosaic genome composed of regions sharing recent ancestry with three bat SCoV2rCs recently discovered in the Yunnan region of China (RmYN02, RpYN06, and RaTG13) or related to more ancient ancestors in bats from Yunnan and Southeast Asia [42], with no evidence of direct recombination with pangolin viruses.
2.2. Super-Infection or Co-Infection with Different SARS-CoV-2 Lineages
SARS-CoV-2 can be named according to different phylogenetic systems, which can often but not always be reconciled. The Global Initiative on Sharing All Influenza Data (GISAID) phylogeny classifies clades with progressive letters (https://www.gisaid.org/index.php?id=208, accessed on 29 April 2022). The Phylogenetic Assignment of Named Global Outbreak LINeages (PANGOLIN) nomenclature uses an alphabetical prefix and a numerical suffix to identify descendants (https://www.pango.network/the-pango-nomenclature-system/statement-of-nomenclature-rules/, accessed on 29 April 2022). NextStrain uses a year-letter system (https://docs.nextstrain.org/projects/ncov/en/latest/reference/naming_clades.html, accessed on 29 April 2022). Finally, the WHO uses progressive Greek letters to dynamically identify variants of interests (VOI) or concern (VOC) (https://www.who.int/activities/tracking-SARS-CoV-2-variants, accessed on 29 April 2022).
A few months after the initiation of the COVID-19 pandemic, co-infections were documented without any evidence of recombination. The first detailed case was described in February 2021 as co-infection from NextStrain 20A and 20B lineages, which was followed up for kinetics of relative abundance: a Portuguese patient had a prolonged viral shedding case (97 days long), first with a severe disease manifestation followed by a short second hospitalization episode, in an otherwise healthy young female [43]. More cases soon followed: e.g., co-infection by B.1.1.248 (either as major or minor haplotype) and B.1.1.33 or B.1.91, respectively [44], or co-infection between B.1.1.7 and B.1.351 [45] or GH and GR [46]. A less conclusive case of co-infection was reported from Iraq, suggesting the need for helper strains from defective co-infective strains [47]. A large study identified 53 (~0.18%) co-infection events (including with two Delta sublineages) out of 29,993 samples: apart from 52 co-infections with two SARS-CoV-2 lineages, one sample with co-infections of three SARS-CoV-2 lineages was firstly identified [48]. Another study identified coinfections in around 0.61% of all samples investigated (nine cases) [49].
Co-infections should be distinguished from subclonal variants (so-called intra-host evolution or quasi-species swarm), which naturally occur during infection, especially long-lasting infections in immunocompromised recipients either spontaneously or after selective pressure from antiviral therapeutics [50].
2.3. Evidence for Recombination in SARS-CoV-2
There is both in silico [51] and in vivo [52] evidence for recombination of different SARS-CoV-2 strains. Studies relying on linkage disequilibrium identified that SARS-CoV-2 recombination occurs at very low levels [52,53,54] or does not occur at all [55,56,57,58,59,60]. Several alternative methods are available for reconstructing genealogies explicitly in the presence of recombination, both with [61] and without [62,63,64] making the parsimony assumption, but none is tailored to the particular problem of detecting recombination in the presence of recurrent mutation. In fact, many tests of recombination assume that all mutations can only occur once at each site, and hence, recurrent mutation from convergent evolution (as it occurs in SARS-CoV-2) and systematic errors can confound signatures of recombination [7,27,36,65].
Hence, novel methodological approaches have been developed to detect recombinant genomes in SARS-CoV-2 lineages. Ignatieva et al. proposed a parsimony-based greedy heuristic algorithm for reconstructing plausible ancestral recombination graphs (KwARG) [66]: it does not scale well to large datasets but was powerful enough for disentangling the effects of recurrent mutation from recombination in the history of a sample [67]. Turakhia et al. developed Recombination Inference using Phylogenetic PLacEmentS (RIPPLES) to break the sequence into distinct segments that are differentiated by mutations on the recombinant sequence and separated by up to two breakpoints: for each set of breakpoints, RIPPLES places each of its corresponding segments using maximum parsimony to find the two parental nodes—hereafter termed donor and acceptor. RIPPLES is very fast with a large dataset but is biased against identifying recombination events near the edges of the viral genome. They identified 606 recombination events by investigating a 1.6M sample tree, showing that approximately 2.7% of sequenced SARS-CoV-2 genomes have recombinant ancestry, that recombination breakpoints occur disproportionately in the Spike protein region, and that cases were coinfected with 2–3 SARS-CoV-2 variants on average [68].
Haddad et al. observed recombination between different strains only in North American and European sequences [69].
Table 1 summarizes the recombinants between VOCs detected in more than one case (generally > 50 GISAID sequences). Many more cases are likely to have occurred between non-VOCs in a pre-VOC era or within individual hosts, such as a recombinant between B.1.160 and Alpha variants in a lymphoma patient chronically infected for 14 months [70]: nevertheless, those recombinant have been not fit enough to spread and outcompete the dominant strain of the moment.
Recombination has been proposed as a mechanism for the generation of B.1.1.7 (Alpha VOC) [71]. Accordingly, further recombination has been detected among B.1.1.7 and other strains (B.1.36.17, B.1.36.28, B.1.177, B.1.177.9, B.1.177.16, and B.1.221.1): interestingly, in six of eight instances (and all four of the transmitting groups, including one transmission cluster of 45 sequenced cases over 2 months), the mosaic viruses contain a Spike gene from the B.1.1.7 lineage, and in four instances, there is a proposed recombination breakpoint at or near the 5′ end of the spike gene [72].
As soon as the possibility of recombination emerged, nomenclature systems started considering how to name these sublineages. In the PANGOLIN phylogeny, all top-level lineages that are recombinants have a prefix that begins with “X” [73]. In most cases, they expect a minimum of 50 sequences to design a novel recombinant linage, but exceptions arise if the recombinant has a particular novelty or significance, with unusual breakpoint and/or parental lineages. As of 5 April 2022, CoV-lineages (https://cov-lineages.org/lineage_list.html) reports lineages from XA to XY, mostly from the UK (which contributes the vast majority of GISAID entries), suggesting new changes to nomenclature will soon be required. To date, neither WHO nor NextStrain phylogenies have a scheming name for SARS-CoV-2 recombinants.
We will here separately discuss recombination between SARS-CoV-2 VOCs.
2.4. Alpha-Delta Recombinants
Recombination between Alpha and Delta SARS-CoV-2 variants has, to date, been reported in a single case despite co-circulation from June 2021 to December 2021. Sekizuka et al. reported a Delta AY.29 and B.1.1.7 (later dubbed XC lineage) [75].
