
I226R Protein of African Swine Fever Virus Is a Suppressor of Innate Antiviral Responses

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus and Plasmids
2.2. Antibodies and Reagents
2.3. Dual-Luciferase Reporter Assay
2.4. Enzyme-Linked Immunosorbent Assay (ELISA)
2.5. RNA Preparation, RT-PCR and RT-qPCR
2.6. Plaque Assay
2.7. Generation of Stable Cell Lines
2.8. Immunofluorescence Assay
2.9. Immunoprecipitation Assay
2.10. Statistical Analysis
3. Results
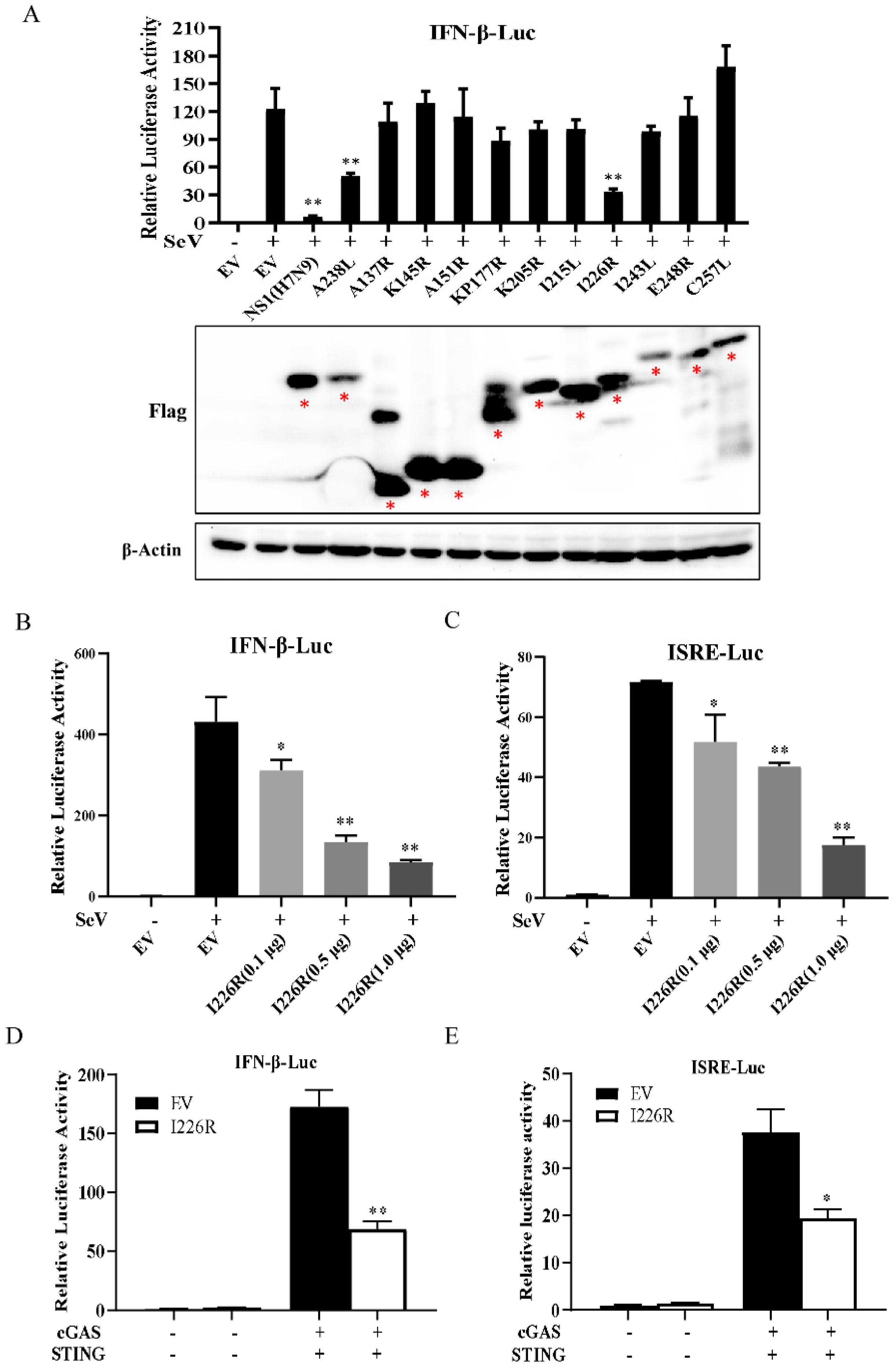
3.1. I226R Protein Is a Key Inhibitor of IFN-β Response
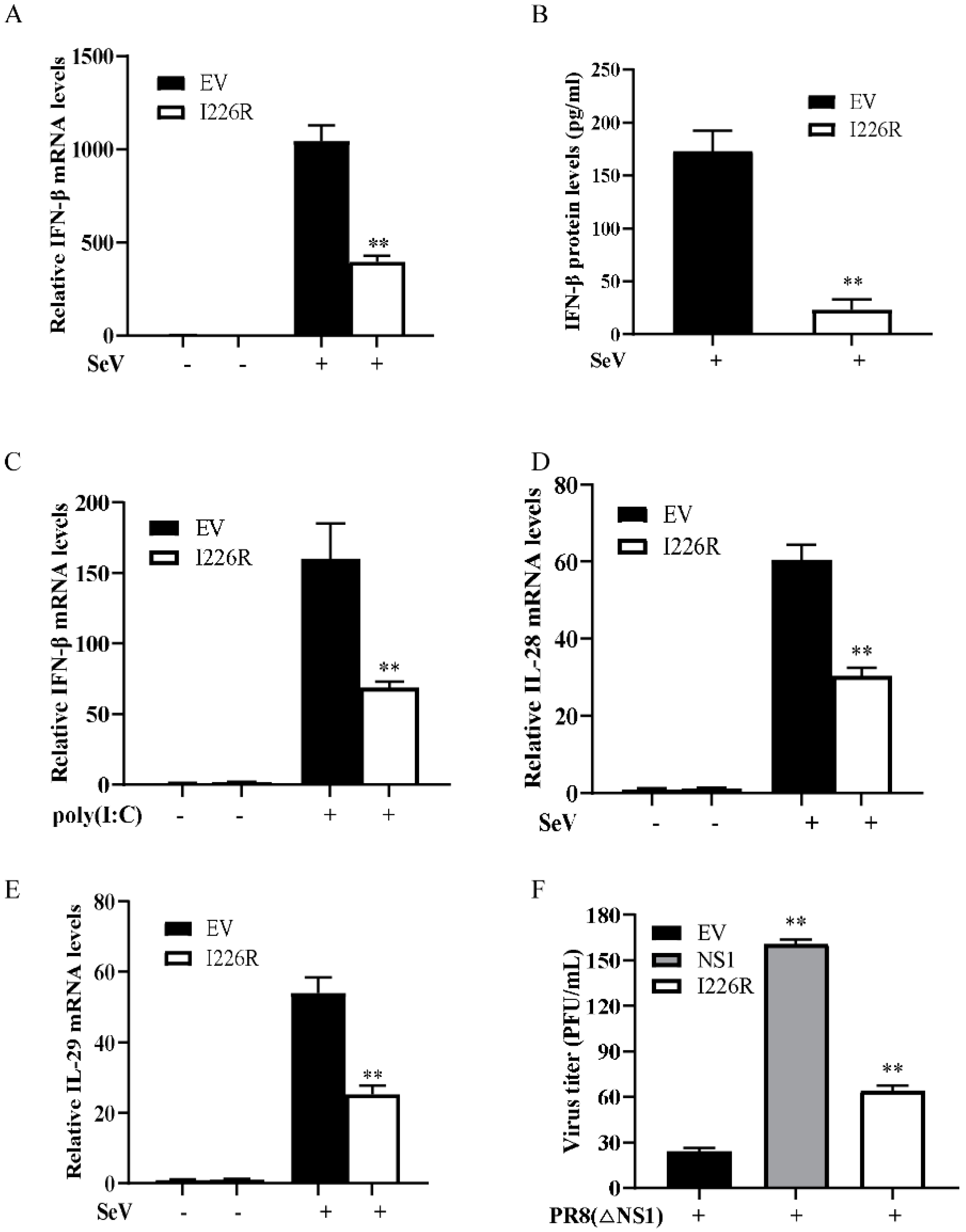
3.2. I226R Protein Reduces the Expression of IFNs Induced by SeV and Poly(I:C)
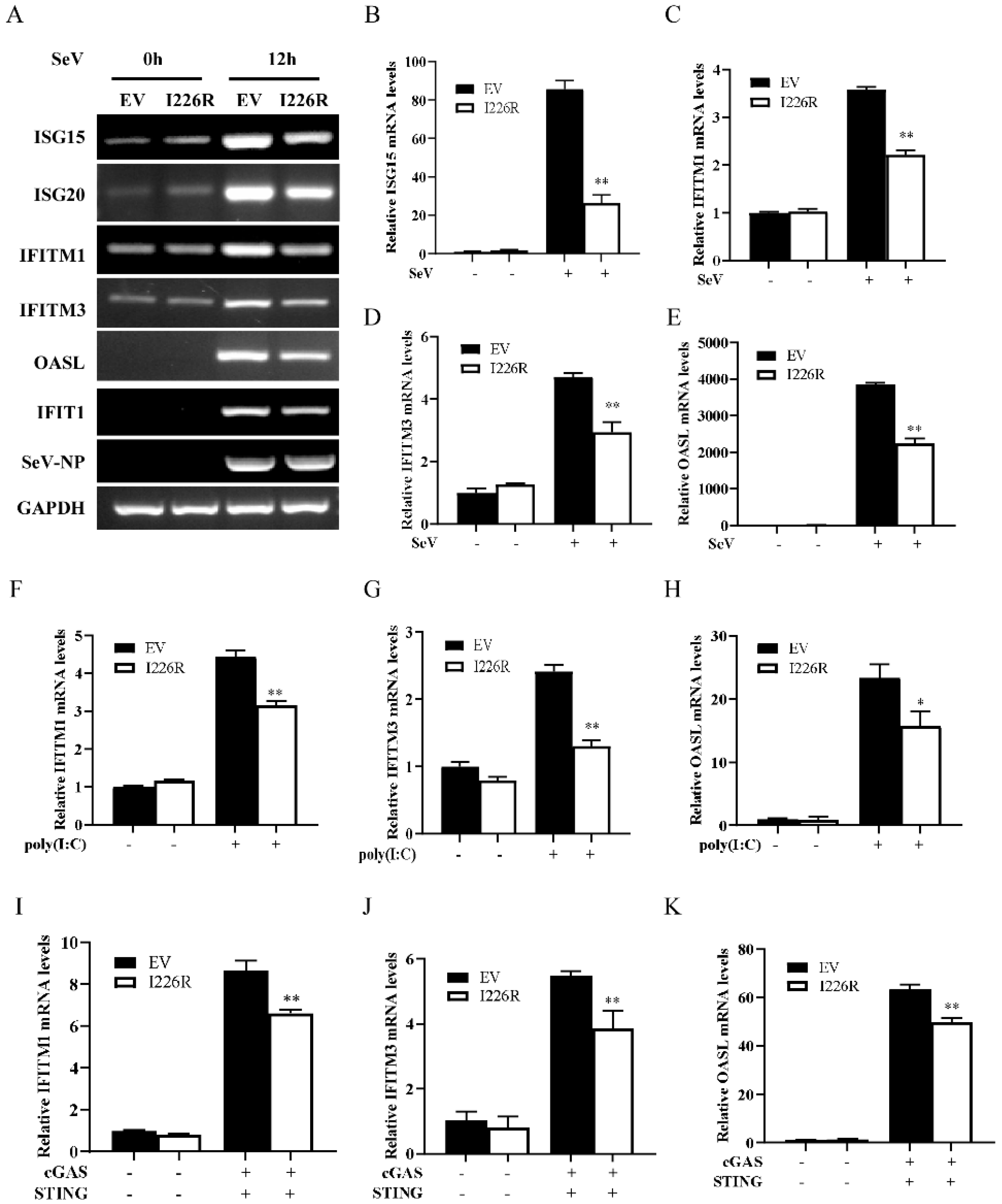
3.3. I226R-Overexpressing Cells Have Impaired Expression of Several Critical ISGs
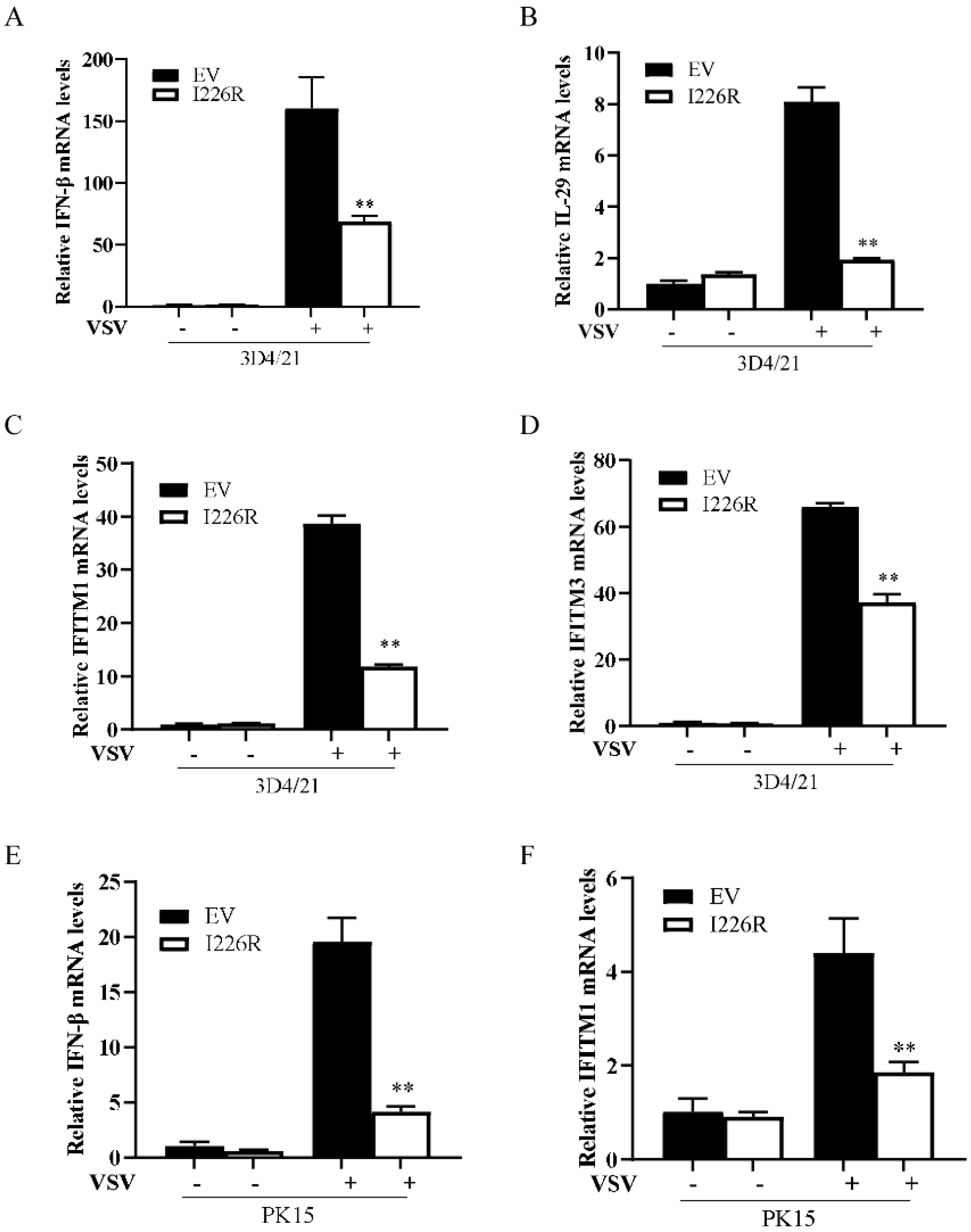
3.4. I226R Protein Inhibits VSV-Induced Expression of IFN-β and ISGs in Swine Cells
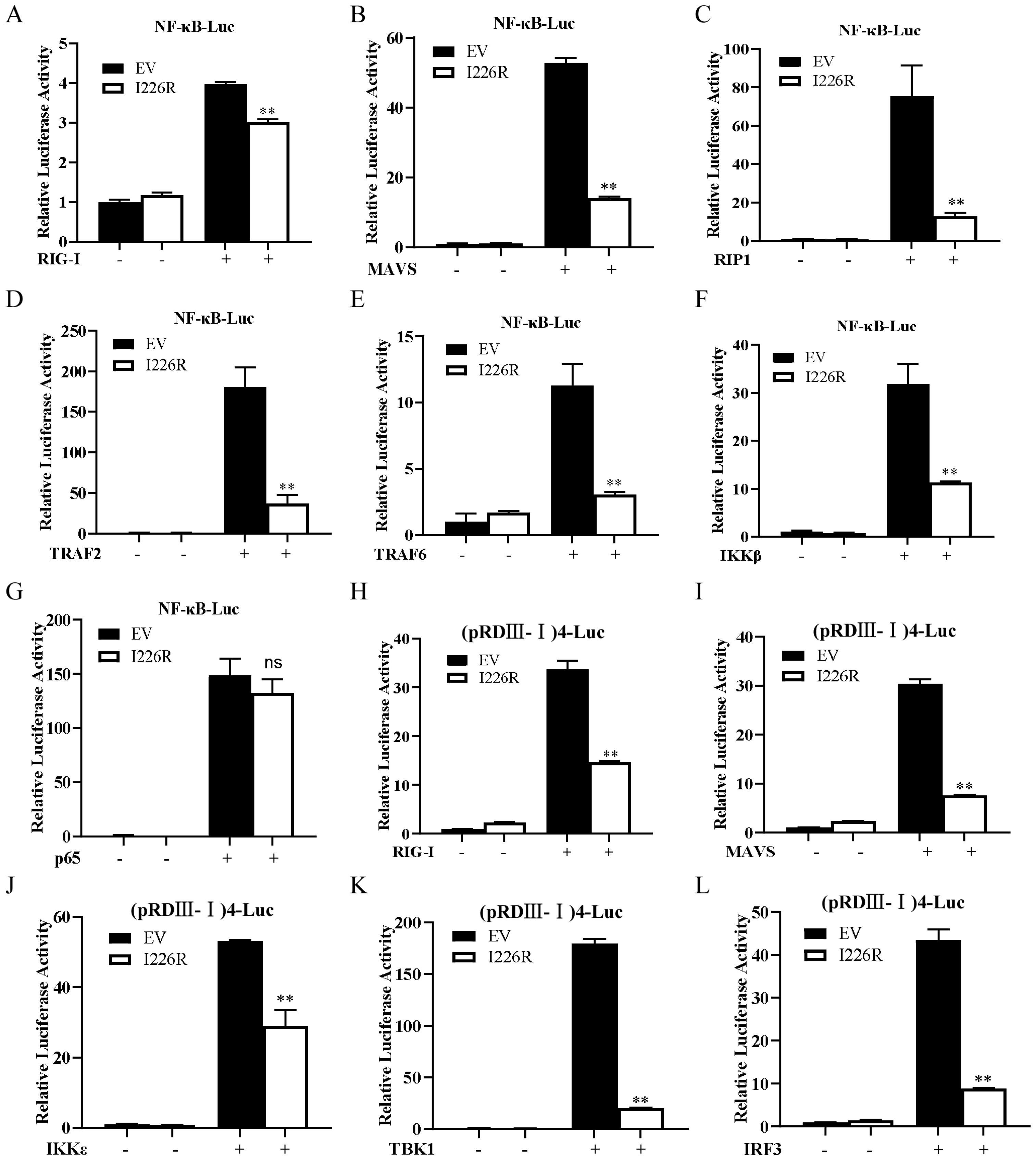
3.5. I226R Protein Impairs the Activation of NF-κB and IRF3 Signaling
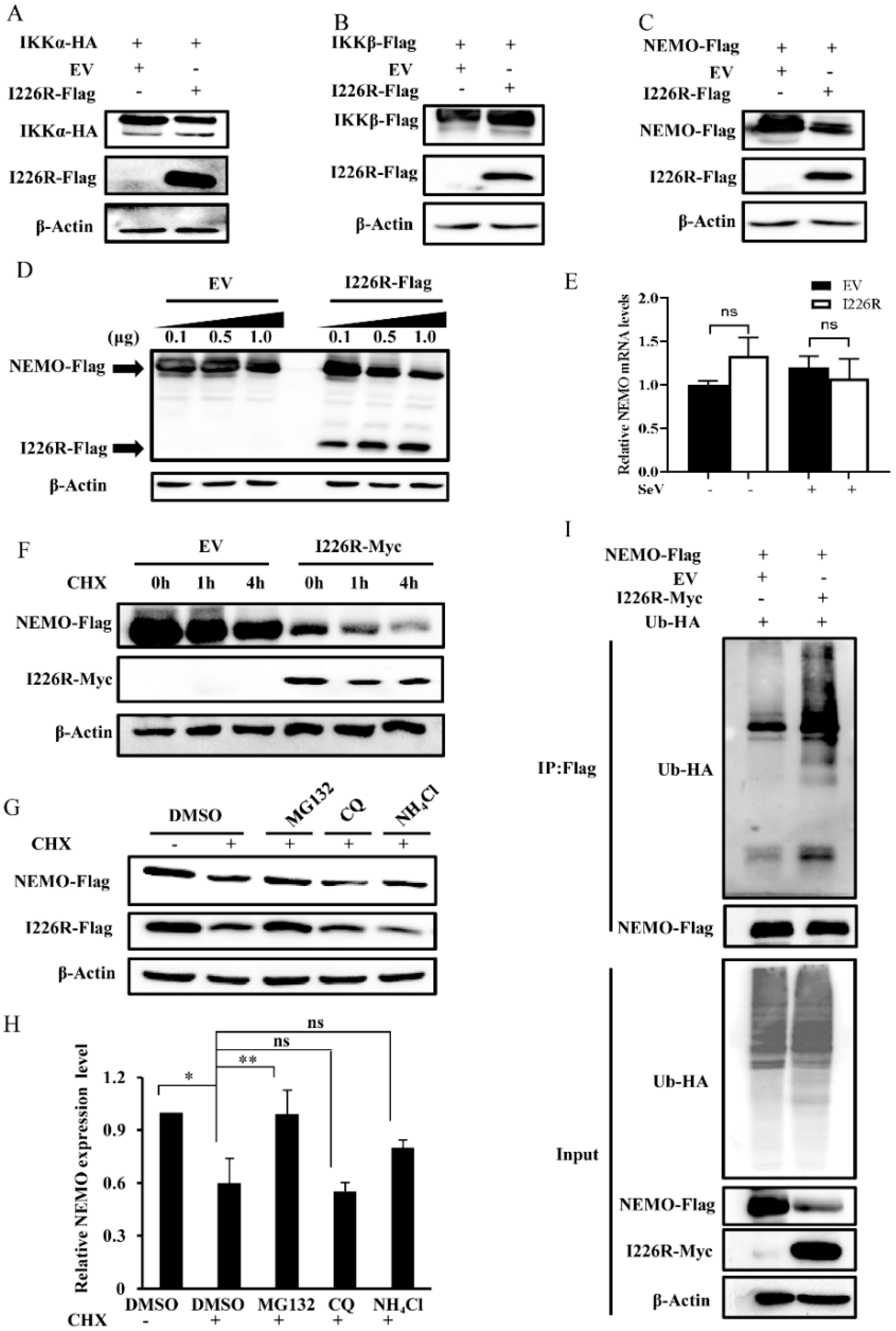
3.6. I226R Protein Inhibits NF-κB through Targeting the IKK Complex
3.7. I226R Protein Inhibits NF-κB Signaling, Likely through Regulation of NEMO
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gallardo, M.C.; Reoyo, A.T.; Fernandez-Pinero, J.; Iglesias, I.; Munoz, M.J.; Arias, M.L. African swine fever: A global view of the current challenge. Porcine Health Manag. 2015, 1, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudreault, N.N.; Madden, D.W.; Wilson, W.C.; Trujillo, J.D.; Richt, J.A. African swine fever virus: An emerging DNA arbovirus. Front. Vet. Sci. 2020, 7, 215. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.E. On a form of swine fever occurring in british east africa (Kenya colony). J. Comp. Pathol. Ther. 1921, 34, 159–191. [Google Scholar] [CrossRef] [Green Version]
