
Human Cytomegalovirus Infection Elicits Global Changes in Host Transcription by RNA Polymerases I, II, and III

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses, Cells, and Conditions of Infection
2.2. PRO-Seq and PRO-Cap
2.3. DFF-ChIP
2.4. Generation of Tracks
2.5. TruQuant Analysis, Gene Body Clustering, FragMaps, and Metaplots
2.6. Bioinformatics Analysis of tRNA Transcription
2.7. Motif Enrichment Analysis
2.8. Statistics
3. Results
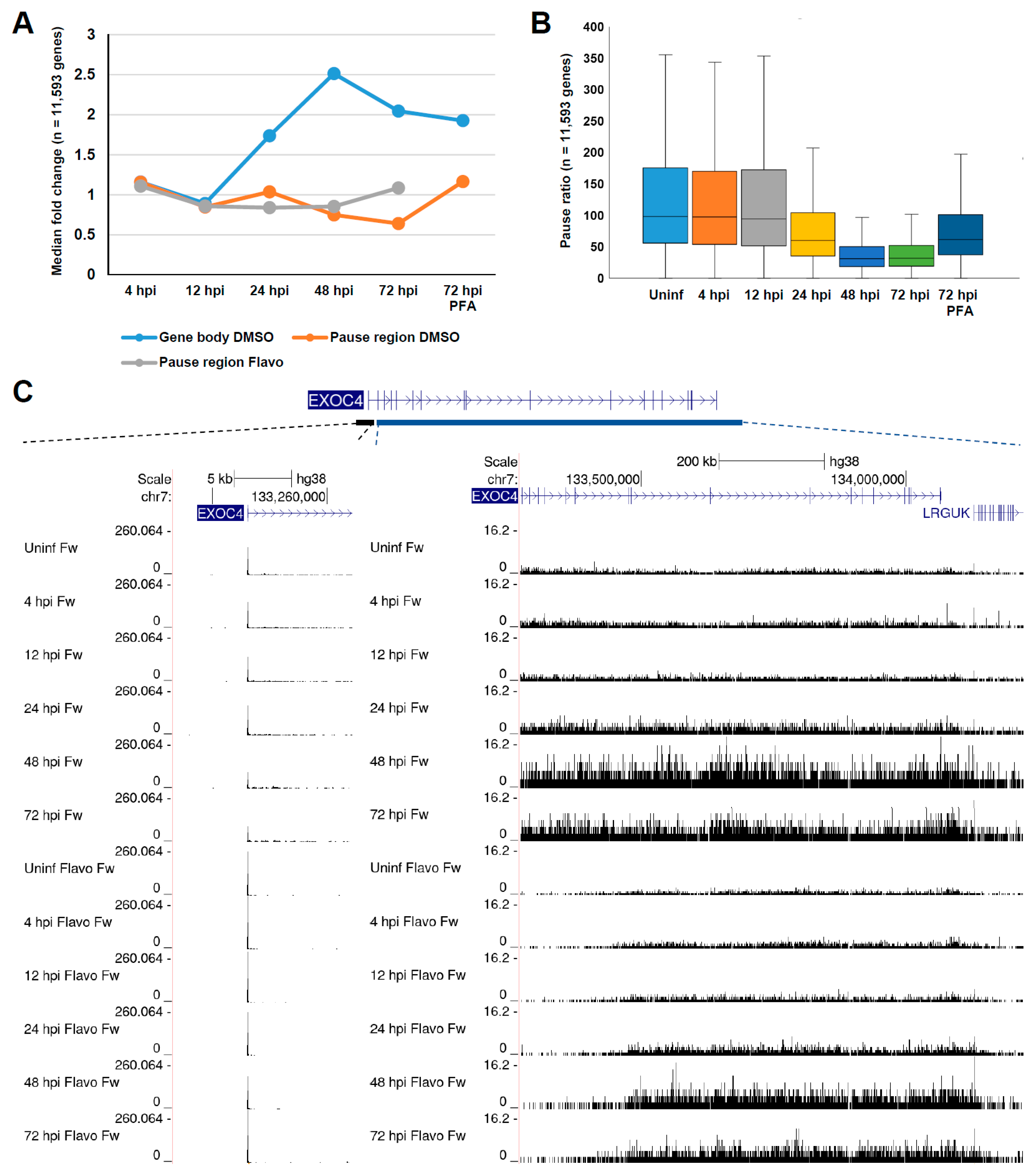
3.1. Global Changes in Pol II Transcription during HCMV Infection
3.2. Late HCMV Infection Is Associated with Increased Rates of Release into Productive Elongation at Host Pol II Genes
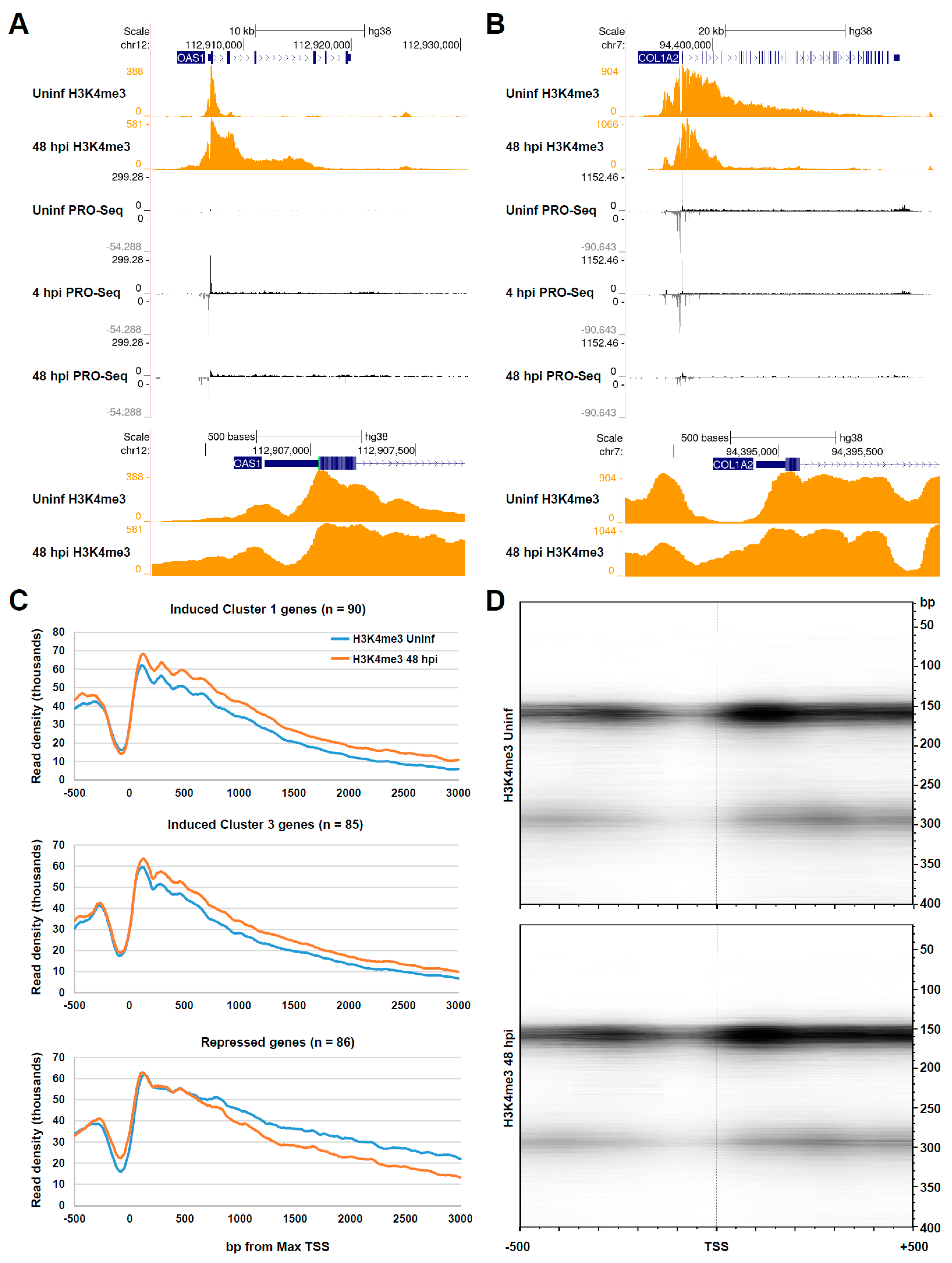
3.3. Chromatin Changes at Pol II Genes Activated and Repressed by HCMV Infection
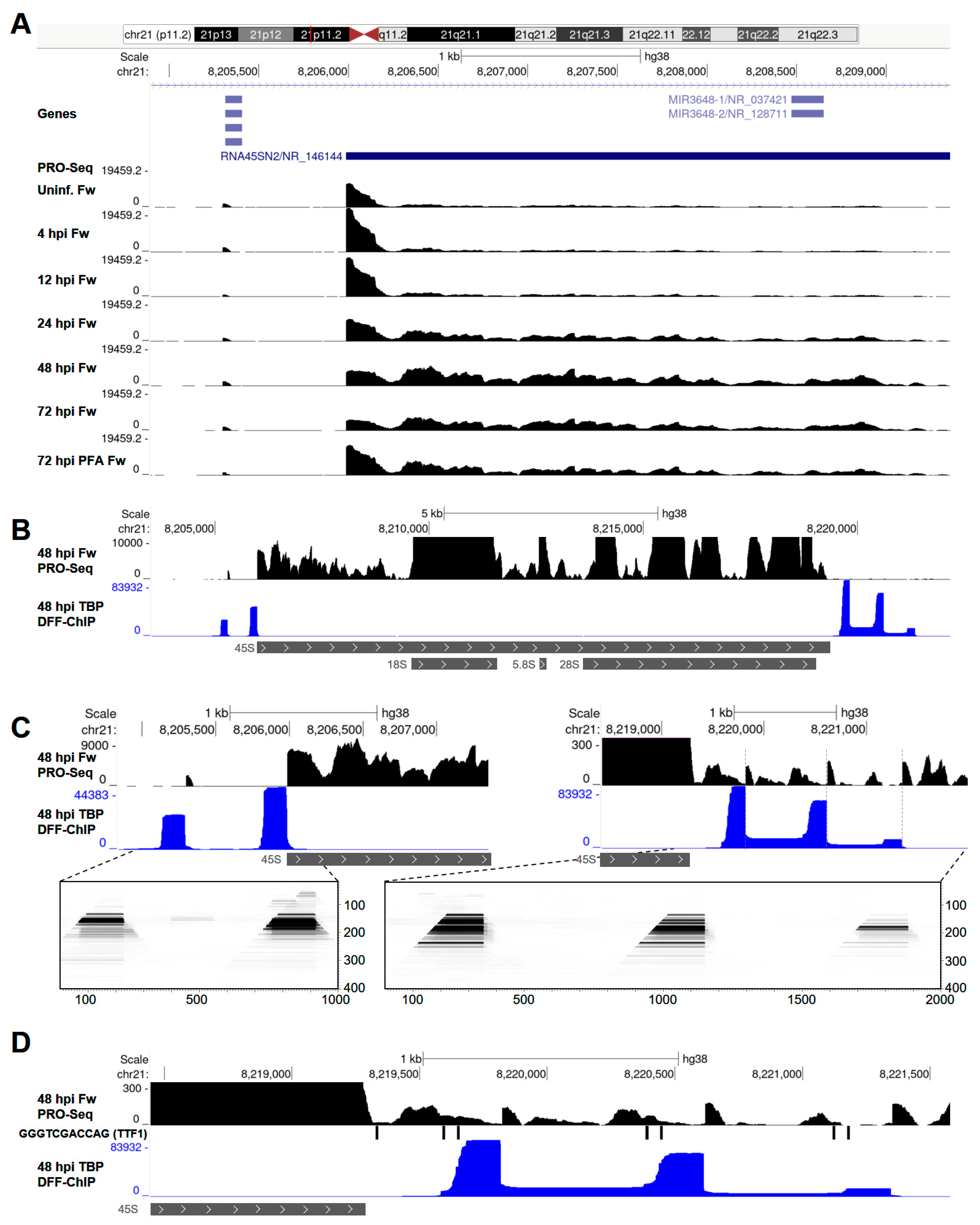
3.4. HCMV Infection Alters Transcription of the 45S rDNA Repeat by Pol I
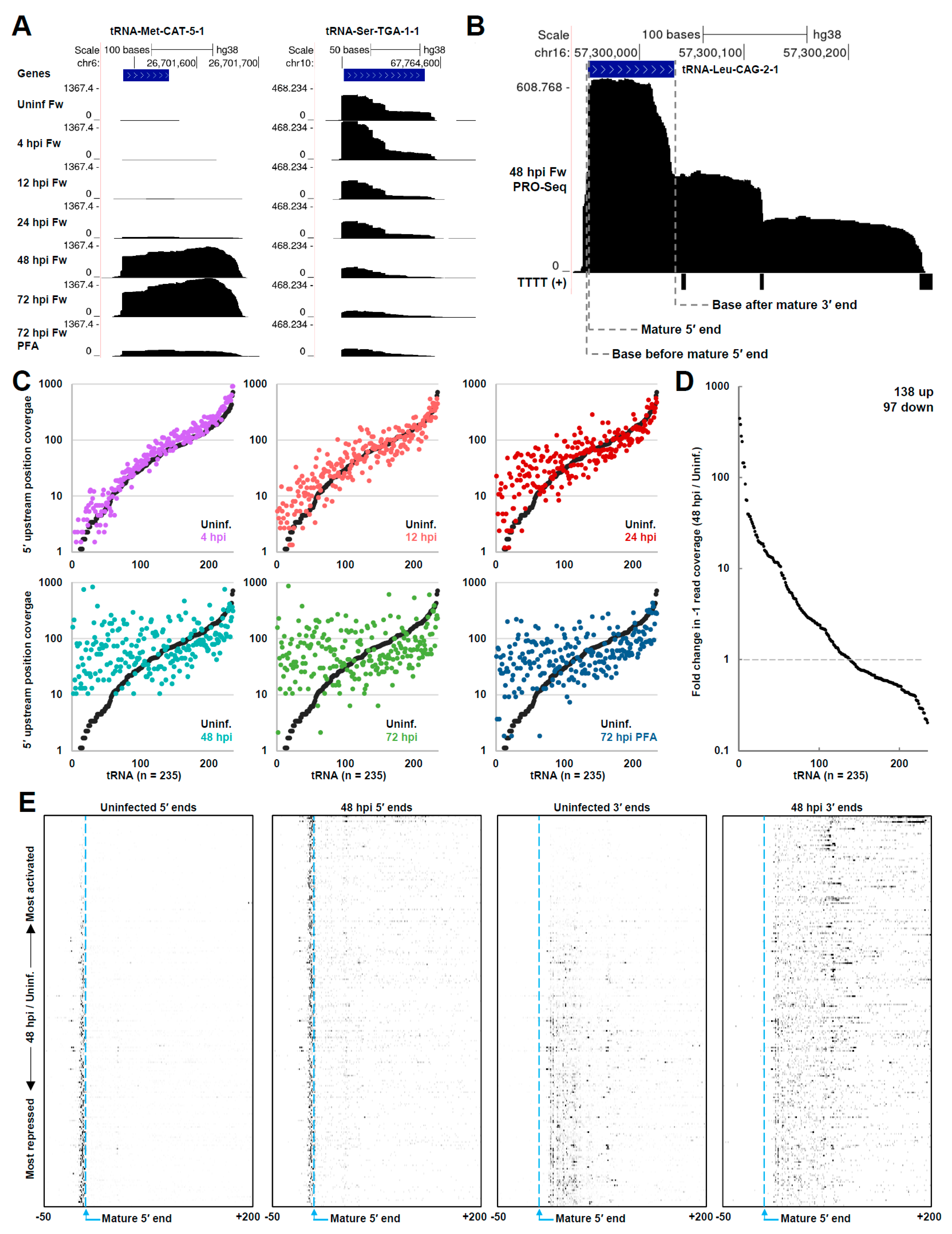
3.5. HCMV Infection Dramatically Impacts tRNA Transcription by Pol III
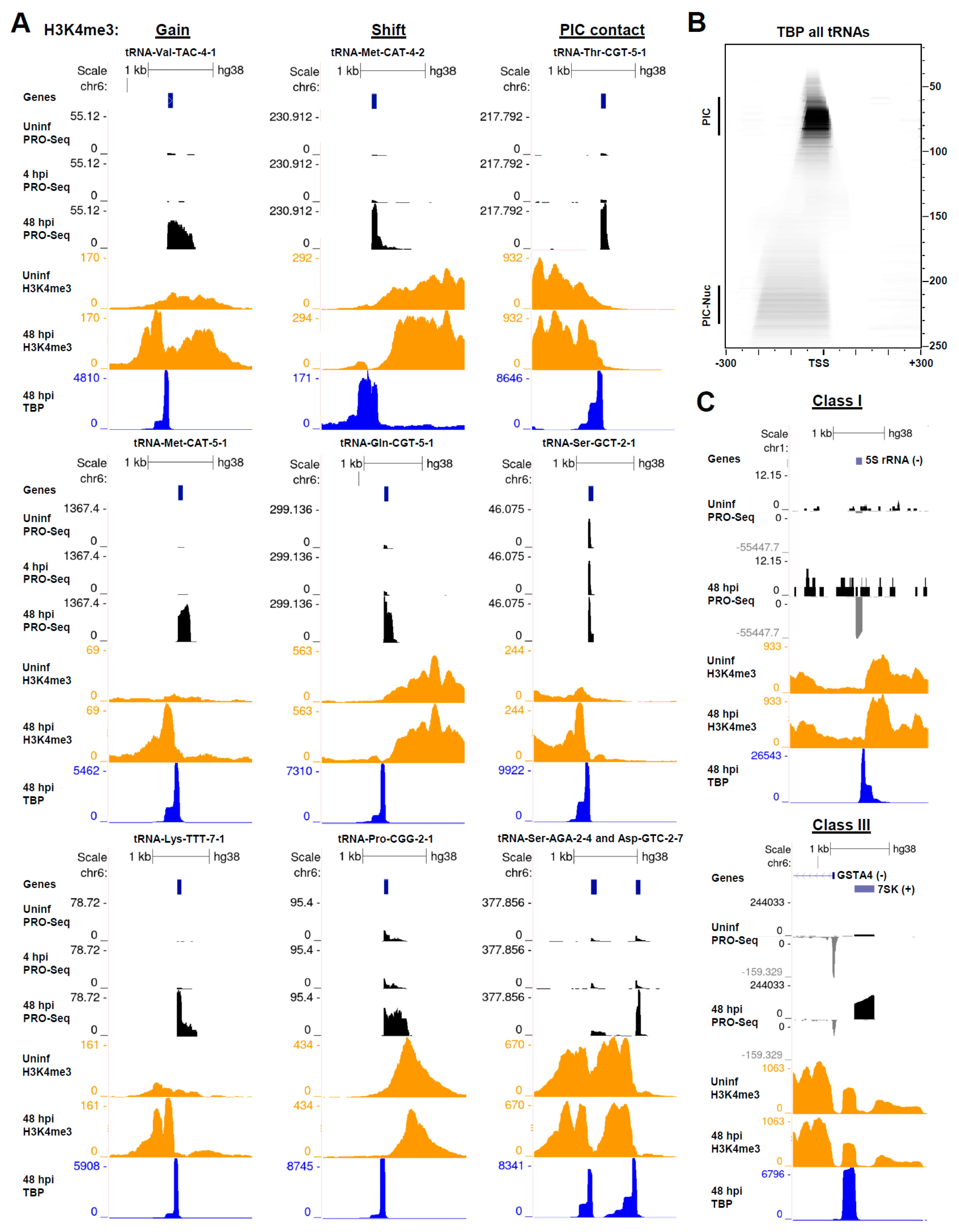
3.6. HCMV Infection Alters the Chromatin State of Pol III-Transcribed Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Russell, J.; Zomerdijk, J.C. The RNA polymerase I transcription machinery. Biochem. Soc. Symp. 2006, 73, 203–216. [Google Scholar] [CrossRef]
- Chen, F.X.; Smith, E.R.; Shilatifard, A. Born to run: Control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2018, 19, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, I.; Lis, J.T. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, A.; Adelman, K. Evaluating Enhancer Function and Transcription. Annu. Rev. Biochem. 2020, 89, 213–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, R.J. Transcription by RNA polymerase III: More complex than we thought. Nat. Rev. Genet. 2011, 12, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Turowski, T.W.; Tollervey, D. Transcription by RNA polymerase III: Insights into mechanism and regulation. Biochem. Soc. Trans. 2016, 44, 1367–1375. [Google Scholar] [CrossRef]
- Dergai, O.; Hernandez, N. How to Recruit the Correct RNA Polymerase? Lessons from snRNA Genes. Trends Genet. 2019, 35, 457–469. [Google Scholar] [CrossRef]
- Willis, I.M.; Moir, R.D. Signaling to and from the RNA Polymerase III Transcription and Processing Machinery. Annu. Rev. Biochem. 2018, 87, 75–100. [Google Scholar] [CrossRef]
- Gerber, A.; Ito, K.; Chu, C.S.; Roeder, R.G. Gene-Specific Control of tRNA Expression by RNA Polymerase II. Mol. Cell 2020, 78, 765–778. [Google Scholar] [CrossRef]
