
Delta Variant with P681R Critical Mutation Revealed by Ultra-Large Atomic-Scale Ab Initio Simulation: Implications for the Fundamentals of Biomolecular Interactions


Abstract
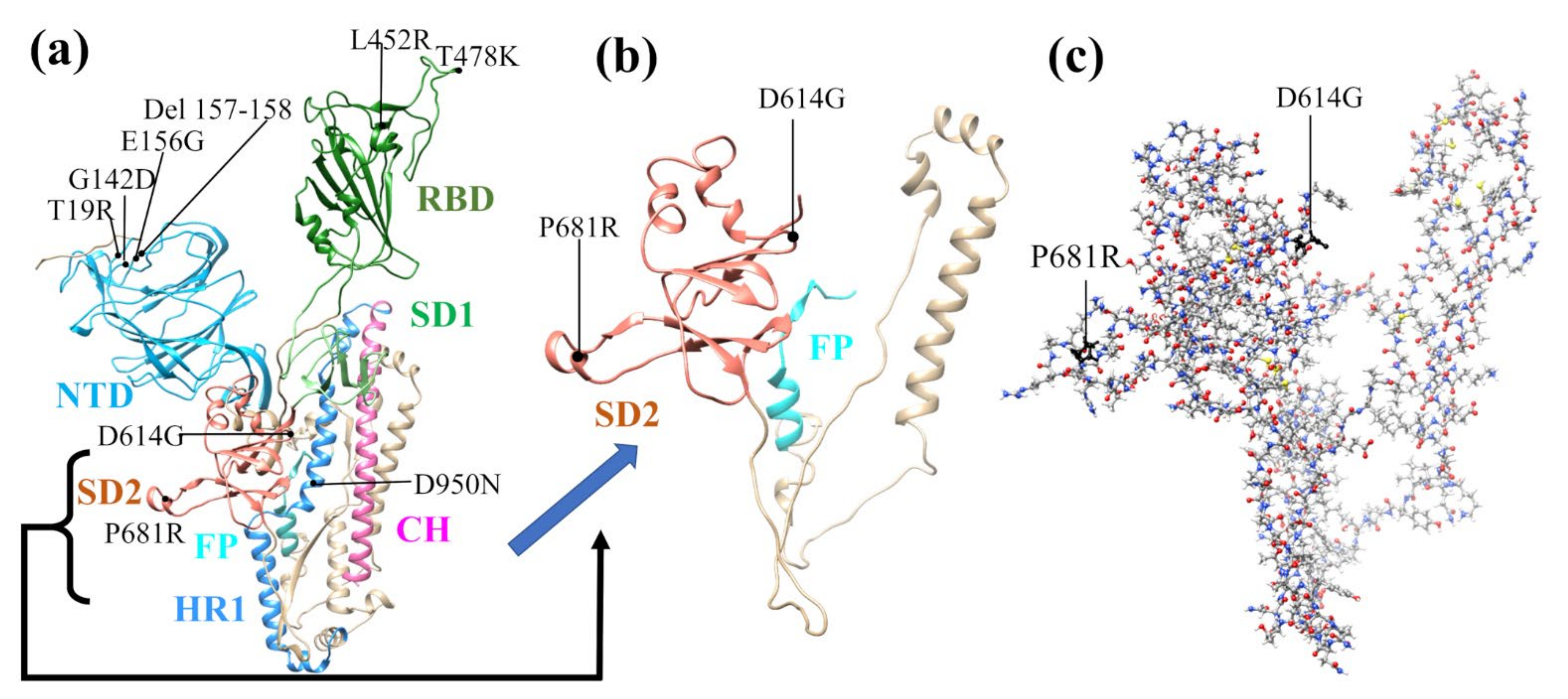
:1. Introduction
2. Model Design and Construction
3. Amino-Acid–Amino-Acid Bond PAIR (AABP)
4. Results
4.1. Electronic Structure
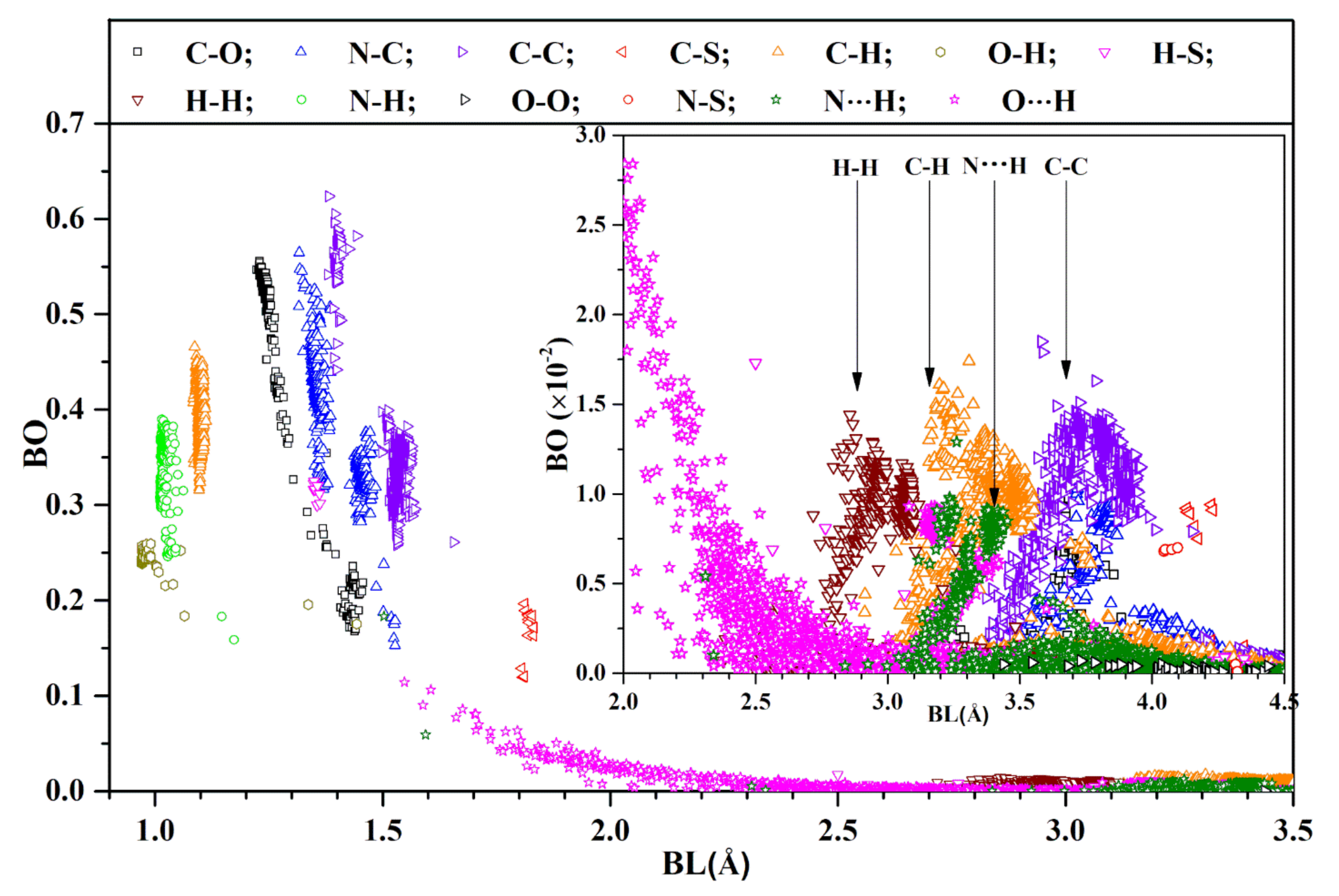
4.2. Interatomic Bonding
- The first group of covalent bonds are O-H, N-H, and C-H, with BL ranging from less than 1 Å to less than 1.2 Å, and with BO ranging from 0.15 e− to 0.48 e−, depending on the actual structure of the AAs listed in Figure S2; it may even be between certain AAs. There are two O-H bonds at 1.33 Å and 1.44 Å. The former bond occurs between two AAs (D808 and K811) and the latter one is from the same AA F592;
- The second group of covalent bonds is the usual covalent bond between C, O, and N. Their BO values can be very large, ranging from 0.16 e− for N-C up to 0.62 e− for C=C, which are strong double bonds. The relatively weaker C-C bonds have a slightly larger BL. A similar observation can be seen for bonds between (C, O) and (N, C) pairs;
- The next important bond is the HBs: mostly O⋯H and a few N⋯H. HBs are much weaker than covalent bonds, but are ubiquitous, ranging all the way to a ‘BL’ of close to 4 Å (see inset). According to a detailed analysis by Lei et al. on a super-cold network of water [51], the maximum BO for O⋯H is around 0.1 e−;
