
Longitudinal Survey of Coronavirus Circulation and Diversity in Insectivorous Bat Colonies in Zimbabwe

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Methods
2.1. Study Sites
2.2. Magweto Cave
2.3. Chirundu Farm
2.4. Sample and Data Collection
2.5. RNA/DNA Extraction
2.6. Reverse Transcription (RT) Using Random Hexamers
2.7. RNA-Dependent RNA-Polymerase (RdRp) Nested PCR on Coronaviruses Using Pan-Coronavirus Primers
2.8. Genetic Analyses
2.8.1. Viruses Sequence Identification and Phylogenetic Analysis
2.8.2. Bat Species Identification
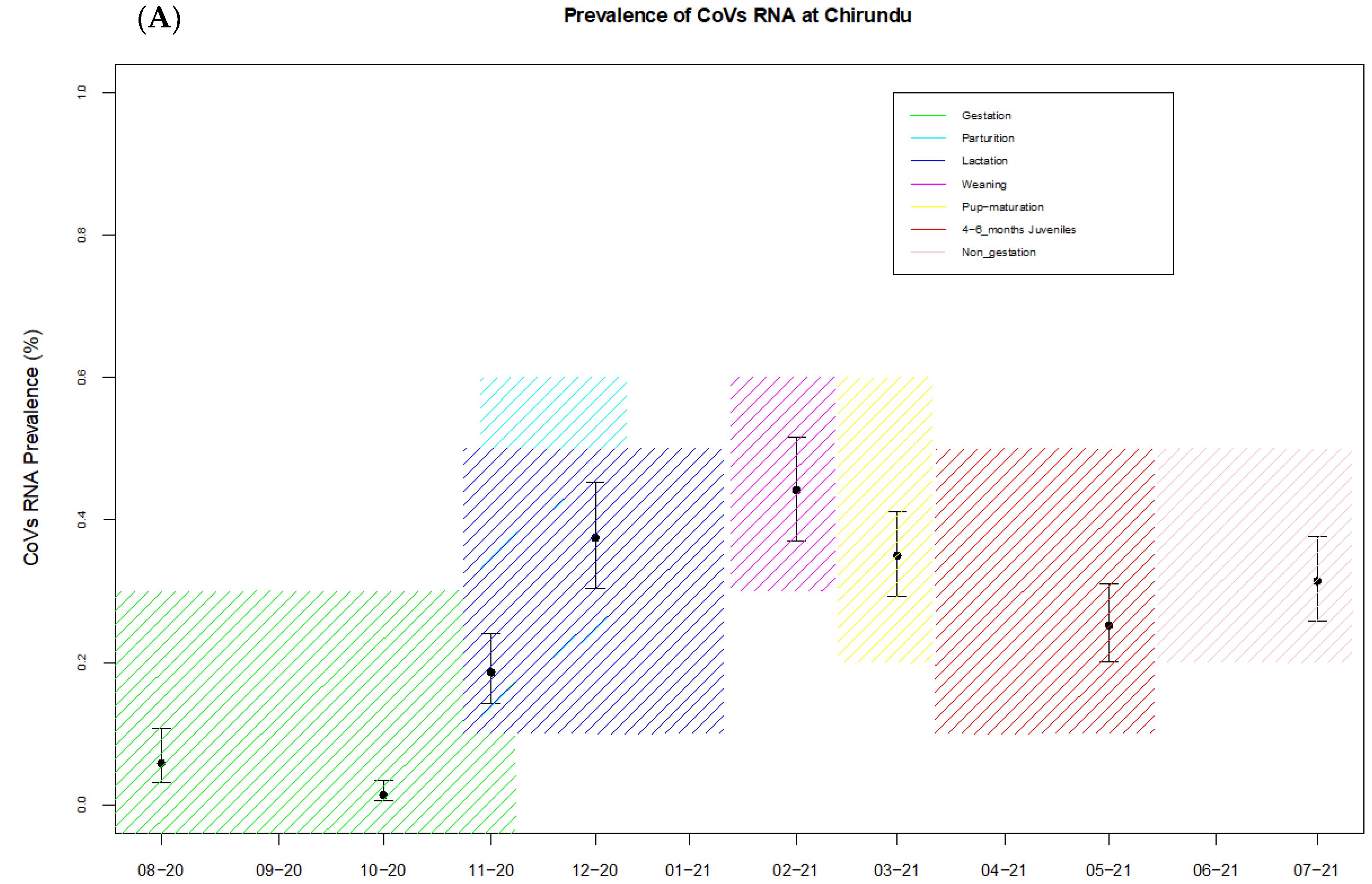
2.8.3. Temporal Variations of Coronavirus Prevalence and Bat Reproduction Phenology
2.8.4. GenBank Accession Numbers
3. Results
3.1. Sampling, Data Collection and Reproductive Phenology
3.2. Bat Species
3.3. Prevalence and Seasonality of RNA Coronaviruses at the Bat Community Level