- Stinski, M.F.; Meier, J.L. Immediate-Early Viral Gene Regulation and Function. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Li, M.; Hu, Q.; Collins, G.; Parida, M.; Ball, C.B.; Price, D.H.; Meier, J.L. Cytomegalovirus late transcription factor target sequence diversity orchestrates viral early to late transcription. PLoS Pathog. 2021, 17, e1009796. [Google Scholar] [CrossRef]
- Parida, M.; Nilson, K.A.; Li, M.; Ball, C.B.; Fuchs, H.A.; Lawson, C.K.; Luse, D.S.; Meier, J.L.; Price, D.H. Nucleotide Resolution Comparison of Transcription of Human Cytomegalovirus and Host Genomes Reveals Universal Use of RNA Polymerase II Elongation Control Driven by Dissimilar Core Promoter Elements. mBio 2019, 10, e02047-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isomura, H.; Stinski, M.F. Coordination of late gene transcription of human cytomegalovirus with viral DNA synthesis: Recombinant viruses as potential therapeutic vaccine candidates. Expert. Opin. Ther. Targets 2013, 17, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Peterlin, B.M.; Price, D.H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Durand, L.O.; Roizman, B. Role of cdk9 in the optimization of expression of the genes regulated by ICP22 of herpes simplex virus 1. J. Virol. 2008, 82, 10591–10599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Tang, K.W.; Muylaert, I.; Samuelsson, T.; Elias, P. CDK9 and SPT5 proteins are specifically required for expression of herpes simplex virus 1 replication-dependent late genes. J. Biol. Chem. 2017, 292, 15489–15500. [Google Scholar] [CrossRef] [Green Version]
- Birkenheuer, C.H.; Baines, J.D. RNA Polymerase II Promoter-Proximal Pausing and Release to Elongation Are Key Steps Regulating Herpes Simplex Virus 1 Transcription. J. Virol. 2020, 94, e02035-19. [Google Scholar] [CrossRef]
- Spector, B.M.; Parida, M.; Li, M.; Ball, C.B.; Meier, J.L.; Luse, D.S.; Price, D.H. Differences in RNA polymerase II complexes and their interactions with surrounding chromatin on human and cytomegalovirus genomes. Nat. Commun. 2022. [Google Scholar] [CrossRef]
- Dremel, S.E.; DeLuca, N.A. Herpes simplex viral nucleoprotein creates a competitive transcriptional environment facilitating robust viral transcription and host shut off. eLife 2019, 8, e51109. [Google Scholar] [CrossRef]
- Dembowski, J.A.; DeLuca, N.A. Selective recruitment of nuclear factors to productively replicating herpes simplex virus genomes. PLoS Pathog. 2015, 11, e1004939. [Google Scholar] [CrossRef] [Green Version]
- Kent, J.R.; Zeng, P.Y.; Atanasiu, D.; Gardner, J.; Fraser, N.W.; Berger, S.L. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J. Virol. 2004, 78, 10178–10186. [Google Scholar] [CrossRef] [Green Version]