- The next interesting bonds are the covalent H-S and C-S bonds from the only S-containing AAs Cys and Met. Some H-S bonds have a short BL (1.4 Å) with a strong BO (0.3 e−), whereas the others have a large BL with a weak BO (more than 2.5 Å and less than 0.05 e−). The C-S bonds are located at a BL of around 1.8 Å and with a relatively strong BO value between 0.1 e− and 0.2 e− at this longer BL. Our results show that there are no disulfide bonds in the SD2-FP model;
- We now focus our observations on the inset of Figure 2 for the BL ranging from 2.0 Å to 4.5 Å. It reveals many weaker HBs, with a BO less than 0.03 e−. Even more surprising is the presence of many atomic pairs (H-H, C-H, C-C, N⋯H) that contribute to BO values with weak but nontrivial values of less than 0.02 e−. These bonds are obviously formed between the non-local AAs, which play a critical role in the total AABP values, to be discussed in the next section;
- One point we must emphasize is that the use of BO is a relatively new concept advocated by us. The BLs must be interpreted as the distance of separations between a pair of atoms, with the proviso that their interatomic interaction can go beyond the actual atomic pairs labeled as ‘BL’ due to the quantum effects arising from overlapping orbitals of their nearby atoms. Such subtle issues are usually ignored in biomolecular systems, since they are seldom discussed in the context of quantum mechanical wave functions, but rely on the distances between two atoms quantified by ‘BL’. Similar issues have been raised recently in the literature regarding the nature of C-H and C-C bonds [52].