2.5. Beta-Delta Recombinants
Recombination between Beta and Delta has, to date, been reported in a single case despite co-circulation since December 2021. He et al. reported possible evidence of recombination in the Orf1ab (174–2692 and 5839) and Spike genes (21,801–22,281, previously proposed as a putative recombination region between the progenitor of SARS-CoV-2 and Bat-SL-CoV) in a patient (dubbed “49H”) maintaining a 1:9 Beta:Delta co-infection ratio for 14 days as part of an outbreak during a flight from South Africa to China [78].
2.6. Delta-BA.1 Recombinants
Delta and Omicron BA.1 co-circulated from November 2021 until February 2022: cases have been reported of Delta and Omicron co-infection [79,80]. Their recombinants are often colloquially referred to as “Deltamicron” or “Deltacron”. They were among the first recombinants to be named by PANGOLIN (XD, XF, and XS), but, as it happened for BA.1, all Deltamicron recombinant were soon out-competed by BA.2.
On 7 January 2022, virologist Leondios Kostrikis at the University of Cyprus in Nicosia deposited 52 sequences in GISAID, which were claimed by media as Deltamicron, but upon further inspection, these appeared to be due to laboratory artifacts (most likely laboratory contamination) or coinfections and were withdrawn from GISAID [81].
Ou et al. reported multiple additional amino acid mutations in the Delta Spike protein were also identified in the recently emerged Omicron isolates, which implied possible recombination events [82].
More individual cases of Deltamicron were reported, which do not have a PANGOLIN name designated yet, e.g.:
- Two clusters of apparent Delta-Omicron recombinants were identified in the United Kingdom (PANGO issue #422 and #441), which have a breakpoint upstream of Spike in orf1ab;
- Lacek et al. reported nine AY.119.2:BA.1.1 cases in the mid-Atlantic region of the USA, with breakpoints within the Spike gene (amino acids 158 to 339), containing substitutions common to Delta lineages at the 5′ end and Omicron lineages at the 3′ end [83];
- Leuking et al. reported two more cases in immunosuppressed patients (a 70-year-old male lung-transplant recipient and a 70-year-old female patient with uncontrolled diabetes) in Texas [84];
- Duerr et al. in New York reported an unvaccinated, immunosuppressed kidney-transplant recipient who had positive COVID-19 tests in December 2021 and February 2022 and was initially treated with sotrovimab. Viral sequencing in February 2022 revealed a 5’ Delta AY.45 portion and a 3’ Omicron BA.1 portion with a recombination breakpoint in the spike N-terminal domain, adjacent to the sotrovimab quaternary binding site [85];
- Bolze et al. identified two independent cases of infection by a Delta-Omicron recombinant virus in USA, where 100% of the viral RNA came from one clonal recombinant. In both cases, the 5′-end of the viral genome was from the Delta genome and the 3′-end from Omicron though the breakpoints were different [80].
Delta and BA.2 co-circulated minimally: accordingly, Delta-BA.2 recombinants only occurred in a doublet from the end of January in Sweden (PANGO issue #519) and a singlet again in January 2022 in Karnataka, India (PANGO issue #484). Another possible explanation for their scarcity is that countries with significant co-circulation (e.g., India and Philippines) do not perform WGS very frequently.
2.7. BA.1-BA.2 Recombinants
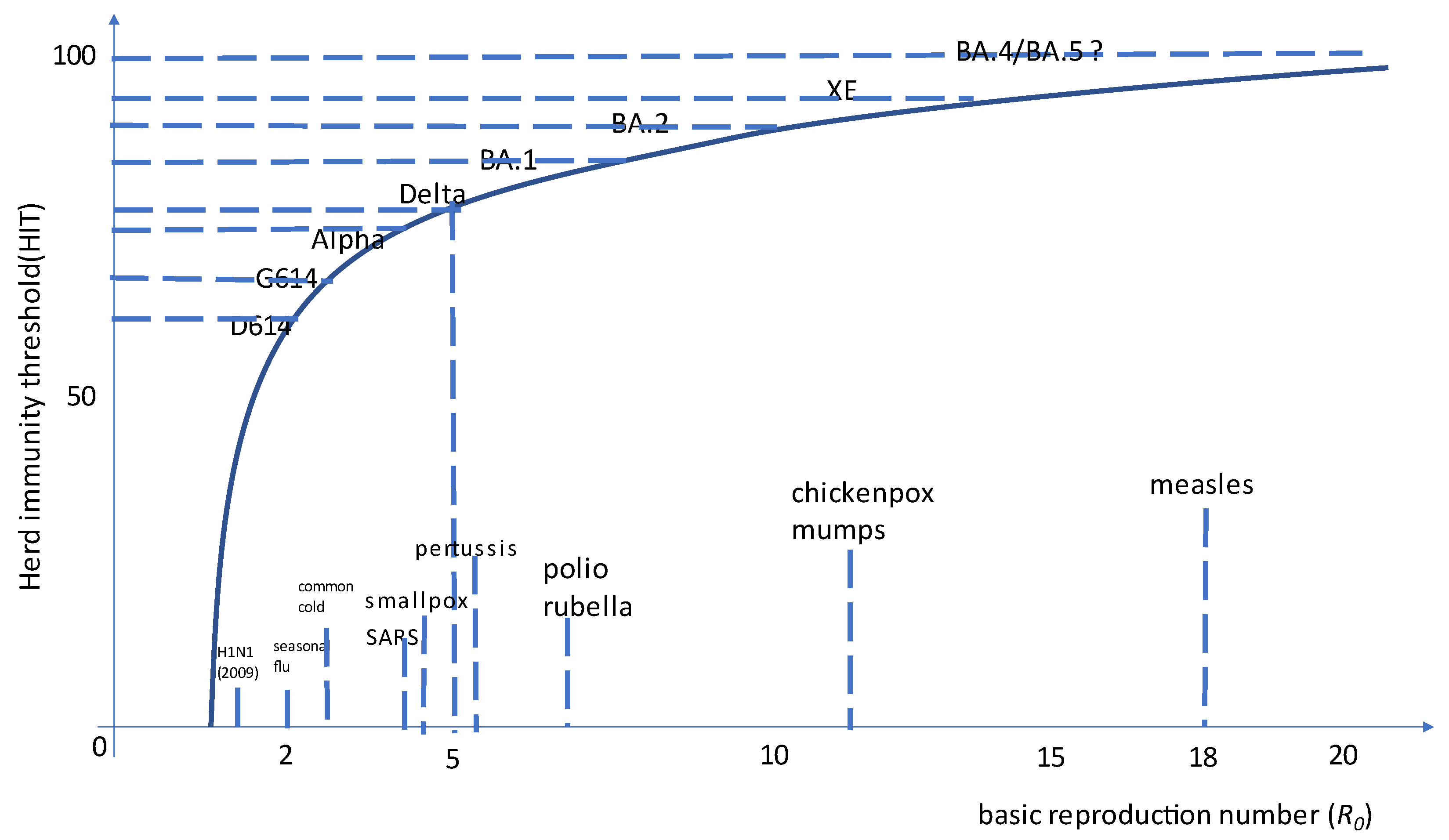
Most Omicron recombinants identified to date have the BA.1 as acceptor and the breakpoint within ORF1ab and hence preserve Spike protein from BA.2 (e.g., XE, XG, XH, XJ, XK, XM, XN, XP, XQ, and XR): this is not surprising since BA.2 currently outcompetes BA.1. XP is the lone exception, having BA.1.1 (the BA.1 sublineage with R346K mutation) as an acceptor (including Spike) and BA.2 as a donor. Among them, XE (also known as V-22APR-02 in Public Health England) is the most concerning, having a growth advantage over BA.2 estimated at first at +9.8% [86] and then raised to +20.9% (largely the same as observed for AY.4.2 over Delta in late 2021) [87]. This further increase in the basic reproductive number approaches SARS-CoV-2 as the most contagious virus in human history (see Figure 1).