- Zalckvar, E.; Paulus, C.; Tillo, D.; Asbach-Nitzsche, A.; Lubling, Y.; Winterling, C.; Strieder, N.; Mucke, K.; Goodrum, F.; Segal, E.; et al. Nucleosome maps of the human cytomegalovirus genome reveal a temporal switch in chromatin organization linked to a major IE protein. Proc. Natl. Acad. Sci. USA 2013, 110, 13126–13131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, M.; Taddeo, B.; Zhang, W.; Roizman, B. Selective degradation of mRNAs by the HSV host shutoff RNase is regulated by the UL47 tegument protein. Proc. Natl. Acad. Sci. USA 2013, 110, E1669–E1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedel, C.C.; Whisnant, A.W.; Djakovic, L.; Rutkowski, A.J.; Friedl, M.S.; Kluge, M.; Williamson, J.C.; Sai, S.; Vidal, R.O.; Sauer, S.; et al. Dissecting Herpes Simplex Virus 1-Induced Host Shutoff at the RNA Level. J. Virol. 2021, 95, e01399-20. [Google Scholar] [CrossRef] [PubMed]
- Hartenian, E.; Gilbertson, S.; Federspiel, J.D.; Cristea, I.M.; Glaunsinger, B.A. RNA decay during gammaherpesvirus infection reduces RNA polymerase II occupancy of host promoters but spares viral promoters. PLoS Pathog. 2020, 16, e1008269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardamean, C.I.; Wu, T.T. Inhibition of Host Gene Expression by KSHV: Sabotaging mRNA Stability and Nuclear Export. Front Cell Infect Microbiol. 2021, 11, 648055. [Google Scholar] [CrossRef]
- Dremel, S.E.; Sivrich, F.L.; Tucker, J.M.; Glaunsinger, B.A.; DeLuca, N.A. Manipulation of RNA polymerase III by Herpes Simplex Virus-1. Nat. Commun. 2022, 13, 623. [Google Scholar] [CrossRef] [PubMed]
- Tucker, J.M.; Schaller, A.M.; Willis, I.; Glaunsinger, B.A. Alteration of the Premature tRNA Landscape by Gammaherpesvirus Infection. mBio 2020, 11, e02664-20. [Google Scholar] [CrossRef]
- White, R.J.; Stott, D.; Rigby, P.W. Regulation of RNA polymerase III transcription in response to Simian virus 40 transformation. EMBO J. 1990, 9, 3713–3721. [Google Scholar] [CrossRef]
- Felton-Edkins, Z.A.; White, R.J. Multiple mechanisms contribute to the activation of RNA polymerase III transcription in cells transformed by papovaviruses. J. Biol. Chem. 2002, 277, 48182–48191. [Google Scholar] [CrossRef] [Green Version]
- Felton-Edkins, Z.A.; Kondrashov, A.; Karali, D.; Fairley, J.A.; Dawson, C.W.; Arrand, J.R.; Young, L.S.; White, R.J. Epstein-Barr virus induces cellular transcription factors to allow active expression of EBER genes by RNA polymerase III. J. Biol. Chem. 2006, 281, 33871–33880. [Google Scholar] [CrossRef] [Green Version]
- Gaynor, R.B.; Feldman, L.T.; Berk, A.J. Transcription of class III genes activated by viral immediate early proteins. Science 1985, 230, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Hoeffler, W.K.; Roeder, R.G. Enhancement of RNA polymerase III transcription by the E1A gene product of adenovirus. Cell 1985, 41, 955–963. [Google Scholar] [CrossRef]