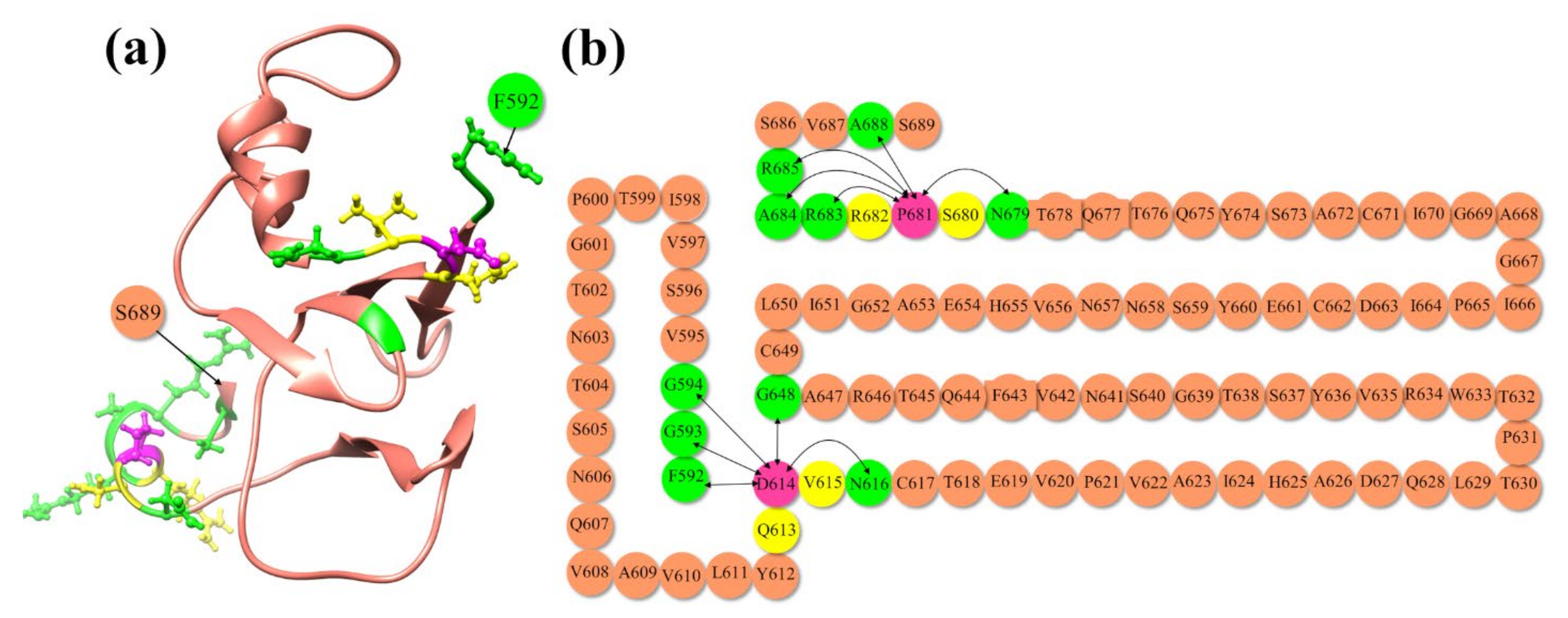
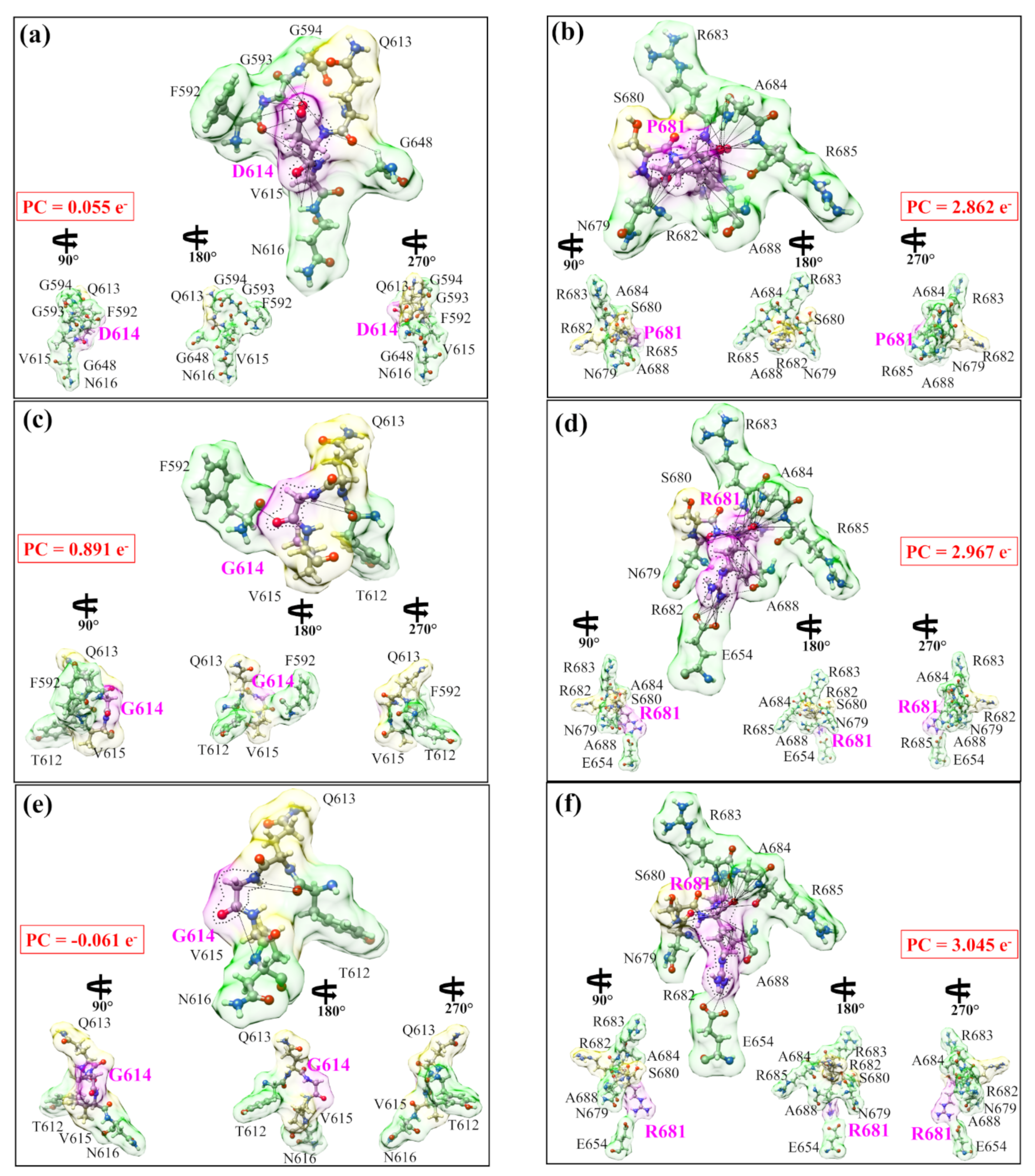
4.3. AABP Data for Mutations in Delta Variant
5. Discussions
5.1. The Origins of Mutation
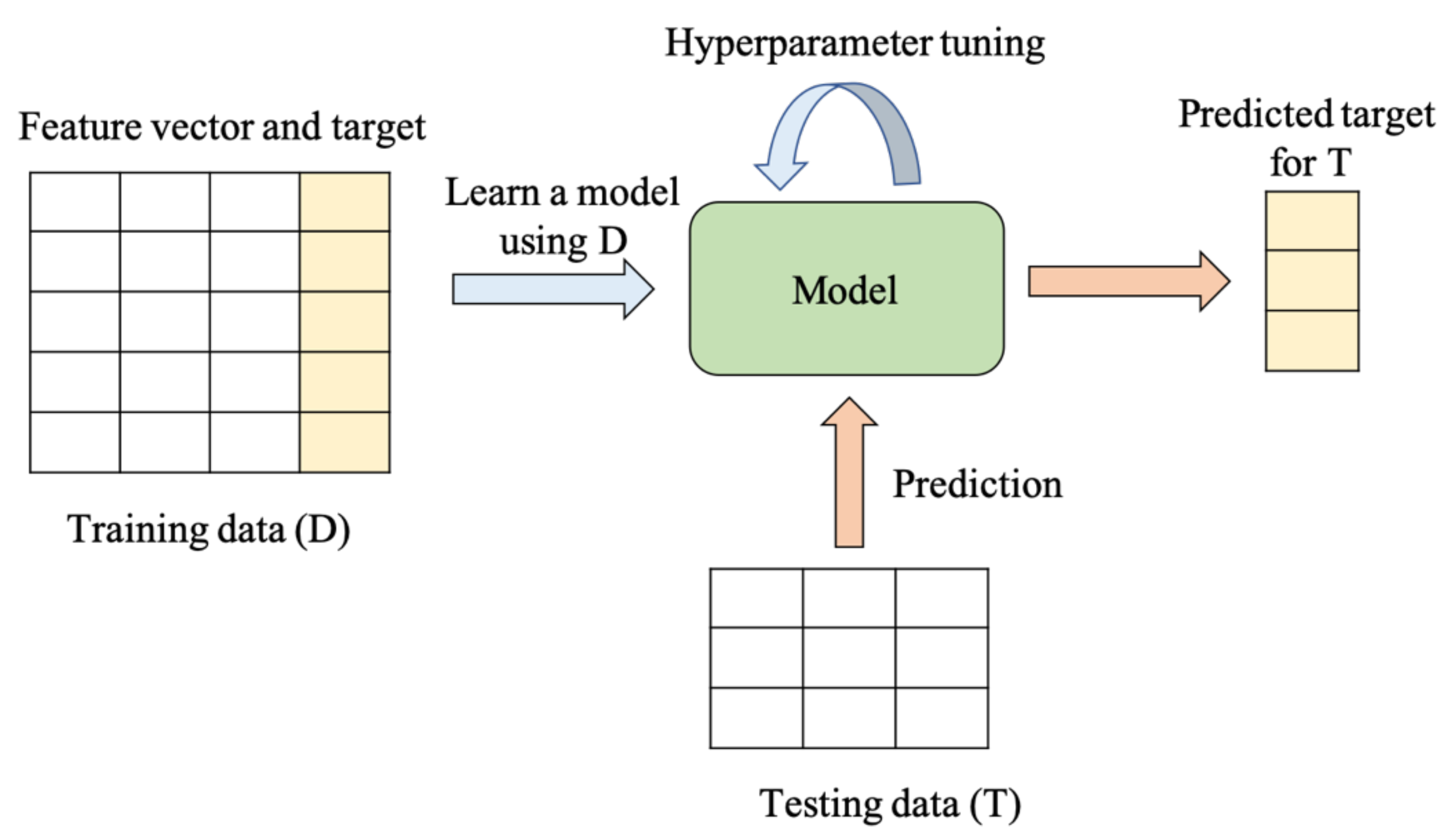
5.2. Extension to Machine Learning (ML)
5.3. Looking Forward
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhong, N.; Zheng, B.; Li, Y.; Poon, L.; Xie, Z.; Chan, K.; Li, P.; Tan, S.; Chang, Q.; Xie, J. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 2003, 362, 1353–1358. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Loman, N.; Pybus, O.; Barclay, W.; Barrett, J.; Carabelli, A.; Connor, T.; Peacock, T.; Robertson, D.L.; Volz, E.; et al. Preliminary Genomic Haracterization of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. SARS-CoV-2 Coronavirus nCoV-2019 Genomic Epidemiology. Virological 2020. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 22 September 2021).