Ou et al. identified, by scanning high-quality completed Omicron Spike gene sequences, 18 core mutations of BA.1 variants (frequency > 99%) (eight in NTD, five near the S1/S2 cleavage site, and five in S2). BA.2 variants share three additional amino acid deletions with the Alpha variants. BA.1 subvariants share nine common amino acid mutations (three more than BA.2) in the Spike protein with most VOCs, suggesting a possible recombination origin of Omicron from these VOCs. There are three more Alpha-related mutations (Δ69–70, Δ144) in BA.1 than in BA.2, and therefore, BA.1 may be phylogenetically closer to the Alpha variant. Revertant mutations are found in some dominant mutations (frequency > 95%) in the BA.1 subvariant [82].
Colson et al. in Marseille detected two samples with a recombinant genome that was mostly that of a BA.2 variant but with a 3′ tip originating from BA.1 [88]. Gu et al. in Japan reported two more cases with a breakpoint near the 5′ end of the Spike gene (nucleotide position 20,055-21,618) [89]. Leuking et al. in Texas reported two more cases in immunosuppressed patients [84].
3. Conclusions
Most recombinants to date have been reported in the UK, Denmark, and the USA mostly because those countries have more dense genomic surveillance programs. None of the recombinants detected so far seems to grow fast enough to become dominant, and greater concern comes from the emerging L452R-carrying BA.2 (e.g., BA.2.12.1 in New York or BA.2.11 in Bretagne) or BA.4/BA.5 sublineages. Albeit recombination is extremely likely to occur between SARS-CoV-2 lineages, several factors limit their generation and spread:
- (1)
- Pandemic waves from recent VOCs are becoming shorter and shorter, minimizing the time of co-circulation of different VOCs.
- (2)
- Apart from immunocompromised hosts, the duration of within-host viral replication is limited, again minimizing the room for co-infection/super-infection.
- (3)
- The increasingly high reproductive number achieved by the currently dominating VOC (BA.2) creates a major barrier for any novel strain to emerge (Figure 2). While approaching the asymptote of the reproductive number, only marginal gains in transmissivity will be possible. In this regard, many GISAID-powered bioinformatics tools are available (e.g., Cov-Spectrum [90] or SARS-CoV-2 Recombinant Finder [91]).
- (4)
- Detecting a recombinant lineage requires WGS efforts to stay in place given that, as for XE, Spike gene sequencing is not enough to detect recombination.
Nevertheless, even extremely rare events are likely to happen under massive viral circulation. In particular, we should not forget that COVID-19 is panzootic, and the possibility of recombination between an animal-adapted lineage and a human-adapted lineage could have unpredictable consequences on the efficacy of current COVID-19 vaccines.
Author Contributions
D.F.: conceptualization and writing—original draft. F.M.: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data are available at PubMed, medRxiv, and bioRxiv.
Conflicts of Interest
We declare we have no conflict of interest to disclose.
Abbreviations
| VOC | variant of concern |
References
- Lai, M.M. Genetic recombination in RNA viruses. Curr. Top. Microbiol. Immunol. 1992, 176, 21–32. [Google Scholar] [CrossRef]
- Bonnet, J.; Fraile, A.; Sacristán, S.; Malpica, J.M.; García-Arenal, F. Role of recombination in the evolution of natural populations of Cucumber mosaic virus, a tripartite RNA plant virus. Virology 2005, 332, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Varsani, A.; Lefeuvre, P.; Roumagnac, P.; Martin, D. Notes on recombination and reassortment in multipartite/segmented viruses. Curr. Opin. Virol. 2018, 33, 156–166. [Google Scholar] [CrossRef] [PubMed]
- González-Candelas, F.; López-Labrador, F.X.; Bracho, M.A. Recombination in hepatitis C virus. Viruses 2011, 3, 2006–2024. [Google Scholar] [CrossRef] [Green Version]
- Yebra, G.; Frampton, D.; Gallo Cassarino, T.; Raffle, J.; Hubb, J.; Ferns, R.B.; Waters, L.; Tong, C.Y.W.; Kozlakidis, Z.; Hayward, A.; et al. A high HIV-1 strain variability in London, UK, revealed by full-genome analysis: Results from the ICONIC project. PLoS ONE 2018, 13, e0192081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bbosa, N.; Kaleebu, P.; Ssemwanga, D. HIV subtype diversity worldwide. Curr. Opin. HIV AIDS 2019, 14, 153–160. [Google Scholar] [CrossRef]
- Bentley, K.; Evans, D.J. Mechanisms and consequences of positive-strand RNA virus recombination. J. Gen. Virol. 2018, 99, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.J. The relation of recombination to mutational advance. Mutat. Res./Fundam. Mol. Mech. Mutagenesis 1964, 1, 2–9. [Google Scholar] [CrossRef]
- NextClade. Quality Control. Available online: https://docs.nextstrain.org/projects/nextclade/en/latest/user/algorithm/07-quality-control.html (accessed on 24 April 2022).
- Faria, N.; Claro, I.; Candido, D.; Franco, L.; Andrade, P.; Coletti, T.; Silva, C.; Sales, F.; Manuli, E.; Aguiar, R.; et al. Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in Manaus: Preliminary Findings. Available online: https://virological.org/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586 (accessed on 27 January 2021).