- Costard, S.; Wieland, B.; de Glanville, W.; Jori, F.; Rowlands, R.; Vosloo, W.; Roger, F.; Pfeiffer, D.U.; Dixon, L.K. African swine fever: How can global spread be prevented? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 2683–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Liu, R.; Zhang, X.; Li, F.; Wang, J.; Zhang, J.; Liu, X.; Wang, L.; Zhang, J.; Wu, X.; et al. Replication and virulence in pigs of the first African swine fever virus isolated in China. Emerg. Microbes Infect. 2019, 8, 438–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Normile, D. African swine fever marches across much of Asia. Science 2019, 364, 617–618. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.; Kim, H.; Lee, J.Y.; Yoo, H.S. African swine fever: Etiology, epidemiological status in Korea, and perspective on control. J. Vet. Sci. 2020, 21, e38. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Kim, J.; Son, K.; Choi, Y.; Jeong, H.S.; Kim, Y.K.; Park, J.E.; Hong, Y.J.; Lee, S.I.; Wang, S.J.; et al. Wild boar harbouring African swine fever virus in the demilitarized zone in South Korea, 2019. Emerg. Microbes Infect. 2020, 9, 628–630. [Google Scholar] [CrossRef]
- Tran, H.T.T.; Truong, A.D.; Dang, A.K.; Ly, D.V.; Nguyen, C.T.; Chu, N.T.; Nguyen, H.T.; Dang, H.V. Genetic characterization of African swine fever viruses circulating in North Central region of Vietnam. Transbound. Emerg. Dis. 2021, 68, 1697–1699. [Google Scholar] [CrossRef]
- Teklue, T.; Sun, Y.; Abid, M.; Luo, Y.; Qiu, H.J. Current status and evolving approaches to African swine fever vaccine development. Transbound. Emerg. Dis. 2020, 67, 529–542. [Google Scholar] [CrossRef]
- Wu, K.; Liu, J.; Wang, L.; Fan, S.; Li, Z.; Li, Y.; Yi, L.; Ding, H.; Zhao, M.; Chen, J. Current state of global african swine fever vaccine development under the prevalence and transmission of ASF in china. Vaccines 2020, 8, 531. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhao, D.; Wang, J.; Zhang, Y.; Wang, M.; Gao, Y.; Li, F.; Wang, J.; Bu, Z.; Rao, Z.; et al. Architecture of African swine fever virus and implications for viral assembly. Science 2019, 366, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Yanez, R.J.; Rodriguez, J.M.; Nogal, M.L.; Yuste, L.; Enriquez, C.; Rodriguez, J.F.; Vinuela, E. Analysis of the complete nucleotide sequence of African swine fever virus. Virology 1995, 208, 249–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, L.K.; Chapman, D.A.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef]