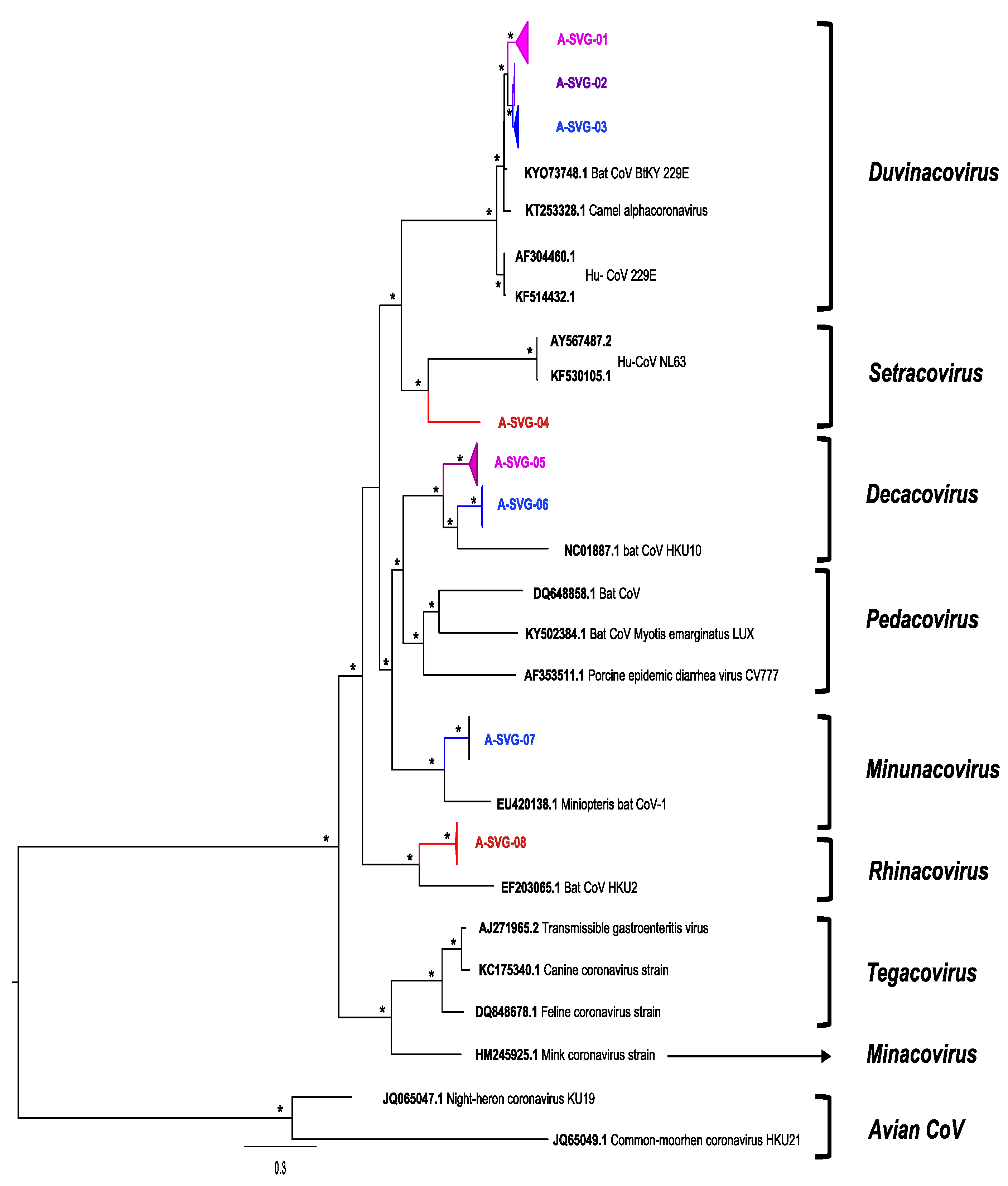
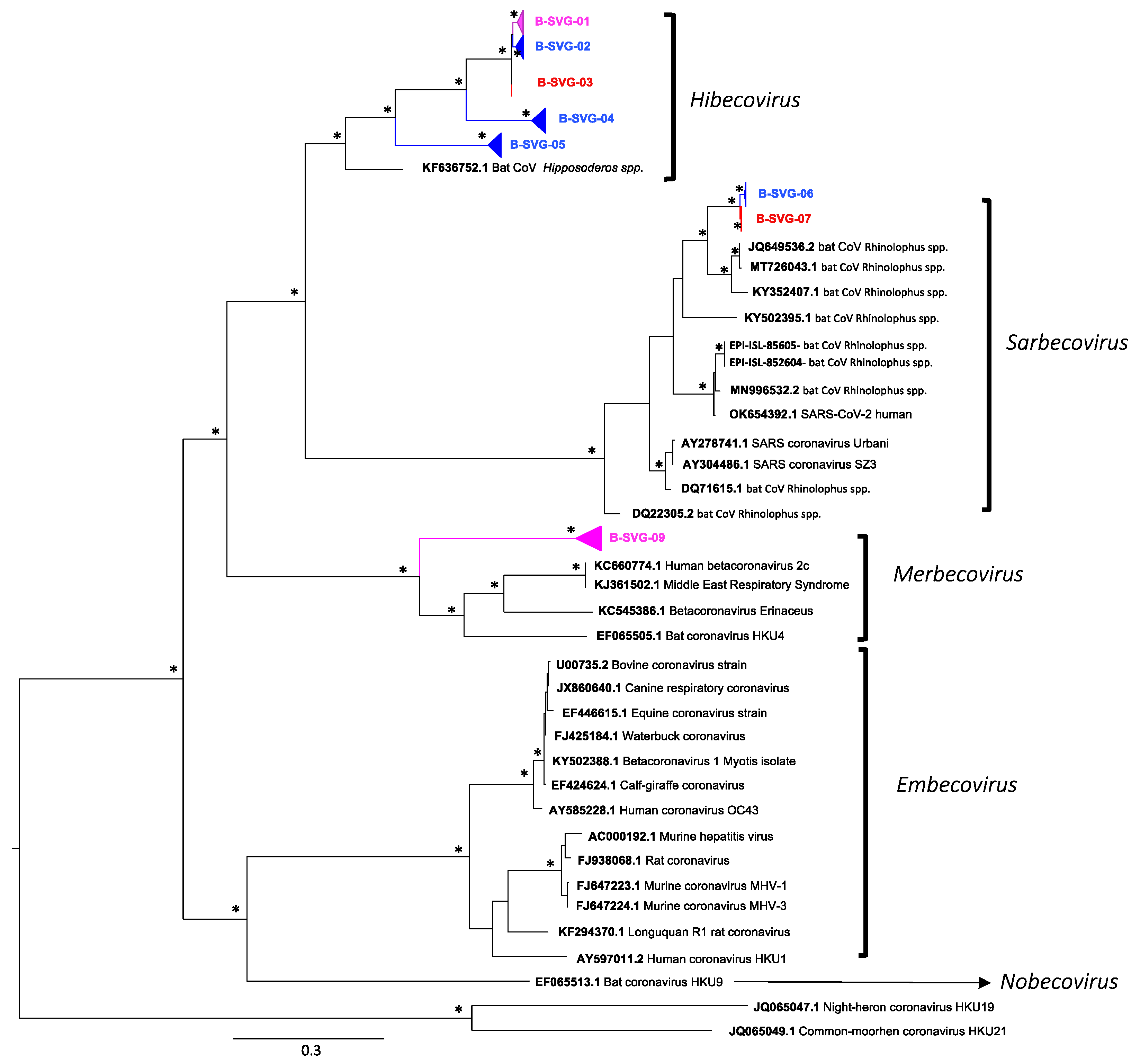
3.4. Genetic Diversity of Coronaviruses at Chirundu and Magweto Sites
3.5. Alphacoronaviruses
3.6. Betacoronaviruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ICTV Positive Sense RNA Viruses: Coronaviridae. VIRUS Taxon. 2020. Available online: https://talk.ictvonline.org/ (accessed on 1 December 2021).