- Wreghitt, T.G.; Abel, S.J.; McNeil, K.; Parameshwar, J.; Stewart, S.; Cary, N.; Sharples, L.; Large, S.; Wallwork, J. Intravenous ganciclovir prophylaxis for cytomegalovirus in heart, heart-lung, and lung transplant recipients. Transpl. Int. 1999, 12, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Dollard, S.C.; Grosse, S.D.; Ross, D.S. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev. Med. Virol. 2007, 17, 355–363. [Google Scholar] [CrossRef]
- Hertel, L.; Mocarski, E.S. Global analysis of host cell gene expression late during cytomegalovirus infection reveals extensive dysregulation of cell cycle gene expression and induction of Pseudomitosis independent of US28 function. J. Virol. 2004, 78, 11988–12011. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Cong, J.P.; Mamtora, G.; Gingeras, T.; Shenk, T. Cellular gene expression altered by human cytomegalovirus: Global monitoring with oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 14470–14475. [Google Scholar] [CrossRef] [Green Version]
- Browne, E.P.; Wing, B.; Coleman, D.; Shenk, T. Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: Viral block to the accumulation of antiviral mRNAs. J. Virol. 2001, 75, 12319–12330. [Google Scholar] [CrossRef] [Green Version]
- Kwak, H.; Fuda, N.J.; Core, L.J.; Lis, J.T. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 2013, 339, 950–953. [Google Scholar] [CrossRef] [Green Version]
- Mahat, D.B.; Kwak, H.; Booth, G.T.; Jonkers, I.H.; Danko, C.G.; Patel, R.K.; Waters, C.T.; Munson, K.; Core, L.J.; Lis, J.T. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc. 2016, 11, 1455–1476. [Google Scholar] [CrossRef]
- Li, M.; Ball, C.B.; Collins, G.; Hu, Q.; Luse, D.S.; Price, D.H.; Meier, J.L. Human cytomegalovirus IE2 drives transcription initiation from a select subset of late infection viral promoters by host RNA polymerase II. PLoS Pathog. 2020, 16, e1008402. [Google Scholar] [CrossRef] [Green Version]
- Ball, C.B.; Nilson, K.A.; Price, D.H. Use of the nuclear walk-on methodology to determine sites of RNA polymerase II initiation and pausing and quantify nascent RNAs in cells. Methods 2019, 159–160, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusmao, E.G.; Allhoff, M.; Zenke, M.; Costa, I.G. Analysis of computational footprinting methods for DNase sequencing experiments. Nat. Methods 2016, 13, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Chao, S.H.; Fujinaga, K.; Marion, J.E.; Taube, R.; Sausville, E.A.; Senderowicz, A.M.; Peterlin, B.M.; Price, D.H. Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J. Biol. Chem. 2000, 275, 28345–28348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurokawa, C.; Iankov, I.D.; Galanis, E. A key anti-viral protein, RSAD2/VIPERIN, restricts the release of measles virus from infected cells. Virus Res. 2019, 263, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Rusinova, I.; Forster, S.; Yu, S.; Kannan, A.; Masse, M.; Cumming, H.; Chapman, R.; Hertzog, P.J. Interferome v2.0: An updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013, 41, D1040–D1046. [Google Scholar] [CrossRef]