- Wang, T.; Sun, Y.; Huang, S.; Qiu, H.J. Multifaceted immune responses to african swine fever virus: Implications for vaccine development. Vet. Microbiol. 2020, 249, 108832. [Google Scholar] [CrossRef]
- Powell, P.P.; Dixon, L.K.; Parkhouse, R.M. An IkappaB homolog encoded by African swine fever virus provides a novel mechanism for downregulation of proinflammatory cytokine responses in host macrophages. J. Virol. 1996, 70, 8527–8533. [Google Scholar] [CrossRef] [Green Version]
- Granja, A.G.; Perkins, N.D.; Revilla, Y. A238L inhibits NF-ATc2, NF-kappa B, and c-Jun activation through a novel mechanism involving protein kinase C-theta-mediated up-regulation of the amino-terminal transactivation domain of p300. J. Immunol. 2008, 180, 2429–2442. [Google Scholar] [CrossRef] [Green Version]
- Granja, A.G.; Sabina, P.; Salas, M.L.; Fresno, M.; Revilla, Y. Regulation of inducible nitric oxide synthase expression by viral A238L-mediated inhibition of p65/RelA acetylation and p300 transactivation. J. Virol. 2006, 80, 10487–10496. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Yang, W.; Li, L.; Li, P.; Ma, Z.; Zhang, J.; Qi, X.; Ren, J.; Ru, Y.; Niu, Q.; et al. African swine fever virus MGF-505-7R negatively regulates cGAS–STING-Mediated signaling pathway. J. Immunol. 2021, 206, 1844–1857. [Google Scholar] [CrossRef]
- Wang, X.; Wu, J.; Wu, Y.; Chen, H.; Zhang, S.; Li, J.; Xin, T.; Jia, H.; Hou, S.; Jiang, Y.; et al. Inhibition of cGAS-STING-TBK1 signaling pathway by DP96R of ASFV China 2018/1. Biochem. Biophys. Res. Commun. 2018, 506, 437–443. [Google Scholar] [CrossRef]
- Hargreaves, D.C.; Medzhitov, R. Innate sensors of microbial infection. J. Clin. Immunol. 2005, 25, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Rai, K.R.; Shrestha, P.; Yang, B.; Chen, Y.; Liu, S.; Maarouf, M.; Chen, J.L. Acute infection of viral pathogens and their innate immune escape. Front. Microbiol. 2021, 12, 672026. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Fujita, T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 2009, 227, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.; Li, X.; Li, P. The mechanism of double-stranded DNA sensing through the cGAS-STING pathway. Cytokine Growth Factor Rev. 2014, 25, 641–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of viral infections by innate immunity. Biochem. Pharmacol. 2021, 183, 114316. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Chen, Z.J. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 2012, 5, ra20. [Google Scholar] [CrossRef] [Green Version]
- Caamano, J.; Hunter, C.A. NF-kappaB family of transcription factors: Central regulators of innate and adaptive immune functions. Clin. Microbiol. Rev. 2002, 15, 414–429. [Google Scholar] [CrossRef] [Green Version]
- Antonia, R.J.; Hagan, R.S.; Baldwin, A.S. Expanding the View of IKK: New Substrates and New Biology. Trends Cell Biol. 2021, 31, 166–178. [Google Scholar] [CrossRef]
- Jefferies, C.A. Regulating IRFs in IFN driven disease. Front. Immunol. 2019, 10, 325. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, J.M.; Salas, M.L.; Vinuela, E. Intermediate class of mRNAs in African swine fever virus. J. Virol. 1996, 70, 8584–8589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch-Camos, L.; Lopez, E.; Navas, M.J.; Pina-Pedrero, S.; Accensi, F.; Correa-Fiz, F.; Park, C.; Carrascal, M.; Dominguez, J.; Salas, M.L.; et al. Identification of Promiscuous African Swine Fever Virus T-Cell Determinants Using a Multiple Technical Approach. Vaccines 2021, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ke, J.; Zhang, J.; Yang, J.; Yue, H.; Zhou, X.; Qi, Y.; Zhu, R.; Miao, F.; Li, Q.; et al. African swine fever virus bearing an I226R gene deletion elicits robust immunity in pigs to african swine fever. J. Virol. 2021, 95, e01199-21. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Pan, W.; Wang, S.; Pan, C.; Ning, H.; Huang, S.; Chiu, S.H.; Chen, J.L. Protein tyrosine phosphatase SHP2 suppresses host innate immunity against influenza a virus by regulating EGFR-Mediated signaling. J. Virol. 2021, 95, e02001-20. [Google Scholar] [CrossRef]