- Keck, J.G.; Stohlman, S.A.; Soe, L.H.; Makino, S.; Lai, M.M. Multiple recombination sites at the 5’-end of murine coronavirus RNA. Virology 1987, 156, 331–341. [Google Scholar] [CrossRef]
- Keck, J.G.; Matsushima, G.K.; Makino, S.; Fleming, J.O.; Vannier, D.M.; Stohlman, S.A.; Lai, M.M. In vivo RNA-RNA recombination of coronavirus in mouse brain. J. Virol. 1988, 62, 1810–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keck, J.G.; Soe, L.H.; Makino, S.; Stohlman, S.A.; Lai, M.M. RNA recombination of murine coronaviruses: Recombination between fusion-positive mouse hepatitis virus A59 and fusion-negative mouse hepatitis virus 2. J. Virol. 1988, 62, 1989–1998. [Google Scholar] [CrossRef] [Green Version]
- Makino, S.; Fleming, J.O.; Keck, J.G.; Stohlman, S.A.; Lai, M.M. RNA recombination of coronaviruses: Localization of neutralizing epitopes and neuropathogenic determinants on the carboxyl terminus of peplomers. Proc. Natl. Acad. Sci. USA 1987, 84, 6567–6571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, C.M.; Izeta, A.; Sánchez-Morgado, J.M.; Alonso, S.; Sola, I.; Balasch, M.; Plana-Durán, J.; Enjuanes, L. Targeted recombination demonstrates that the spike gene of transmissible gastroenteritis coronavirus is a determinant of its enteric tropism and virulence. J. Virol. 1999, 73, 7607–7618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, R.Y.; Krishnan, R.; Brian, D.A. The UCUAAAC promoter motif is not required for high-frequency leader recombination in bovine coronavirus defective interfering RNA. J. Virol. 1996, 70, 2720–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haijema, B.J.; Volders, H.; Rottier, P.J. Switching species tropism: An effective way to manipulate the feline coronavirus genome. J. Virol. 2003, 77, 4528–4538. [Google Scholar] [CrossRef] [Green Version]
- Herrewegh, A.A.; Smeenk, I.; Horzinek, M.C.; Rottier, P.J.; de Groot, R.J. Feline coronavirus type II strains 79-1683 and 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J. Virol. 1998, 72, 4508–4514. [Google Scholar] [CrossRef] [Green Version]
- Cavanagh, D.; Davis, P.J.; Cook, J.K. Infectious bronchitis virus: Evidence for recombination within the Massachusetts serotype. Avian Pathol. J. WVPA 1992, 21, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Kottier, S.A.; Cavanagh, D.; Britton, P. Experimental evidence of recombination in coronavirus infectious bronchitis virus. Virology 1995, 213, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Kusters, J.G.; Jager, E.J.; Niesters, H.G.; van der Zeijst, B.A. Sequence evidence for RNA recombination in field isolates of avian coronavirus infectious bronchitis virus. Vaccine 1990, 8, 605–608. [Google Scholar] [CrossRef]
- Wang, L.; Junker, D.; Collisson, E.W. Evidence of natural recombination within the S1 gene of infectious bronchitis virus. Virology 1993, 192, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, K.; Baltimore, D. The mechanism of RNA recombination in poliovirus. Cell 1986, 47, 433–443. [Google Scholar] [CrossRef]
- Baric, R.S.; Fu, K.; Schaad, M.C.; Stohlman, S.A. Establishing a genetic recombination map for murine coronavirus strain A59 complementation groups. Virology 1990, 177, 646–656. [Google Scholar] [CrossRef]
- Kuo, L.; Masters, P.S. Genetic evidence for a structural interaction between the carboxy termini of the membrane and nucleocapsid proteins of mouse hepatitis virus. J. Virol. 2002, 76, 4987–4999. [Google Scholar] [CrossRef] [Green Version]
- Mounir, S.; Talbot, P.J. Human coronavirus OC43 RNA 4 lacks two open reading frames located downstream of the S gene of bovine coronavirus. Virology 1993, 192, 355–360. [Google Scholar] [CrossRef]
- Wu, H.-Y.; Brian, D.A. Subgenomic messenger RNA amplification in coronaviruses. Proc. Natl. Acad. Sci. USA 2010, 107, 12257–12262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. PLoS Pathog. 2021, 17, e1009226. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.L.; Baric, R.S. Recombination, reservoirs, and the modular spike: Mechanisms of coronavirus cross-species transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.; Lau, S.K.; Yip, C.C.; Huang, Y.; Tsoi, H.W.; Chan, K.H.; Yuen, K.Y. Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J. Virol. 2006, 80, 7136–7145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyrc, K.; Dijkman, R.; Deng, L.; Jebbink, M.F.; Ross, H.A.; Berkhout, B.; van der Hoek, L. Mosaic structure of human coronavirus NL63, one thousand years of evolution. J. Mol. Biol. 2006, 364, 964–973. [Google Scholar] [CrossRef]
- Abdul-Rasool, S.; Fielding, B.C. Understanding Human Coronavirus HCoV-NL63. Open Virol. J. 2010, 4, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Lee, P.; Tsang, A.K.; Yip, C.C.; Tse, H.; Lee, R.A.; So, L.Y.; Lau, Y.L.; Chan, K.H.; Woo, P.C.; et al. Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J. Virol. 2011, 85, 11325–11337. [Google Scholar] [CrossRef] [Green Version]
- Holmes, E.C.; Rambaut, A. Viral evolution and the emergence of SARS coronavirus. Philos. Trans. R Soc. Lond. Ser. B Biol. Sci. 2004, 359, 1059–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.W.; Yap, Y.L.; Danchin, A. Testing the hypothesis of a recombinant origin of the SARS-associated coronavirus. Arch. Virol. 2005, 150, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Corman, V.M.; Ithete, N.L.; Richards, L.R.; Schoeman, M.C.; Preiser, W.; Drosten, C.; Drexler, J.F. Rooting the phylogenetic tree of middle East respiratory syndrome coronavirus by characterization of a conspecific virus from an African bat. J. Virol. 2014, 88, 11297–11303. [Google Scholar] [CrossRef] [Green Version]