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. MedRxiv 2020. [Google Scholar] [CrossRef]
- Singh, J.; Rahman, S.A.; Ehtesham, N.Z.; Hira, S.; Hasnain, S.E. SARS-CoV-2 variants of concern are emerging in India. Nat. Med. 2021, 27, 1131–1133. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Claro, I.M.; Candido, D.; Franco, L.M.; Andrade, P.S.; Coletti, T.M.; Silva, C.A.; Sales, F.C.; Manuli, E.R.; Aguiar, R.S.; et al. Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: Preliminary findings. Virological 2021, 372, 815–821. [Google Scholar]
- Ozer, E.A.; Simons, L.M.; Adewumi, O.M.; Fowotade, A.A.; Omoruyi, E.C.; Adeniji, J.A.; Dean, T.J.; Taiwo, B.O.; Hultquist, J.F.; Lorenzo-Redondo, R. High prevalence of SARS-CoV-2 B. 1.1. 7 (UK variant) and the novel B. 1.5. 2.5 lineage in Oyo State, Nigeria. MedRxiv 2021. [Google Scholar] [CrossRef]
- Annavajhala, M.K.; Mohri, H.; Zucker, J.E.; Sheng, Z.; Wang, P.; Gomez-Simmonds, A.; Ho, D.D.; Uhlemann, A.-C. A novel SARS-CoV-2 variant of concern, B. 1.526, identified in New York. MedRxiv 2021. [Google Scholar] [CrossRef]
- Liu, C.; Ginn, H.M.; Dejnirattisai, W.; Supasa, P.; Wang, B.; Tuekprakhon, A.; Nutalai, R.; Zhou, D.; Mentzer, A.J.; Zhao, Y. Reduced neutralization of SARS-CoV-2 B. 1.617 by vaccine and convalescent serum. Cell 2021, 184, 4220.e13–4236.e13. [Google Scholar] [CrossRef]
- Kimura, I.; Kosugi, Y.; Wu, J.; Yamasoba, D.; Butlertanaka, E.P.; Tanaka, Y.L.; Liu, Y.; Shirakawa, K.; Kazuma, Y.; Nomura, R.; et al. SARS-CoV-2 Lambda variant exhibits higher infectivity and immune resistance. BioRxiv 2021. [Google Scholar] [CrossRef]
- Laiton-Donato, K.; Franco-Munoz, C.; Alvarez-Diaz, D.A.; Ruiz-Moreno, H.; Usme-Ciro, J.; Prada, D.; Reales, J.; Corchuelo, S.; Herrera-sepulveda, M.; Naizaque, J.; et al. Characterization of the emerging B. 1.621 variant of interest of SARS-CoV-2. MedRxiv 2021. [Google Scholar] [CrossRef]
- Reardon, S. How the Delta variant achieves its ultrafast spread. Nature 2021, 21. [Google Scholar] [CrossRef]
- Krishnan, L.; Ogunwole, S.M.; Cooper, L.A. Historical Insights on Coronavirus Disease 2019 (COVID-19), the 1918 Influenza Pandemic, and Racial Disparities: Illuminating a Path Forward. Ann. Intern. Med. 2020, 173, 474–481. [Google Scholar] [CrossRef] [PubMed]
- A Timeline of HIV and AIDS. Available online: https://www.hiv.gov/hiv-basics/overview/history/hiv-and-aids-timeline (accessed on 22 September 2021).
- Hemida, M.; Perera, R.; Wang, P.; Alhammadi, M.; Siu, L.; Li, M.; Poon, L.; Saif, L.; Alnaeem, A.; Peiris, M. Middle East Respiratory Syndrome (MERS) coronavirus seroprevalence in domestic livestock in Saudi Arabia, 2010 to 2013. Eurosurveillance 2013, 18, 20659. [Google Scholar] [CrossRef]
- Preventing the Spread of the Coronavirus. Available online: https://www.health.harvard.edu/diseases-and-conditions/preventing-the-spread-of-the-coronavirus (accessed on 9 December 2020).
- Understanding How COVID-19 Vaccines Work. Available online: https://www.cdc.gov/coronavirus/2019-ncov/vaccines/different-vaccines/how-they-work.html (accessed on 2 November 2020).
- COVID-19 Genomic Surveillance. Available online: https://covid19.sanger.ac.uk/lineages/raw (accessed on 1 November 2021).