- Nilson, K.A.; Lawson, C.K.; Mullen, N.J.; Ball, C.B.; Spector, B.M.; Meier, J.L.; Price, D.H. Oxidative stress rapidly stabilizes promoter-proximal paused Pol II across the human genome. Nucleic Acids Res. 2017, 45, 11088–11105. [Google Scholar] [CrossRef] [Green Version]
- Platanitis, E.; Decker, T. Regulatory Networks Involving STATs, IRFs, and NFkappaB in Inflammation. Front. Immunol. 2018, 9, 2542. [Google Scholar] [CrossRef] [Green Version]
- DeFilippis, V.R.; Robinson, B.; Keck, T.M.; Hansen, S.G.; Nelson, J.A.; Fruh, K.J. Interferon regulatory factor 3 is necessary for induction of antiviral genes during human cytomegalovirus infection. J. Virol. 2006, 80, 1032–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Xu, L.; Su, J.; Peppelenbosch, M.P.; Pan, Q. Transcriptional Regulation of Antiviral Interferon-Stimulated Genes. Trends Microbiol. 2017, 25, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Paulus, C.; Krauss, S.; Nevels, M. A human cytomegalovirus antagonist of type I IFN-dependent signal transducer and activator of transcription signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 3840–3845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Lee, S.; Cho, D.Y.; Lee, S.; Kim, H.; Yu, N.; Lee, S.; Ahn, K. Human cytomegalovirus induces and exploits Roquin to counteract the IRF1-mediated antiviral state. Proc. Natl. Acad. Sci. USA 2019, 116, 18619–18628. [Google Scholar] [CrossRef] [Green Version]
- Ziehr, B.; Vincent, H.A.; Moorman, N.J. Human Cytomegalovirus pTRS1 and pIRS1 Antagonize Protein Kinase R To Facilitate Virus Replication. J. Virol. 2016, 90, 3839–3848. [Google Scholar] [CrossRef] [Green Version]
- Luse, D.S.; Parida, M.; Spector, B.M.; Nilson, K.A.; Price, D.H. A unified view of the sequence and functional organization of the human RNA polymerase II promoter. Nucleic Acids Res. 2020, 48, 7767–7785. [Google Scholar] [CrossRef]
- Cui, K.; Tailor, P.; Liu, H.; Chen, X.; Ozato, K.; Zhao, K. The chromatin-remodeling BAF complex mediates cellular antiviral activities by promoter priming. Mol. Cell Biol. 2004, 24, 4476–4486. [Google Scholar] [CrossRef] [Green Version]
- Soares, L.M.; He, P.C.; Chun, Y.; Suh, H.; Kim, T.; Buratowski, S. Determinants of Histone H3K4 Methylation Patterns. Mol. Cell 2017, 68, 773–785. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Chen, Z.; Wu, D.; Zhang, L.; Lin, X.; Su, J.; Rodriguez, B.; Xi, Y.; Xia, Z.; Chen, X.; et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat. Genet. 2015, 47, 1149–1157. [Google Scholar] [CrossRef]