- Dudas, G.; Rambaut, A. MERS-CoV recombination: Implications about the reservoir and potential for adaptation. Virus Evol. 2016, 2, vev023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabir, J.S.; Lam, T.T.; Ahmed, M.M.; Li, L.; Shen, Y.; Abo-Aba, S.E.; Qureshi, M.I.; Abu-Zeid, M.; Zhang, Y.; Khiyami, M.A.; et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 2016, 351, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Müller, N.F.; Kistler, K.; Bedford, T. Recombination patterns in coronaviruses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Li, X.; Giorgi, E.E.; Marichannegowda, M.H.; Foley, B.; Xiao, C.; Kong, X.P.; Chen, Y.; Gnanakaran, S.; Korber, B.; Gao, F. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci. Adv. 2020, 6, eabb9153. [Google Scholar] [CrossRef]
- Zhu, Z.; Meng, K.; Meng, G. Genomic recombination events may reveal the evolution of coronavirus and the origin of SARS-CoV-2. Sci. Rep. 2020, 10, 21617. [Google Scholar] [CrossRef]
- Hassanin, A.; Rambaud, O.; Klein, D. Genomic Bootstrap Barcodes and Their Application to Study the Evolution of Sarbecoviruses. Viruses 2022, 14, 440. [Google Scholar]
- Pedro, N.; Silva, C.N.; Magalhães, A.C.; Cavadas, B.; Rocha, A.M.; Moreira, A.C.; Gomes, M.S.; Silva, D.; Sobrinho-Simões, J.; Ramos, A.; et al. Dynamics of a Dual SARS-CoV-2 Lineage Co-Infection on a Prolonged Viral Shedding COVID-19 Case: Insights into Clinical Severity and Disease Duration. Microorganisms 2021, 9, 300. [Google Scholar] [CrossRef] [PubMed]
- Francisco Junior, R.d.S.; Benites, L.F.; Lamarca, A.P.; de Almeida, L.G.P.; Hansen, A.W.; Gularte, J.S.; Demoliner, M.; Gerber, A.L.; Guimaraes, A.P.d.C.; Antunes, A.K.E.; et al. Pervasive transmission of E484K and emergence of VUI-NP13L with evidence of SARS-CoV-2 co-infection events by two different lineages in Rio Grande do Sul, Brazil. Virus Res. 2021, 296, 198345. [Google Scholar] [CrossRef]
- Vankeerberghen, A.; Holderberke, A.; Boel, A.; Van Vaerenbergh, K.; De Beehouwer, H.; Cattoir, L. Case Report: A 90 year-old lady infected withb two COVID19 VoCs: 20I/501Y.V1 and 20H/501Y.V2. In Proceedings of the ECCMID, Vienna, Austria, 9–12 July 2021; p. 04978. [Google Scholar]
- Samoilov, A.E.; Kaptelova, V.V.; Bukharina, A.Y.; Shipulina, O.Y.; Korneenko, E.V.; Saenko, S.S.; Lukyanov, A.V.; Grishaeva, A.A.; PLoSkireva, A.A.; Speranskaya, A.S.; et al. Case report: Change of dominant strain during dual SARS-CoV-2 infection. BMC Infect. Dis. 2021, 21, 959. [Google Scholar] [CrossRef] [PubMed]
- Hashim, H.O.; Mohammed, M.K.; Mousa, M.J.; Abdulameer, H.H.; Alhassnawi, A.T.S.; Hassan, S.A.; Al-Shuhaib, M.B.S. Infection with different strains of SARS-CoV-2 in patients with COVID-19. Arch. Biol. Sci. 2020, 72, 575–585. [Google Scholar] [CrossRef]
- Zhou, H.-Y.; Cheng, Y.-X.; Xu, L.; Li, J.-Y.; Tao, C.-Y.; Ji, C.-Y.; Han, N.; Yang, R.; Li, Y.; Wu, A. Genomic evidence for divergent co-infections of SARS-CoV-2 lineages. bioRxiv 2021. [Google Scholar] [CrossRef]
- Dezordi, F.Z.; Resende, P.C.; Naveca, F.G.; do Nascimento, V.A.; de Souza, V.C.; Paixão, A.C.D.; Appolinario, L.; Lopes, R.S.; Mendonça, A.C.d.F.; da Rocha, A.S.B.; et al. Unusual SARS-CoV-2 intra-host diversity reveals lineages superinfection. Microb. Genom. 2021, 8. [Google Scholar] [CrossRef]
- Focosi, D.; Maggi, F.; Franchini, M.; McConnell, S.; Casadevall, A. Analysis of Immune Escape Variants from Antibody-Based Therapeutics against COVID-19: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 29. [Google Scholar] [CrossRef]
- Tonkin-Hill, G.; Martincorena, I.; Amato, R.; Lawson, A.R.J.; Gerstung, M.; Johnston, I.; Jackson, D.K.; Park, N.R.; Lensing, S.V.; Quail, M.A.; et al. Patterns of within-host genetic diversity in SARS-CoV-2. Elife 2020, 10, e66857. [Google Scholar] [CrossRef]
- Varabyou, A.; Pockrandt, C.; Salzberg, S.L.; Pertea, M. Rapid detection of inter-clade recombination in SARS-CoV-2 with Bolotie. Genetics 2020, 218, iyab074. [Google Scholar] [CrossRef]
- VanInsberghe, D.; Neish, A.; Lowen, A.C.; Koelle, K. Identification of SARS-CoV-2 recombinant genomes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zaman, S.; Sledzieski, S.; Berger, B.; Wu, Y.-C.; Bansal, M.S. Phylogenetic reconciliation reveals extensive ancestral recombination in Sarbecoviruses and the SARS-CoV-2 lineage. bioRxiv 2021. [Google Scholar] [CrossRef]
- De Maio, N.; Walker, C.; Borges, R.; Weilguny, L. Issues with SARS-CoV-2 Sequencing Data. Available online: https://virological.org/t/issues-with-sars-cov-2-sequencing-data/473 (accessed on 7 June 2022).
- van Dorp, L.; Richard, D.; Tan, C.C.S.; Shaw, L.P.; Acman, M.; Balloux, F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat. Commun. 2020, 11, 5986. [Google Scholar] [CrossRef] [PubMed]
- Nie, Q.; Li, X.; Chen, W.; Liu, D.; Chen, Y.; Li, H.; Li, D.; Tian, M.; Tan, W.; Zai, J. Phylogenetic and phylodynamic analyses of SARS-CoV-2. Virus Res. 2020, 287, 198098. [Google Scholar] [CrossRef]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z.; et al. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Kosakovsky Pond, S.; Nekrutenko, A.; Nielsen, R. Testing Recombination in the Pandemic SARS-CoV-2 Strains. Available online: https://virological.org/t/testing-recombination-inthe-pandemic-sars-cov-2-strains/492 (accessed on 7 June 2022).