- Al-Qahtani, A.A. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): Emergence, history, basic and clinical aspects. Saudi J. Biol. Sci. 2020, 27, 2531–2538. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and Sources of Endemic Human Coronaviruses. Adv. Virus Res. 2018, 100, 163–188. [Google Scholar] [PubMed]
- Li, K.; Lin, X.; Wang, W.; Shi, M.; Guo, W.; Zhang, X.; Xing, J.; He, J.; Wang, K.; Li, M.; et al. Isolation and characterization of a novel arenavirus harbored by Rodents and Shrews in Zhejiang province, China. Virology 2015, 476, 37–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monchatre-Leroy, E.; Boué, F.; Boucher, J.M.; Renault, C.; Moutou, F.; Gouilh, M.A.; Umhang, G. Identification of alpha and beta coronavirus in wildlife species in france: Bats, rodents, rabbits, and hedgehogs. Viruses 2017, 9, 364. [Google Scholar] [CrossRef] [Green Version]
- Wertheim, J.O.; Chu, D.K.W.; Peiris, J.S.M.; Kosakovsky Pond, S.L.; Poon, L.L.M. A Case for the Ancient Origin of Coronaviruses. J. Virol. 2013, 87, 7039–7045. [Google Scholar] [CrossRef] [Green Version]
- Macnaughton, M.R.; Thomas, B.J.; Davies, H.A.; Patterson, S. Infectivity of human coronavirus strain 229E. J. Clin. Microbiol. 1980, 12, 462–468. [Google Scholar] [CrossRef] [Green Version]
- Van Der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.M.; Wolthers, K.C.; Wertheim-Van Dillen, P.M.E.; Kaandorp, J.; Spaargaren, J.; Berkhout, B. Identification of a new human coronavirus. Nat. Med. 2004, 10, 368–373. [Google Scholar] [CrossRef]
- Razanajatovo, N.H.; Nomenjanahary, L.A.; Wilkinson, D.A.; Razafimanahaka, J.H.; Goodman, S.M.; Jenkins, R.K.; Jones, J.P.G.; Heraud, J.-M. Detection of new genetic variants of Betacoronaviruses in Endemic Frugivorous Bats of Madagascar. Virol. J. 2015, 12, 42. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.Y.; Lau, S.K.P.; Li, K.S.M.; Poon, R.W.S.; Wong, B.H.L.; Tsoi, H.; Yip, B.C.K.; Huang, Y.; Chan, K.; Yuen, K. Molecular diversity of coronaviruses in bats. Virology 2006, 351, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Ge, X.; Wang, L.; Shi, Z. Bat origin of human coronaviruses: Emerging and re-emerging pathogens in humans and animals Susanna Lau Positive-strand RNA viruses. Virol. J. 2015, 12, 209. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.; Wang, X.; Hu, B.; Zhang, L.; Zhang, W.; Guo, H.; Jiang, R.; Liu, M.; Chen, Y.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlasova, A.; Saif, L.J. Bovine Coronavirus and the Associated Diseases. Front. Vet. Sci. 2021, 8, 1–14. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Huang, Y.; Yuen, K.Y. Coronavirus Diversity, Phylogeny and Interspecies Jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luis, A.D.; O’Shea, T.J.; Hayman, D.T.S.; Wood, J.L.N.; Cunningham, A.A.; Gilbert, A.T.; Mills, J.N.; Webb, C.T. Network analysis of host—Virus communities in bats and rodents reveals determinants of cross-species transmission. Ecol. Lett. 2015, 18, 1153–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Cottontail, V.M.; Rasche, A.; Yordanov, S.; et al. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peiris, J.S.M.; Guan, Y.; Yuen, K.Y. Severe acute respiratory syndrome. Nat. Med. 2004, 10, S88–S97. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, A.; Kamel, M. Coronaviruses in humans and animals: The role of bats in viral evolution. Environ. Sci. Pollut. Res. Int. 2021, 28, 19589–19600. [Google Scholar] [CrossRef]