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271.e8–280.e8. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Xia, S.; Lan, Q.; Su, S.; Wang, X.; Xu, W.; Liu, Z.; Zhu, Y.; Wang, Q.; Lu, L.; Jiang, S. The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal. Transduct. Target. Ther. 2020, 5, 1–3. [Google Scholar] [CrossRef]
- Jaimes, J.A.; Millet, J.K.; Whittaker, G.R. Proteolytic cleavage of the SARS-CoV-2 spike protein and the role of the novel S1/S2 site. iScience 2020, 23, 101212. [Google Scholar] [CrossRef]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortolá, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. BioRxiv 2021. [Google Scholar] [CrossRef]
- Peacock, T.P.; Sheppard, C.M.; Brown, J.C.; Goonawardane, N.; Zhou, J.; Whiteley, M.; de Silva, T.I.; Barclay, W.S.; Consortium, P.V. The SARS-CoV-2 variants associated with infections in India, B. 1.617, show enhanced spike cleavage by furin. BioRxiv 2021. [Google Scholar] [CrossRef]
- Saito, A.; Nasser, H.; Uriu, K.; Kosugi, Y.; Irie, T.; Shirakawa, K. SARS-CoV-2 spike P681R mutation enhances and accelerates viral fusion. BioRxiv 2021, 10, 17.448820. [Google Scholar]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Gobeil, S.M.-C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Manne, K.; Stalls, V.; Kopp, M.F.; Henderson, R.; Edwards, R.J.; et al. D614G mutation alters SARS-CoV-2 spike conformation and enhances protease cleavage at the S1/S2 junction. Cell Rep. 2021, 34, 108630. [Google Scholar] [CrossRef]
- Adhikari, P.; Ching, W.-Y. Amino acid interacting network in the receptor-binding domain of SARS-CoV-2 spike protein. RSC Adv. 2020, 10, 39831–39841. [Google Scholar] [CrossRef]
- Woo, H.; Park, S.-J.; Choi, Y.K.; Park, T.; Tanveer, M.; Cao, Y.; Kern, N.R.; Lee, J.; Yeom, M.S.; Croll, T.I. Developing a fully glycosylated full-length SARS-CoV-2 spike protein model in a viral membrane. J. Phys. Chem. B 2020, 124, 7128–7137. [Google Scholar] [CrossRef]
- CHARMM-GUI Archive—COVID-19 Proteins Library. Available online: https://charmm-gui.org/?doc=archive&lib=covid19 (accessed on 1 November 2021).
- Case, D.A.; Betz, R.; Cerutti, D.; Cheatham, T.; Darden, T.; Duke, R.; Giese, T.; Gohlke, H.; Goetz, A.; Homeyer, N. AMBER 2020 Reference Manual. University of California, San Francisco, 2020. Available online: https://ambermd.org/Manuals.php (accessed on 1 November 2021).
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E., III; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Dunbrack, R.L., Jr. Rotamer libraries in the 21st century. Curr. Opin. Struct. Biol. 2002, 12, 431–440. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VASP—Vienna Ab Initio Simulation Package. Available online: https://www.vasp.at/ (accessed on 1 November 2021).
- Ching, W.-Y.; Rulis, P. Electronic Structure Methods for Complex Materials: The Orthogonalized Linear Combination of Atomic Orbitals; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Ryadnov, M.; Hudecz, F. Amino Acids, Peptides and Proteins; Royal Society of Chemistry: Cambridge, UK, 2017; Volume 42. [Google Scholar]
- Khan, R.J.; Jha, R.K.; Amera, G.M.; Jain, M.; Singh, E.; Pathak, A.; Singh, R.P.; Muthukumaran, J.; Singh, A.K. Targeting SARS-CoV-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2′-O-ribose methyltransferase. J. Biomol. Struct. Dyn. 2021, 39, 2679–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, M.; Gao, J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 13967–13974. [Google Scholar] [CrossRef] [PubMed]
- Marx, D.; Hutter, J. Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods; Cambridge University Press: New York, NY, USA, 2009. [Google Scholar]
- Adhikari, P.; Li, N.; Shin, M.; Steinmetz, N.F.; Twarock, R.; Podgornik, R.; Ching, W.-Y. Intra- and intermolecular atomic-scale interactions in the receptor binding domain of SARS-CoV-2 spike protein: Implication for ACE2 receptor binding. Phys. Chem. Chem. Phys. 2020, 22, 18272–18283. [Google Scholar] [CrossRef]
- Ching, W.-Y.; Adhikari, P.; Jawad, B.; Podgornik, R. Ultra-Large-Scale Ab Initio Quantum Chemical Computation of Bio-Molecular Systems: The Case of Spike Protein of SARS-CoV-2 Virus. Comput. Struct. Biotechnol. J. 2021, 19, 1288–1301. [Google Scholar] [CrossRef]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.-Y. Key interacting residues between RBD of SARS-CoV-2 and ACE2 receptor: Combination of molecular dynamic simulation and density functional calculation. J. Chem. Inf. Model. 2021, 61, 4425–4441. [Google Scholar] [CrossRef]