- Richard, D.; Owen, C.J.; van Dorp, L.; Balloux, F. No detectable signal for ongoing genetic recombination in SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lyngsø, R.; Song, Y.; Hein, J. Minimum recombination histories by branch and bound. In Proceedings of the International Workshop on Algorithms in Bioinformatics, Mallorca, Spain, 3–6 October 2005. [Google Scholar]
- Rasmussen, M.D.; Hubisz, M.J.; Gronau, I.; Siepel, A. Genome-wide inference of ancestral recombination graphs. PLoS Genet. 2014, 10, e1004342. [Google Scholar] [CrossRef] [Green Version]
- Kelleher, J.; Wong, Y.; Wohns, A.W.; Fadil, C.; Albers, P.K.; McVean, G. Inferring whole-genome histories in large population datasets. Nat. Genet. 2019, 51, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Speidel, L.; Forest, M.; Shi, S.; Myers, S.R. A method for genome-wide genealogy estimation for thousands of samples. Nat. Genet. 2019, 51, 1321–1329. [Google Scholar] [CrossRef]
- Yi, H. 2019 Novel Coronavirus Is Undergoing Active Recombination. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2020, 71, 884–887. [Google Scholar] [CrossRef]
- Ignatieva, A.; Lyngsø, R.B.; Jenkins, P.A.; Hein, J. KwARG: Parsimonious reconstruction of ancestral recombination graphs with recurrent mutation. Bioinformatics 2021, 37, 3277–3284. [Google Scholar] [CrossRef] [PubMed]
- Ignatieva, A.; Hein, J.; Jenkins, P.A. Ongoing Recombination in SARS-CoV-2 Revealed through Genealogical Reconstruction. Mol. Biol. Evol. 2022, 39. [Google Scholar] [CrossRef] [PubMed]
- Turakhia, Y.; Thornlow, B.; Hinrichs, A.S.; Mcbroome, J.; Ayala, N.; Ye, C.; De Maio, N.; Haussler, D.; Lanfear, R.; Corbett-Detig, R. Pandemic-Scale Phylogenomics Reveals Elevated Recombination Rates in the SARS-CoV-2 Spike Region. bioRxiv 2021. [Google Scholar] [CrossRef]
- Haddad, D.; John, S.E.; Mohammad, A.; Hammad, M.M.; Hebbar, P.; Channanath, A.; Nizam, R.; Al-Qabandi, S.; Al Madhoun, A.; Alshukry, A.; et al. SARS-CoV-2: Possible recombination and emergence of potentially more virulent strains. PLoS ONE 2021, 16, e0251368. [Google Scholar] [CrossRef]
- Burel, E.; Colson, P.; Lagier, J.-C.; Levasseur, A.; Bedotto, M.; Lavrard, P.; Fournier, P.-E.; La Scola, B.; Raoult, D. Sequential appearance and isolation of a SARS-CoV-2 recombinant between two major SARS-CoV-2 variants in a chronically infected immunocompromised patient. medRxiv 2022. [Google Scholar] [CrossRef]
- Xie, X.; Lewis, T.-J.; Green, N.; Wang, Z. Phylogenetic network analysis revealed the recombinant origin of the SARS-CoV-2 VOC202012/01 (B.1.1.7) variant first discovered in U.K. bioRxiv. [CrossRef]
- Jackson, B.; Boni, M.F.; Bull, M.J.; Colleran, A.; Colquhoun, R.M.; Darby, A.C.; Haldenby, S.; Hill, V.; Lucaci, A.; McCrone, J.T.; et al. Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic. Cell 2021, 184, 5179–5188. [Google Scholar] [CrossRef]
- O’Toole, A.N.; Pybus, O.; Abram, M.E.; Kelly, E.J.; Rambaut, A. Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences. BMC Genom. 2021, 23, 121. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, B.; Castelan, H.G.; da Silva Candido, D.; Jackson, B.; Fleishon, S.; Ruis, C.; Delaye, L.; Rambaut, A.; Pybus, O.; Escalera-Zamudio, M. Emergence and widespread circulation of a recombinant SARS-CoV-2 lineage in North America. medRxiv 2021. [Google Scholar] [CrossRef]
- Sekizuka, T.; Itokawa, K.; Saito, M.; Shimatani, M.; Matsuyama, S.; Hasegawa, H.; Saito, T.; Kuroda, M.; Japan, C.-G.S.N.i. Genome Recombination between Delta and Alpha Variants of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). medRxiv 2021. [Google Scholar] [CrossRef]
- Colson, P.; Fournier, P.-E.; Delerce, J.; Million, M.; Bedotto, M.; Houhamdi, L.; Yahi, N.; Bayette, J.; Levasseur, A.; Fantini, J.; et al. Culture and identification of a “Deltamicron” SARS-CoV-2 in a three cases cluster in southern France. J. Med. Virol. 2022. [Google Scholar] [CrossRef]
- Simon-Loriere, E.; Montagutelli, X.; Lemoine, F.; Donati, F.; Touret, F.; Bourret, J.; Prot, M.; Munier, S.; Attia, M.; Conquet, L.; et al. Rapid characterization of a Delta-Omicron SARS-CoV-2 recombinant detected in Europe. Res. Sq. 2022. [Google Scholar] [CrossRef]
- He, Y.; Ma, W.; Dang, S.; Chen, L.; Zhang, R.; Mei, S.; Wei, X.; Lv, Q.; Peng, B.; Chen, J.; et al. Possible recombination between two variants of concern in a COVID-19 patient. Emerg. Microbes Infect. 2022, 11, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Rockett, R.J.; Draper, J.; Gall, M.; Sim, E.M.; Arnott, A.; Agius, J.E.; Johnson-Mackinnon, J.; Martinez, E.; Drew, A.P.; Lee, C.; et al. Co-infection with SARS-COV-2 Omicron and Delta Variants Revealed by Genomic Surveillance. medRxiv 2022. [Google Scholar] [CrossRef]
- Bolze, A.; Basler, T.; White, S.; Rossi, A.D.; Wyman, D.; Roychoudhury, P.; Greninger, A.L.; Hayashibara, K.; Beatty, M.; Shah, S.; et al. Evidence for SARS-CoV-2 Delta and Omicron co-infections and recombination. medRxiv 2022. [Google Scholar] [CrossRef]
- Kreier, F. Deltacron: The story of the variant that wasn’t. Nature 2022, 602, 19. [Google Scholar] [CrossRef]
- Ou, J.; Lan, W.; Wu, X.; Zhao, T.; Duan, B.; Yang, P.; Ren, Y.; Quan, L.; Zhao, W.; Seto, D.; et al. Tracking SARS-CoV-2 Omicron diverse spike gene mutations identifies multiple inter-variant recombination events. Signal Transduct Target 2022, 7, 138. [Google Scholar] [CrossRef]
- Lacek, K.A.; Rambo-Martin, B.L.; Batra, D.; Zheng, X.-y.; Sakaguchi, H.; Peacock, T.; Keller, M.; Wilson, M.M.; Sheth, M.; Davis, M.L.; et al. Identification of a Novel SARS-CoV-2 Delta-Omicron Recombinant Virus in the United States. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Maejima, M.; Shibusawa, M.; Natori, Y.; Nagakubo, Y.; Hosaka, K.; Sueki, H.; Mochizuki, H.; Tsutsui, T.; Kakizaki, Y.; et al. Classification of Omicron BA.1, BA.1.1 and BA.2 sublineages by TaqMan assay consistent with whole genome analysis data. medRxiv. [CrossRef]
- Duerr, R.; Dimartino, D.; Marier, C.; Zappile, P.; Wang, G.; Plitnick, J.; Griesemer, S.; Lasek-Nesselquist, E.; Dittman, M.; Ortigoza, M.B.; et al. Delta-Omicron recombinant SARS-CoV-2 in a transplant patient treated with Sotrovimab. bioRxiv 2022. [Google Scholar] [CrossRef]
- UK Health Security Agency. SARS-CoV-2 Variants of Concern and Variants under Investigation in England Technical Briefing 39. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1063424/tech-briefing-39-25march2022_final.pdf (accessed on 12 April 2022).