- Li, X.; Zai, J.; Zhao, Q.; Nie, Q.; Li, Y.; Foley, B.T.; Chaillon, A. Evolutionary history, potential intermediate animal host, and cross-species analyses of SARS-CoV-2. J. Med. Virol. 2020, 92, 602–611. [Google Scholar] [CrossRef]
- Delaune, D.; Hul, V.; Karlsson, E.A.; Hassanin, A.; Ou, T.P.; Baidaliuk, A.; Gámbaro, F.; Prot, M.; Tu, V.T.; Chea, S.; et al. A novel SARS-CoV-2 related coronavirus in bats from Cambodia. Nat. Commun. 2021, 12, 6563. [Google Scholar] [CrossRef]
- Coertse, J.; De Vries, L.; Geldenhuys, M.; Mortlock, M. Bat-borne viruses in Africa: A critical review. J. Zool. 2020, 311, 77–98. [Google Scholar]
- Ar Gouilh, M.; Puechmaille, S.J.; Diancourt, L.; Vandenbogaert, M.; Serra-Cobo, J.; Lopez Roïg, M.; Brown, P.; Moutou, F.; Caro, V.; Vabret, A.; et al. SARS-CoV related Betacoronavirus and diverse Alphacoronavirus members found in western old-world. Virology 2018, 517, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.P. Viruses and Bats. Viruses 2019, 11, 884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latinne, A.; Hu, B.; Olival, K.J.; Zhu, G.; Zhang, L.; Li, H.; Chmura, A.A.; Field, H.E.; Zambrana-Torrelio, C.; Epstein, J.H.; et al. Origin and cross-species transmission of bat coronaviruses in China. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important Reservoir Hosts of Emerging Viruses. Clin. Microbiol. Rev. 2006, 19, 531–545. [Google Scholar] [CrossRef] [Green Version]
- Role, T.H.E.; Bats, O.F.; Zoonoses, I.N.E. Investigating the Role of Bats in Emerging Zoonoses: Balancing Ecology, Conservation and Public Health Interests; Newman, S., Field, H., de Jong, C., Epstein, J.H., Eds.; FAO Animal; FAO: Rome, Italy, 2011; ISBN 1810-1119. [Google Scholar]
- Alfelt, A.; Devaux, C.; Serra-Cobo, J.; Frutos, R. Bats, Bat-Borne Viruses, and Environmental Changes. In Bats; IntechOpen: London, UK, 2018; pp. 113–132. [Google Scholar]
- Wacharapluesadee, S.; Duengkae, P.; Chaiyes, A.; Kaewpom, T.; Rodpan, A.; Yingsakmongkon, S.; Petcharat, S.; Phengsakul, P.; Maneeorn, P.; Hemachudha, T. Longitudinal study of age-specific pattern of coronavirus infection in Lyle’s flying fox (Pteropus lylei) in Thailand. Virol. J. 2018, 15, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Anthony, S.J.; Johnson, C.K.; Greig, D.J.; Kramer, S.; Che, X.; Wells, H.; Hicks, A.L.; Joly, D.O.; Wolfe, N.D.; Daszak, P.; et al. Global patterns in coronavirus diversity. Virus Evol. 2017, 3, 1–15. [Google Scholar] [CrossRef]
- Cappelle, J.; Furey, N.; Hoem, T.; Ou, T.P.; Lim, T.; Hul, V.; Heng, O.; Chevalier, V.; Dussart, P.; Duong, V. Longitudinal monitoring in Cambodia suggests higher circulation of alpha and betacoronaviruses in juvenile and immature bats of three species. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Banerjee, A.; Kulcsar, K.; Misra, V.; Frieman, M.; Mossman, K. Bats and coronaviruses. Viruses 2019, 11, 41. [Google Scholar] [CrossRef] [Green Version]
- Bourgarel, M.; Pfukenyi, D.M.; Boué, V.; Talignani, L.; Chiweshe, N.; Diop, F.; Caron, A.; Matope, G.; Missé, D.; Liégeois, F. Circulation of Alphacoronavirus, Betacoronavirus and Paramyxovirus in Hipposideros bat species in Zimbabwe. Infect. Genet. Evol. 2018, 58, 253–257. [Google Scholar] [CrossRef]