- Liu, S.; Liao, Y.; Chen, B.; Chen, Y.; Yu, Z.; Wei, H.; Zhang, L.; Huang, S.; Rothman, P.B.; Gao, G.F.; et al. Critical role of Syk-dependent STAT1 activation in innate antiviral immunity. Cell Rep. 2021, 34, 108627. [Google Scholar] [CrossRef]
- Wang, S.; Li, H.; Chen, Y.; Wei, H.; Gao, G.F.; Liu, H.; Huang, S.; Chen, J.L. Transport of influenza virus neuraminidase (NA) to host cell surface is regulated by ARHGAP21 and Cdc42 proteins. J. Biol. Chem. 2012, 287, 9804–9816. [Google Scholar] [CrossRef] [Green Version]
- Rai, K.R.; Chen, B.; Zhao, Z.; Chen, Y.; Hu, J.; Liu, S.; Maarouf, M.; Li, Y.; Xiao, M.; Liao, Y.; et al. Robust expression of p27Kip1 induced by viral infection is critical for antiviral innate immunity. Cell. Microbiol. 2020, 22, e13242. [Google Scholar] [CrossRef]
- Xing, J.; Wang, S.; Lin, R.; Mossman, K.L.; Zheng, C. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J. Virol. 2012, 86, 3528–3540. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Zheng, C.; Xing, J.; Wang, S.; Li, S.; Lin, R.; Mossman, K.L. Varicella-zoster virus immediate-early protein ORF61 abrogates the IRF3-mediated innate immune response through degradation of activated IRF3. J. Virol. 2011, 85, 11079–11089. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Chen, Y.; Zhang, Z.; Ouyang, J.; Wang, Y.; Yan, R.; Huang, S.; Gao, G.F.; Guo, G.; Chen, J.L. Robust expression of vault RNAs induced by influenza A virus plays a critical role in suppression of PKR-mediated innate immunity. Nucleic Acids Res. 2015, 43, 10321–10337. [Google Scholar] [CrossRef]
- Wang, S.; Chi, X.; Wei, H.; Chen, Y.; Chen, Z.; Huang, S.; Chen, J.L. Influenza A virus-induced degradation of eukaryotic translation initiation factor 4B contributes to viral replication by suppressing IFITM3 protein expression. J. Virol. 2014, 88, 8375–8385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Wang, S.; Chen, Q.; Chen, Y.; Chi, X.; Zhang, L.; Huang, S.; Gao, G.F.; Chen, J.L. Suppression of interferon lambda signaling by SOCS-1 results in their excessive production during influenza virus infection. PLoS Pathog. 2014, 10, e1003845. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, L.; Zhang, R.; Chi, X.; Yang, Z.; Xie, Y.; Shu, S.; Liao, Y.; Chen, J.L. Identification of two residues within the NS1 of H7N9 influenza A virus that critically affect the protein stability and function. Vet. Res. 2018, 49, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia, S.; Ventura, S.; Parkhouse, R.M. Identification and utility of innate immune system evasion mechanisms of ASFV. Virus Res. 2013, 173, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Riera Romo, M.; Perez-Martinez, D.; Castillo Ferrer, C. Innate immunity in vertebrates: An overview. Immunology 2016, 148, 125–139. [Google Scholar] [CrossRef]
- Zhuo, Y.; Guo, Z.; Ba, T.; Zhang, C.; He, L.; Zeng, C.; Dai, H. African Swine Fever Virus MGF360-12L Inhibits Type I Interferon Production by Blocking the Interaction of Importin alpha and NF-kappaB Signaling Pathway. Virol. Sin. 2021, 36, 176–186. [Google Scholar] [CrossRef]
- Garcia-Belmonte, R.; Perez-Nunez, D.; Pittau, M.; Richt, J.A.; Revilla, Y. African Swine Fever Virus Armenia/07 Virulent Strain Controls Interferon Beta Production through the cGAS-STING Pathway. J. Virol. 2019, 93, e02298-18. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Xu, W.; Liu, H.; Xue, M.; Liu, X.; Zhang, K.; Hu, L.; Li, J.; Liu, X.; Xiang, Z.; et al. African swine fever virus pI215L negatively regulates cGAS-STING signaling pathway through recruiting RNF138 to inhibit K63-Linked ubiquitination of TBK1. J. Immunol. 2021, 207, 2754–2769. [Google Scholar] [CrossRef]
- Barrado-Gil, L.; Del Puerto, A.; Galindo, I.; Cuesta-Geijo, M.A.; Garcia-Dorival, I.; de Motes, C.M.; Alonso, C. African swine fever virus Ubiquitin-Conjugating enzyme is an immunomodulator targeting NF-κB activation. Viruses 2021, 13, 1160. [Google Scholar] [CrossRef]
- Song, K.; Li, S. The Role of Ubiquitination in NF-κB Signaling during Virus Infection. Viruses 2021, 13, 145. [Google Scholar] [CrossRef]