- UK Health Security Agency. SARS-CoV-2 Variants of Concern and Variants under Investigation in England. Technical Briefing 40. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1067672/Technical-Briefing-40-8April2022.pdf (accessed on 12 April 2022).
- Colson, P.; Delerce, J.; Marion-Paris, E.; Lagier, J.-C.; Bedotto, M.; Levasseur, A.; Fournier, P.-E.; LA Scola, B.; Raoult, D. A 21L/BA.2-21K/BA.1 MixOmicron SARS-CoV-2 hybrid undetected by qPCR that screen for variant in routine diagnosis. medRxiv. [CrossRef]
- Gu, H.; Ng, D.; Liu, G.; Cheng, S.; Krishnan, P.; Chang, L.; Cheuk, S.; Hui, M.; Lam, T.T.-Y.; Peiris, J.S.M.; et al. Detection of a BA.1/BA.2 recombinant in travelers arriving in Hong Kong, February 2022. Emerg. Infect. Dis. 2022, 28, 1276–1278. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Nadeau, S.; Yared, M.; Voinov, P.; Xie, N.; Roemer, C.; Stadler, T. CoV-Spectrum: Analysis of globally shared SARS-CoV-2 data to identify and characterize new variants. Bioinformatics 2021, 38, 1735–1737. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, L. SARS-CoV-2 Recombinant Finder (sc2rf). Available online: https://github.com/lenaschimmel/sc2rf (accessed on 24 April 2022).
- Focosi, D.; Maggi, F.; Casadevall, A. Mucosal Vaccines, Sterilizing Immunity, and the Future of SARS-CoV-2 Virulence. Viruses 2022, 14, 187. [Google Scholar] [CrossRef] [PubMed]
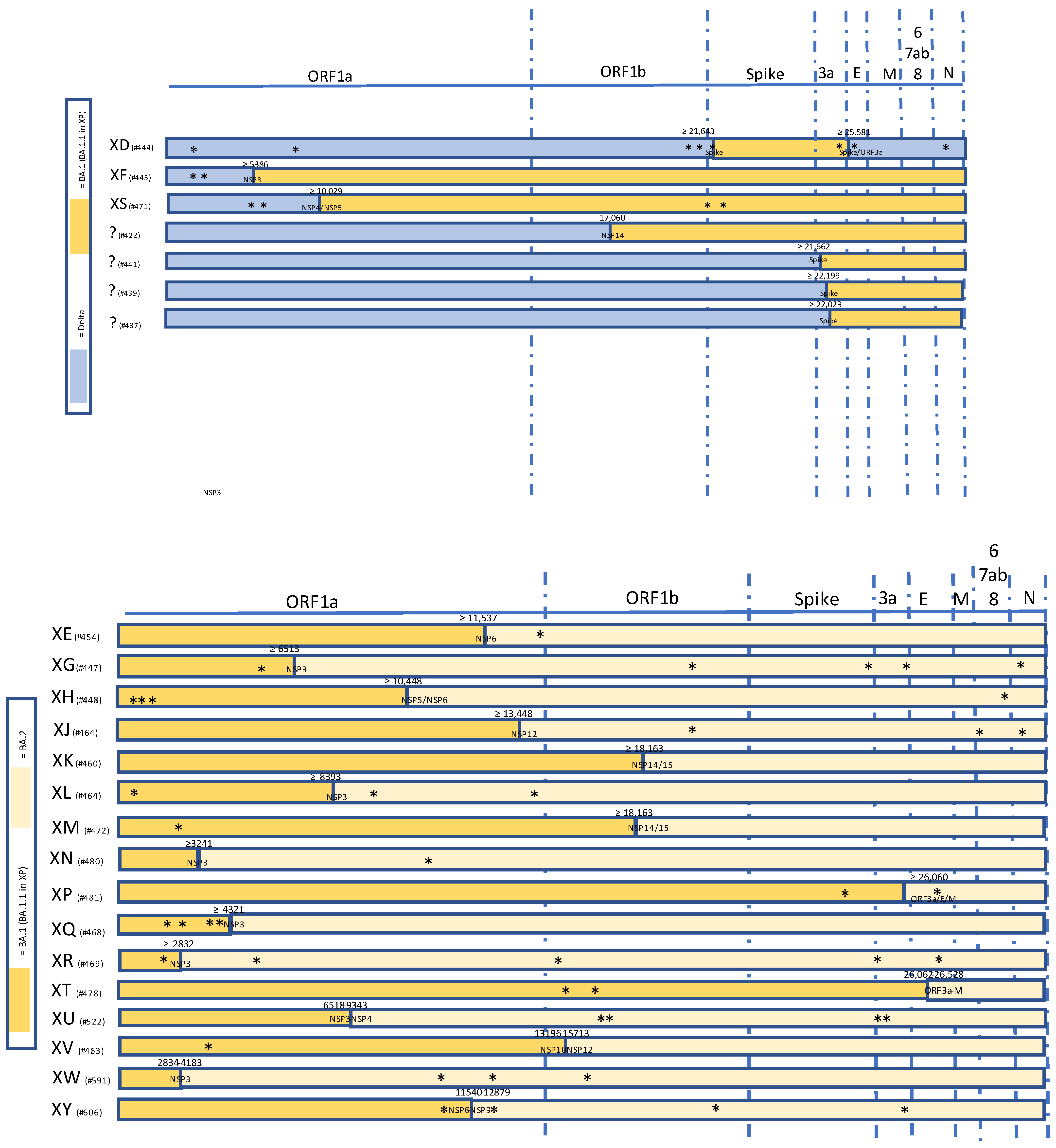
Figure 1.
Summary of selected recombinant SARS-CoV-2 lineages of interest between VOC Delta and Omicron (upper panel) and within Omicron VOC sublineages (lower panel). Unlabeled private mutations are marked with *.
Figure 1.
Summary of selected recombinant SARS-CoV-2 lineages of interest between VOC Delta and Omicron (upper panel) and within Omicron VOC sublineages (lower panel). Unlabeled private mutations are marked with *.

Figure 2.
Basic reproductive number (R0) and estimated herd immunity threshold for SARS-CoV-2 variants of concern and the XE recombinant compared to other human pathogens. Please note herd immunity cannot be currently achieved with the current generation of systemically delivered vaccines [92].
Figure 2.