- Lauberth, S.M.; Nakayama, T.; Wu, X.; Ferris, A.L.; Tang, Z.; Hughes, S.H.; Roeder, R.G. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell 2013, 152, 1021–1036. [Google Scholar] [CrossRef] [Green Version]
- Sims, R.J., 3rd; Millhouse, S.; Chen, C.F.; Lewis, B.A.; Erdjument-Bromage, H.; Tempst, P.; Manley, J.L.; Reinberg, D. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol. Cell 2007, 28, 665–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhardt, B.; Winkler, M.; Schaarschmidt, P.; Pretsch, R.; Zhou, S.; Vaida, B.; Schmid-Kotsas, A.; Michel, D.; Walther, P.; Bachem, M.; et al. Human cytomegalovirus-induced reduction of extracellular matrix proteins in vascular smooth muscle cell cultures: A pathomechanism in vasculopathies? J. Gen. Virol. 2006, 87, 2849–2858. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.J.; McSharry, B.P.; Rickards, C.R.; Wang, E.C.; Tomasec, P.; Wilkinson, G.W. Cytomegalovirus destruction of focal adhesions revealed in a high-throughput Western blot analysis of cellular protein expression. J. Virol. 2007, 81, 7860–7872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wroblewska, Z.; Wellish, M.C.; Wolinsky, J.S.; Gilden, D. Comparison of human cytomegalovirus growth in MRC-5 human fibroblasts, brain, and choroid plexus cells in vitro. J. Med. Virol. 1981, 8, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Moss, T.; Mars, J.C.; Tremblay, M.G.; Sabourin-Felix, M. The chromatin landscape of the ribosomal RNA genes in mouse and human. Chromosome Res. 2019, 27, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Mars, J.C.; Sabourin-Felix, M.; Tremblay, M.G.; Moss, T. A Deconvolution Protocol for ChIP-Seq Reveals Analogous Enhancer Structures on the Mouse and Human Ribosomal RNA Genes. G3 (Bethesda) 2018, 8, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Herdman, C.; Mars, J.C.; Stefanovsky, V.Y.; Tremblay, M.G.; Sabourin-Felix, M.; Lindsay, H.; Robinson, M.D.; Moss, T. A unique enhancer boundary complex on the mouse ribosomal RNA genes persists after loss of Rrn3 or UBF and the inactivation of RNA polymerase I transcription. PLoS Genet. 2017, 13, e1006899. [Google Scholar] [CrossRef]
- Zheng, G.; Qin, Y.; Clark, W.C.; Dai, Q.; Yi, C.; He, C.; Lambowitz, A.M.; Pan, T. Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods 2015, 12, 835–837. [Google Scholar] [CrossRef]
- Gogakos, T.; Brown, M.; Garzia, A.; Meyer, C.; Hafner, M.; Tuschl, T. Characterizing Expression and Processing of Precursor and Mature Human tRNAs by Hydro-tRNAseq and PAR-CLIP. Cell Rep. 2017, 20, 1463–1475. [Google Scholar] [CrossRef] [Green Version]