- Romero, N.; Van Waesberghe, C.; Favoreel, H.W. Pseudorabies Virus Infection of Epithelial Cells Leads to Persistent but Aberrant Activation of the NF-kappaB Pathway, Inhibiting Hallmark NF-kappaB-Induced Proinflammatory Gene Expression. J. Virol. 2020, 94, e00196-20. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Liao, Z.; Zou, X.; Xu, Z.; Wang, Y.; Li, T.; Li, Y.; Ou, X.; Deng, Y.; Guo, Y.; et al. Herpes Simplex Virus 1 UL2 Inhibits the TNF-α-Mediated NF-κB Activity by Interacting with p65/p50. Front. Immunol. 2020, 11, 549. [Google Scholar] [CrossRef] [PubMed]
- Maubach, G.; Schmadicke, A.C.; Naumann, M. NEMO links nuclear Factor-κB to human diseases. Trends Mol. Med. 2017, 23, 1138–1155. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; Casanova, J.L.; Puel, A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency. Clin. Microbiol. Rev. 2011, 24, 490–497. [Google Scholar] [CrossRef] [Green Version]
- Fang, R.; Wang, C.; Jiang, Q.; Lv, M.; Gao, P.; Yu, X.; Mu, P.; Zhang, R.; Bi, S.; Feng, J.M.; et al. NEMO-IKKbeta are essential for IRF3 and NF-kappaB activation in the cGAS-STING pathway. J. Immunol. 2017, 199, 3222–3233. [Google Scholar] [CrossRef]
- Zhao, T.; Yang, L.; Sun, Q.; Arguello, M.; Ballard, D.W.; Hiscott, J.; Lin, R. The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat. Immunol. 2007, 8, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, S.; Dorf, M.E. NEMO binds ubiquitinated TANK-binding kinase 1 (TBK1) to regulate innate immune responses to RNA viruses. PLoS ONE 2012, 7, e43756. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Tian, J.; Li, Z.; Kang, H.; Zhang, J.; Huang, J.; Yin, H.; Hu, X.; Qu, L. Feline infectious peritonitis virus nsp5 inhibits type i interferon production by cleaving NEMO at multiple sites. Viruses 2019, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Fang, L.; Wang, D.; Yang, Y.; Chen, J.; Ye, X.; Foda, M.F.; Xiao, S. Porcine deltacoronavirus nsp5 inhibits interferon-beta production through the cleavage of NEMO. Virology 2017, 502, 33–38. [Google Scholar] [CrossRef]
- Biswas, S.; Shisler, J.L. Molluscum contagiosum virus MC159 abrogates cIAP1-NEMO interactions and inhibits NEMO polyubiquitination. J. Virol. 2017, 91, e00276-17. [Google Scholar] [CrossRef] [Green Version]