- Bourgarel, M.; Noël, V.; Pfukenyi, D.; Michaux, J.; André, A.; Becquart, P.; Cerqueira, F.; Barrachina, C.; Boué, V.; Talignani, L.; et al. Next-generation sequencing on insectivorous bat guano: An accurate tool to identify arthropod viruses of potential agricultural concern. Viruses 2019, 11, 1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monadjem, A.; Taylor, P.J.; Cotterill, F.P.D.; Schoeman, M. Bats of Southern and Central Africa: A Biogeographic and Taxonomic Synthesis; Wits University Press: Johannesburg, South Africa, 2010. [Google Scholar]
- De Nys, H.M.; Mbala Kingebeni, P.; Keita, A.K.; Butel, C.; Thaurignac, G.; Villabona-Arenas, C.J.; Lemarcis, T.; Geraerts, M.; Vidal, N.; Esteban, A.; et al. Survey of ebola viruses in frugivorous and insectivorous bats in Guinea, Cameroon, and the democratic republic of the Congo, 2015–2017. Emerg. Infect. Dis. 2018, 24, 2228–2240. [Google Scholar] [CrossRef]
- Chu, D.K.W.; Leung, C.Y.H.; Gilbert, M.; Joyner, P.H.; Ng, E.M.; Tse, T.M.; Guan, Y.; Peiris, J.S.M.; Poon, L.L.M. Avian coronavirus in wild aquatic birds. J. Virol. 2011, 85, 12815–12820. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoder, A.D.; Yang, Z. Divergence dates for Malagasy lemurs estimated from multiple gene loci: Geological and evolutionary context. Mol. Ecol. 2004, 13, 757–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocher, T.D.; Thomas, W.K.; Meyer, A.; Edwards, S.V.; Paabo, S.; Villablanca, F.X.; Wilson, A.C. Dynamics of mitochondrial DNA evolution in animals: Amplification and sequencing with conserved primers. Proc. Natl. Acad. Sci. USA 1989, 86, 6196–6200. [Google Scholar] [CrossRef] [Green Version]
- Agresti, A.; Coull, B.A. Approximate Is Better than “Exact” for Interval Estimation of Binomial Proportions. Am. Stat. 1998, 52, 119–126. [Google Scholar]
- Geldenhuys, M.; Mortlock, M.; Epstein, J.H.; Pawęska, J.T.; Weyer, J.; Markotter, W. Overview of Bat and Wildlife Coronavirus Surveillance in Africa: A Framework for Global Investigations. Viruses 2021, 13, 936. [Google Scholar] [CrossRef]
- Seltmann, A.; Corman, V.M.; Rasche, A.; Drosten, C. Seasonal Fluctuations of Astrovirus, But Not Coronavirus Shedding in Bats Inhabiting Human-Modified Tropical Forests. EcoHealth 2017, 14, 272–284. [Google Scholar] [CrossRef] [Green Version]
- Drexler, J.F.; Corman, V.M.; Wegner, T.; Tateno, A.F.; Zerbinati, R.M.; Gloza-Rausch, F.; Seebens, A.; Müller, M.A.; Drosten, C. Amplification of emerging viruses in a bat colony. Emerg. Infect. Dis. 2011, 17, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Montecino-latorre, D.; Goldstein, T.; Gilardi, K.; Wolking, D.; Wormer, E.V.; Kazwala, R.; Ssebide, B.; Nziza, J.; Sijali, Z.; Cranfield, M.; et al. Reproduction of East-African bats may guide risk mitigation for coronavirus spillover. One Health Outlook 2020, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cakenberghe, V.; Seamark, E. African Chiroptera Report 2020; AfricanBats NPC: Pretoria, South Africa, 2020. [Google Scholar]
- Wong, A.C.P.; Li, X.; Lau, S.K.P.; Woo, P.C.Y. Global epidemiology of bat coronaviruses. Viruses 2019, 11, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anthony, S.J.; Ojeda-Flores, R.; Rico-Chávez, O.; Navarrete-Macias, I.; Zambrana-Torrelio, C.M.; Rostal, M.K.; Epstein, J.H.; Tipps, T.; Liang, E.; Sanchez-Leon, M.; et al. Coronaviruses in bats from Mexico. J. Gen. Virol. 2013, 94, 1028–1038. [Google Scholar] [CrossRef]
- Annan, A.; Baldwin, H.J.; Corman, V.M.; Klose, S.M.; Owusu, M.; Nkrumah, E.E.; Badu, E.K.; Anti, P.; Agbenyega, O.; Meyer, B.; et al. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 2013, 19, 456–459. [Google Scholar] [CrossRef]