- Behrens, A.; Rodschinka, G.; Nedialkova, D.D. High-resolution quantitative profiling of tRNA abundance and modification status in eukaryotes by mim-tRNAseq. Mol. Cell 2021, 81, 1802–1815. [Google Scholar] [CrossRef]
- Pan, T. Modifications and functional genomics of human transfer RNA. Cell Res. 2018, 28, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Arimbasseri, A.G.; Rijal, K.; Maraia, R.J. Transcription termination by the eukaryotic RNA polymerase III. Biochim. Biophys. Acta 2013, 1829, 318–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, Y.; Bhargava, P. A unique nucleosome arrangement, maintained actively by chromatin remodelers facilitates transcription of yeast tRNA genes. BMC Genom. 2013, 14, 402. [Google Scholar] [CrossRef] [Green Version]
- Zumer, K.; Maier, K.C.; Farnung, L.; Jaeger, M.G.; Rus, P.; Winter, G.; Cramer, P. Two distinct mechanisms of RNA polymerase II elongation stimulation in vivo. Mol. Cell 2021, 81, 3096–3109. [Google Scholar] [CrossRef] [PubMed]
- Nilson, K.A.; Price, D.H. The Role of RNA Polymerase II Elongation Control in HIV-1 Gene Expression, Replication, and Latency. Genet. Res. Int. 2011, 2011, 726901. [Google Scholar] [CrossRef] [Green Version]
- Alfonso-Dunn, R.; Turner, A.W.; Jean Beltran, P.M.; Arbuckle, J.H.; Budayeva, H.G.; Cristea, I.M.; Kristie, T.M. Transcriptional Elongation of HSV Immediate Early Genes by the Super Elongation Complex Drives Lytic Infection and Reactivation from Latency. Cell Host Microbe 2017, 21, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Tamrakar, S.; Kapasi, A.J.; Spector, D.H. Human cytomegalovirus infection induces specific hyperphosphorylation of the carboxyl-terminal domain of the large subunit of RNA polymerase II that is associated with changes in the abundance, activity, and localization of cdk9 and cdk7. J. Virol. 2005, 79, 15477–15493. [Google Scholar] [CrossRef] [Green Version]
- Shigematsu, M.; Honda, S.; Loher, P.; Telonis, A.G.; Rigoutsos, I.; Kirino, Y. YAMAT-seq: An efficient method for high-throughput sequencing of mature transfer RNAs. Nucleic Acids Res. 2017, 45, e70. [Google Scholar] [CrossRef]
- Pinkard, O.; McFarland, S.; Sweet, T.; Coller, J. Quantitative tRNA-sequencing uncovers metazoan tissue-specific tRNA regulation. Nat. Commun. 2020, 11, 4104. [Google Scholar] [CrossRef]
- Krishna, S.; Raghavan, S.; DasGupta, R.; Palakodeti, D. tRNA-derived fragments (tRFs): Establishing their turf in post-transcriptional gene regulation. Cell Mol. Life Sci. 2021, 78, 2607–2619. [Google Scholar] [CrossRef]