- Bodda, C.; Reinert, L.S.; Fruhwurth, S.; Richardo, T.; Sun, C.; Zhang, B.C.; Kalamvoki, M.; Pohlmann, A.; Mogensen, T.H.; Bergstrom, P.; et al. HSV1 VP1-2 deubiquitinates STING to block type I interferon expression and promote brain infection. J. Exp. Med. 2020, 217, e20191422. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zheng, H.; Yu, S.; Ding, Y.; Wu, W.; Mao, X.; Liao, Y.; Meng, C.; Ur Rehman, Z.; Tan, L.; et al. Newcastle disease virus v protein degrades mitochondrial antiviral signaling protein to inhibit host type i interferon pro-duction via e3 ubiquitin ligase RNF5. J. Virol. 2019, 93, e00322-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Wang, R.; Ma, Z.; Xiao, Y.; Nan, Y.; Wang, Y.; Lin, S.; Zhang, Y.J. Porcine reproductive and respiratory syndrome virus antagonizes JAK/STAT3 signaling via nsp5, which induces STAT3 degradation. J. Virol. 2017, 91, e02087-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashida, H.; Kim, M.; Schmidt-Supprian, M.; Ma, A.; Ogawa, M.; Sasakawa, C. A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKgamma to dampen the host NF-kappaB-mediated inflammatory response. Nat. Cell. Biol. 2010, 12, 66–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chathuranga, K.; Kim, T.H.; Lee, H.; Park, J.S.; Kim, J.H.; Chathuranga, W.A.G.; Ekanayaka, P.; Choi, Y.J.; Lee, C.H.; Kim, C.J.; et al. Negative regulation of NEMO signaling by the ubiquitin E3 ligase MARCH2. EMBO J. 2020, 39, e105139. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, J.; Chi, X.; Yuan, X.; Wen, F.; Rai, K.R.; Wu, L.; Song, Z.; Wang, S.; Guo, G.; Chen, J.-L. I226R Protein of African Swine Fever Virus Is a Suppressor of Innate Antiviral Responses. Viruses 2022, 14, 575. https://doi.org/10.3390/v14030575
Hong J, Chi X, Yuan X, Wen F, Rai KR, Wu L, Song Z, Wang S, Guo G, Chen J-L. I226R Protein of African Swine Fever Virus Is a Suppressor of Innate Antiviral Responses. Viruses. 2022; 14(3):575. https://doi.org/10.3390/v14030575
Chicago/Turabian StyleHong, Jinxuan, Xiaojuan Chi, Xu Yuan, Faxin Wen, Kul Raj Rai, Lei Wu, Zhongbao Song, Song Wang, Guijie Guo, and Ji-Long Chen. 2022. "I226R Protein of African Swine Fever Virus Is a Suppressor of Innate Antiviral Responses" Viruses 14, no. 3: 575. https://doi.org/10.3390/v14030575