- Memish, Z.A.; Mishra, N.; Olival, K.J.; Fagbo, S.F.; Kapoor, V.; Epstein, J.H.; AlHakeem, R.; Al Asmari, M.; Islam, A.; Kapoor, A.; et al. Middle East respiratory syndrome coronavirus in Bats, Saudi Arabia. Emerg. Infect. Dis. 2013, 19, 1819–1823. [Google Scholar] [CrossRef] [Green Version]
- Monadjem, A.; Soisook, P.; Thong, V.; Kingston, T. Family Hipposideridae (Old World Leaf-nosed Bats). Handb. Mamm. World 2019, 9, 210–227. [Google Scholar]
- Plowright, R.K.; Eby, P.; Hudson, P.J.; Smith, I.L.; Westcott, D.; Bryden, W.L.; Middleton, D.; Reid, P.A.; Mcfarlane, R.A.; Martin, G.; et al. Ecological dynamics of emerging bat virus spillover. Proc. R. Soc. B Biol. Sci. 2014, 282, 20142124. [Google Scholar] [CrossRef] [Green Version]
- Hayman, D.T.S.; Bowen, R.A.; Cryan, P.M.; Mccracken, G.F.; O’Shea, T.J.; Peel, A.J.; Gilbert, A.; Webb, C.T.; Wood, J.L.N. Ecology of Zoonotic Infectious Diseases in Bats: Current Knowledge and Future Directions. Zoonoses Public Health 2013, 60, 2–21. [Google Scholar] [CrossRef] [Green Version]
- Bergner, L.M.; Orton, R.J.; Benavides, J.A.; Becker, D.J.; Tello, C.; Biek, R.; Streicker, D.G. Demographic and environmental drivers of metagenomic viral diversity in vampire bats. Mol. Ecol. 2020, 29, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Menachery, V.D.; Yount, B.L.; Debbink, K.; Agnihothram, S.; Gralinski, L.E.; Plante, J.A.; Graham, R.L.; Scobey, T.; Ge, X.Y.; Donaldson, E.F.; et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2015, 21, 1508–1513. [Google Scholar] [CrossRef] [PubMed]
- Plowright, R.K.; Parrish, C.R.; Mccallum, H.; Hudson, P.J.; Ko, A.I.; Graham, A.L.; Lloyd-smith, J.O. Pathways to zoonotic spillover. Nat. Rev. Microbiol. 2017, 15, 502–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrish, C.R.; Holmes, E.C.; Morens, D.M.; Park, E.; Burke, D.S.; Calisher, C.H.; Laughlin, C.A.; Saif, L.J.; Daszak, P. Cross-Species Virus Transmission and the Emergence of New Epidemic Diseases. Microbiol. Mol. Biol. Rev. 2008, 72, 457–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Reproduction Cycle | Month Sampled | No of Samples Tested | No of Coronavirus Positives | Prevalence (%) + CI (95%) |
|---|---|---|---|---|---|
| Chirundu | Non-gestation | August 2020 | 154 | 9 | 5.8 (3.1–10.7) |
| Pregnancy | October 2020 | 296 | 4 | 1.35 (0.5–3.4) | |
| Parturition | November 2020 | 242 | 45 | 18 (14.2–23.9) | |
| Lactation | December 2020 | 160 | 60 | 37 (30.4–45.2) | |
| Weaning | Februar 2021 | 172 | 76 | 44.2 (37–51.7) | |
| 4–6 months juveniles | March 2021 | 240 | 84 | 35 (29.2–41.2) | |
| 4–6 months juveniles | May 2021 | 242 | 61 | 25.2 (20.2–31) | |
| Non-gestation | July 2021 | 226 | 71 | 31 (25.7–37.7) | |
| Overall prevalence | 1732 | 410 | 23.7 (21.73–25.73) | ||
| Magweto | Non-gestation | September 2020 | 348 | 7 | 2.0 (0.98–4.1) |
| Pregnancy | October 2020 | 257 | 27 | 10.5 (7.3–14.9) | |
| Parturition | November 2020 | 228 | 82 | 35.9 (30–42.4) | |
| 4–6 months juveniles | March 2021 | 242 | 84 | 34.7 (28.9–40.9) | |
| 4–6 months juveniles | April 2021 | 241 | 40 | 16.6 (12.4–21.8) | |
| Non-gestation | June 2021 | 242 | 17 | 7.02 (4.4–10.9) | |
| Overall prevalence | 1558 | 257 | 16.5 (14.74–18.42) | ||
| Magweto M. gigas | Non-gestation | September 2020 | 2 | 2 | 100 (34.2–100) |
| Pregnancy | Octobe 2020 | 124 | 70 | 56.4 (47.7–64.9) | |