- Desai, N.; Lee, J.; Upadhya, R.; Chu, Y.; Moir, R.D.; Willis, I.M. Two steps in Maf1-dependent repression of transcription by RNA polymerase III. J. Biol. Chem. 2005, 280, 6455–6462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodfellow, S.J.; Graham, E.L.; Kantidakis, T.; Marshall, L.; Coppins, B.A.; Oficjalska-Pham, D.; Gerard, M.; Lefebvre, O.; White, R.J. Regulation of RNA polymerase III transcription by Maf1 in mammalian cells. J. Mol. Biol. 2008, 378, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Michels, A.A.; Robitaille, A.M.; Buczynski-Ruchonnet, D.; Hodroj, W.; Reina, J.H.; Hall, M.N.; Hernandez, N. mTORC1 directly phosphorylates and regulates human MAF1. Mol. Cell Biol. 2010, 30, 3749–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantidakis, T.; Ramsbottom, B.A.; Birch, J.L.; Dowding, S.N.; White, R.J. mTOR associates with TFIIIC, is found at tRNA and 5S rRNA genes, and targets their repressor Maf1. Proc. Natl. Acad. Sci. USA 2010, 107, 11823–11828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.; Saha, B.; Riley, J.L. The battle over mTOR: An emerging theatre in host-pathogen immunity. PLoS Pathog. 2012, 8, e1002894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Xuan, B.; Liu, H.; Zhong, J.; Yu, D.; Qian, Z. Tuberous Sclerosis Complex Protein 2-Independent Activation of mTORC1 by Human Cytomegalovirus pUL38. J. Virol. 2015, 89, 7625–7635. [Google Scholar] [CrossRef] [Green Version]
- Moorman, N.J.; Cristea, I.M.; Terhune, S.S.; Rout, M.P.; Chait, B.T.; Shenk, T. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 2008, 3, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Vink, E.I.; Lee, S.; Smiley, J.R.; Mohr, I. Remodeling mTORC1 Responsiveness to Amino Acids by the Herpes Simplex Virus UL46 and Us3 Gene Products Supports Replication during Nutrient Insufficiency. J. Virol. 2018, 92, e01377-18. [Google Scholar] [CrossRef] [Green Version]
- Chuluunbaatar, U.; Roller, R.; Feldman, M.E.; Brown, S.; Shokat, K.M.; Mohr, I. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev. 2010, 24, 2627–2639. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ball, C.B.; Parida, M.; Li, M.; Spector, B.M.; Suarez, G.A.; Meier, J.L.; Price, D.H. Human Cytomegalovirus Infection Elicits Global Changes in Host Transcription by RNA Polymerases I, II, and III. Viruses 2022, 14, 779. https://doi.org/10.3390/v14040779
Ball CB, Parida M, Li M, Spector BM, Suarez GA, Meier JL, Price DH. Human Cytomegalovirus Infection Elicits Global Changes in Host Transcription by RNA Polymerases I, II, and III. Viruses. 2022; 14(4):779. https://doi.org/10.3390/v14040779
Chicago/Turabian StyleBall, Christopher B., Mrutyunjaya Parida, Ming Li, Benjamin M. Spector, Gustavo A. Suarez, Jeffery L. Meier, and David H. Price. 2022. "Human Cytomegalovirus Infection Elicits Global Changes in Host Transcription by RNA Polymerases I, II, and III" Viruses 14, no. 4: 779. https://doi.org/10.3390/v14040779