Basic reproductive number (R0) and estimated herd immunity threshold for SARS-CoV-2 variants of concern and the XE recombinant compared to other human pathogens. Please note herd immunity cannot be currently achieved with the current generation of systemically delivered vaccines [92].


{kind=link}
{kind=link}
Table 1.
Details of PANGO-assigned recombinant SARS-CoV-2 lineages (modified from https://cov-lineages.org/lineage_list.html). More lineages can be found in Sakaguchi Hitochi’s Google Spreadsheet freely available online at https://docs.google.com/spreadsheets/d/1cQILRxXD756gJoRsaqMdJkxZm7sEjhV7ceY398Iz7gI/htmlview#gid=0 (accessed on 7 Jun 2022).
Table 1.
Details of PANGO-assigned recombinant SARS-CoV-2 lineages (modified from https://cov-lineages.org/lineage_list.html). More lineages can be found in Sakaguchi Hitochi’s Google Spreadsheet freely available online at https://docs.google.com/spreadsheets/d/1cQILRxXD756gJoRsaqMdJkxZm7sEjhV7ceY398Iz7gI/htmlview#gid=0 (accessed on 7 Jun 2022).
| Name | Most Common Countries | Earliest Date | Parental Lineages | PANGO Designation Issue (Ref) | Unlabeled Private Mutations | |
|---|---|---|---|---|---|---|
| Donor | Acceptor | |||||
| XA | UK 51.0%, USA 42.0%, Czech Republic 2.0%, Sweden 1.0%, Switzerland 1.0% | 18 December 2020 | B.1.1.7 and B.1.177 (20E/EU.1) | n.a. | n.a. | |
| XB | USA, Mexico, Guatemala, Honduras, El Salvador | 8 July 2020 | B.1.634 and B.1.631. Formally B.1.628 | #189 [74] | n.a. | |
| XC | Japan | 12 August 2021 | Delta AY.29 and B.1.1.7 | #263 [75] | C27972T, G28048T, A28111G (ORF8: Q27, R52I, Y73C) | |
| XD | France (40), Denmark (8), Belgium (1) | 13 December 2021 | Delta AY.4 | Omicron BA.1 Spike (nt 21,643 to 25,581; codons 156–179) | #444 [76,77] | A1321C, A8723G, C20032T, G21641T, T21760C, C25667T, G25855T, G29540A (NSP2: E172D) |
| XE | UK (763) and Ireland, growth rate of +9.8% per week, with a growth advantage over BA.2 of ~ 10% | 16 January 2022 | Omicron BA.1 | Omicron BA.2 | #454 | C14599T (NSP12) |
| XF | UK (39), no sample after 14 February 2022 | 7 January 2022 | Delta AY.4 (or AY.4.x) | Omicron BA.1 (break point near the end of NSP3 at nt 5386) | #445 | T1390C A2255G |
| XG | Denmark, UK, USA, Germany | 11 January 2022 | Omicron BA.1 | Omicron BA.2 (likely breakpoint between 5927 and 6511 (NSP3)) | #447 | G5672T, A19855G, C25672T, G26062C, dG29140T |
| XH | Denmark | 30 December 2021 | Omicron BA.1 | Omicron BA.2 (likely breakpoint between 10,448 and 11,287 (NSP5 or NSP6)) | #448 | T902C, C904A, G1244A, C28435T, (ORF1a:C213R, ORF1a:G327S, Nuc T902C) |
| XJ | Finland (Sweden, France, UK, Israel) | ??-??-2022 | Omicron BA.1 | Omicron BA.2 (breakpoint seems to be between nt 13,200 and 17,400) | #449 | T19857A, C27495T, -29759C |
| XK | Belgium (20) | 13 February 2022 | Omicron BA.1.1 | Omicron BA.2 | #460 | (ORF1a:R1628C, ORF1b:Q866R) |
| XL | UK | 6 February 2022 | Omicron BA.1 | Omicron BA.2 | #464 | G875T, T9208C, G14229A |
| XM | Europe | 21 February 2022 | Omicron BA.1.1 | Omicron BA.2 | #472 | C2470T |
| XN | UK, Italy | 1 February 2022 | Omicron BA.1 | Omicron BA.2 (likely breakpoint: between 2834 and 4183 at NSP3) | #480 | G10986A |
| XP | UK | 26 February 2022 | Omicron BA.2 | Omicron BA.1.1 | #481 | A24190C, C26880C |
| XQ | UK | 13 February 2022 | Omicron BA.1.1 | Omicron BA.2 | #468 | A17615G, C2470T, (ORF3a:T223I, ORF1a:K856R, ORF1a:L3027F |
| XR | UK | 13 February 2022 | Omicron BA.1.1 | Omicron BA.2 (likely breakpoint between 4322 and 4891 at NSP3) | #469 | (ORF1a:K856R, ORF1a:T1543I, ORF1a:D4344N, A29510C) |
| XS | USA | 19 January 2022 | Delta AY.x | Omicron BA.1.1 | #471 | C5365T, C6196T, T13195C, C15240T, C21595T, C27807T |
| XT | South Africa (Gauteng, Limpopo and North-West) | 13 December 2021 | BA.1 | BA.2 (likely breakpoint between 26,062 and 26,258 at ORF3a/M) | #478 | C13994T, C16386T (S:G75V) |
| XU | India (Gurajat, Maharashtra), Japan, Australia | 20 January 2022 | BA.1 | BA.2 (likely breakpoint between 6518 and 9343 at NSP3 or NSP4) | #522 | C16887T, C17012T, C25416T, G25471C |
| XV | Denmark, Italy | 31 January 2022 | BA.1 | BA.2 (likely breakpoint between 13,196 and 15,713 (NSP10 to NSP12)) | #463 | C3583T |
| XW | USA, Germany, England, Canada, Japan (ex-Finland) | 13 March 2022 | BA.1 | BA.2 (likely breakpoint between 2834 and 4183 (NSP3) | #591 | C10507T, C12756T, G16020T (ORF1a:T4164I) |
| XY | France, Israel, Scotland, USA | 28 February 2022 | BA.1 | BA.2 (likely breakpoint between 11,540 and 12,879 (NSP6-NSP9) | #606 | A1585G, T11049C (ORF1a:V3595A) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Focosi, D.; Maggi, F. Recombination in Coronaviruses, with a Focus on SARS-CoV-2. Viruses 2022, 14, 1239. https://doi.org/10.3390/v14061239
AMA Style
Focosi D, Maggi F. Recombination in Coronaviruses, with a Focus on SARS-CoV-2. Viruses. 2022; 14(6):1239. https://doi.org/10.3390/v14061239
Chicago/Turabian StyleFocosi, Daniele, and Fabrizio Maggi. 2022. "Recombination in Coronaviruses, with a Focus on SARS-CoV-2" Viruses 14, no. 6: 1239. https://doi.org/10.3390/v14061239
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.