| Parturition | November 2020 | 74 | 26 | 35.1 (25.2–46.5) | |
| 4–6 months juveniles | March 2021 | 42 | 8 | 19.1 (9.98–33.3) | |
| 4–6 months juveniles | April 2021 | 39 | 9 | 23.1 (12.7–38.3) | |
| Non-gestation | June 2021 | 27 | 5 | 18.5 (8.18–36.7) | |
| Overall prevalence | 308 | 120 | 38.96 (33.68–44.51) | ||
| Number of Sequence per Site | Number of Sequences per Bat Species | Longitudinal Detection of the Different Viral Groups | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Viral Group | Total Number of Sequences | Magweto | Chirundu | Hipposideros spp. | Macronycteris spp. | Rhinolophe spp. | Nycteris spp. | Miniopterus spp. | Unknown | Magweto | Chirundu |
| A-SVG-01 | 114 | 8 | 106 | 89 | 4 | 3 | 7 | 0 | 11 | March 2021 to June 2021 | August 2020 to May 2021 |
| A-SVG-02 | 45 | 42 | 3 | 0 | 41 | 2 | 0 | 0 | 2 | September 2020 to November 2020 | October 2020 to November 2020 |
| A-SVG-03 | 32 | 32 | 0 | 29 | 0 | 2 | 0 | 0 | 1 | September 2020 to March 2021 | - |
| A-SVG-04 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | - | May 2021 |
| A-SVG-05 | 84 | 6 | 78 | 7 | 4 | 52 | 1 | 3 | 17 | June 2021 | September 2020 to July 2021 |
| A-SVG-06 | 13 | 13 | 0 | 1 | 0 | 9 | 0 | 1 | 2 | September 2020 to November 2020 | - |
| A-SVG-07 | 2 | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | October 2020 | - |
| A-SVG-08 | 16 | 0 | 16 | 0 | 0 | 8 | 1 | 0 | 7 | - | August-November-December 2020-July 2021 |
| sub Total | 307 | 103 | 204 | 126 | 49 | 76 | 9 | 6 | 41 | ||
| Number of Sequence per site | Number of sequences per Bat species | Longitudinal Detection of the different viral groups | |||||||||
| B-SVG-01 | 105 | 5 | 100 | 76 | 2 | 4 | 1 | 0 | 22 | March to April 2021 | November 2020 to July 2021 |
| B-SVG-02 | 21 | 21 | 0 | 19 | 0 | 0 | 0 | 0 | 2 | November 2020 to April 2021 | - |
| B-SVG-03 | 6 | 0 | 6 | 2 | 0 | 1 | 0 | 0 | 3 | - | February 2021 May and June 2021 |
| B-SVG-04 | 13 | 13 | 0 | 0 | 13 | 0 | 0 | 0 | 0 | September 2020 to June 2021 | - |
| B-SVG-05 | 51 | 51 | 0 | 48 | 0 | 0 | 0 | 0 | 3 | November 2020 to July 2021 | March 2021 |
| B-SVG-06 | 8 | 8 | 0 | 0 | 0 | 5 | 1 | 0 | 2 | October 2020 to March 2021 | - |
| B-SVG-07 | 4 | 0 | 4 | 0 | 0 | 2 | 0 | 0 | 2 | - | July 2021 |
| B-SVG-08 | 17 | 3 | 14 | 8 | 0 | 1 | 6 | 0 | 2 | April 2021 | August 2020 to Mach 2021 |
| sub Total | 225 | 101 | 124 | 153 | 15 | 13 | 8 | 0 | 36 | ||
| TOTAL | 532 | 204 | 328 | 279 | 64 | 89 | 17 | 6 | 77 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chidoti, V.; De Nys, H.; Pinarello, V.; Mashura, G.; Missé, D.; Guerrini, L.; Pfukenyi, D.; Cappelle, J.; Chiweshe, N.; Ayouba, A.; et al. Longitudinal Survey of Coronavirus Circulation and Diversity in Insectivorous Bat Colonies in Zimbabwe. Viruses 2022, 14, 781. https://doi.org/10.3390/v14040781
Chidoti V, De Nys H, Pinarello V, Mashura G, Missé D, Guerrini L, Pfukenyi D, Cappelle J, Chiweshe N, Ayouba A, et al. Longitudinal Survey of Coronavirus Circulation and Diversity in Insectivorous Bat Colonies in Zimbabwe. Viruses. 2022; 14(4):781. https://doi.org/10.3390/v14040781
Chicago/Turabian StyleChidoti, Vimbiso, Hélène De Nys, Valérie Pinarello, Getrude Mashura, Dorothée Missé, Laure Guerrini, Davies Pfukenyi, Julien Cappelle, Ngoni Chiweshe, Ahidjo Ayouba, and et al. 2022. "Longitudinal Survey of Coronavirus Circulation and Diversity in Insectivorous Bat Colonies in Zimbabwe" Viruses 14, no. 4: 781. https://doi.org/10.3390/v14040781