- Adhikari, P.; Podgornik, R.; Jawad, B.; Ching, W.-Y. First-Principles Simulation of Dielectric Function in Biomolecules. Materials 2021, 14, 5774. [Google Scholar] [CrossRef]
- Baral, K.; Adhikari, P.; Jawad, B.; Podgornik, R.; Ching, W.-Y. Solvent Effect on the Structure and Properties of RGD Peptide (1FUV) at Body Temperature (310 K) Using Ab Initio Molecular Dynamics. Polymers 2021, 13, 3434. [Google Scholar] [CrossRef]
- Liang, L.; Rulis, P.; Ouyang, L.; Ching, W. Ab initio investigation of hydrogen bonding and network structure in a supercooled model of water. Phys. Rev. B 2011, 83, 024201. [Google Scholar] [CrossRef] [Green Version]
- Vermeeren, P.; van Zeist, W.-J.; Hamlin, T.A.; Guerra, C.F.; Bickelhaupt, F.M.; Bickelhaupt, F.; Guerra, C.F. Not Carbon s–p Hybridization, but Coordination Number Determines C−H and C−C Bond Length. Chem. A Eur. J. 2021, 27, 7074–7079. [Google Scholar] [CrossRef] [PubMed]
- Mlcochova, P.; Kemp, S.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S. SARS-CoV-2 B. 1.617. 2 Delta variant replication and immune evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Lopez Bernal, J.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G. Effectiveness of COVID-19 vaccines against the B. 1.617. 2 (Delta) variant. N. Engl. J. Med. 2021, 385, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.R.; Spratt, A.N.; Cohen, A.R.; Naqvi, S.H.; Chand, H.S.; Quinn, T.P.; Lorson, C.L.; Byrareddy, S.N.; Singh, K. Evolutionary analysis of the Delta and Delta Plus variants of the SARS-CoV-2 viruses. J. Autoimmun. 2021, 124, 102715. [Google Scholar] [CrossRef]
- Rajah, M.M.; Hubert, M.; Bishop, E.; Saunders, N.; Robinot, R.; Grzelak, L.; Planas, D.; Dufloo, J.; Gellenoncourt, S.; Bongers, A. SARS-CoV-2 Alpha, Beta, and Delta variants display enhanced Spike-mediated syncytia formation. EMBO J. 2021, 40, e108944. [Google Scholar] [CrossRef]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef]
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Palero, F.; González-Candelas, F. Recombination in viruses: Mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 2015, 30, 296–307. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Graham, R.L.; Baric, R.S. Recombination, reservoirs, and the modular spike: Mechanisms of coronavirus cross-species transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef] [Green Version]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.-Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- On the origin of Species. The Economist, 25 August 2021.
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef] [PubMed]
- Eshete, B. Making machine learning trustworthy. Science 2021, 373, 743–744. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, I.; McDaniel, P.; Papernot, N. Making machine learning robust against adversarial inputs. Commun. ACM 2018, 61, 56–66. [Google Scholar] [CrossRef]
- James, G.; Witten, D.; Hastie, T.; Tibshirani, R. An Introduction to Statistical Learning with Applications in R; Springer: New York, NY, USA, 2013; Volume 112. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Quinlan, J.R. Induction of decision trees. Mach. Learn. 1986, 1, 81–106. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 13 August 2016. [Google Scholar]
- Richardson, M.; Domingos, P. Markov logic networks. Mach. Learn. 2006, 62, 107–136. [Google Scholar] [CrossRef] [Green Version]
- Pearl, J. Causality: Models, Reasoning and Inference; Cambridge University Press: Cambridge, UK, 2000; p. 19. [Google Scholar]
- Tsamardinos, I.; Brown, L.E.; Aliferis, C.F. The max-min hill-climbing Bayesian network structure learning algorithm. Mach. Learn. 2006, 65, 31–78. [Google Scholar] [CrossRef] [Green Version]
- Classification of Omicron (B.1.1.529): SARS-CoV-2 Variant of Concern. 2021. Available online: https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern (accessed on 28 November 2021).
- NERSC Perlmutter. 2021. Available online: https://www.nersc.gov/systems/perlmutter/ (accessed on 28 November 2021).
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Poudel, L.; Steinmetz, N.F.; French, R.H.; Parsegian, V.A.; Podgornik, R.; Ching, W.-Y. Implication of the solvent effect, metal ions and topology in the electronic structure and hydrogen bonding of human telomeric G-quadruplex DNA. Phys. Chem. Chem. Phys. 2016, 18, 21573–21585. [Google Scholar] [CrossRef] [PubMed]
- Poudel, L.; Twarock, R.; Steinmetz, N.F.; Podgornik, R.; Ching, W.-Y. Impact of hydrogen bonding in the binding site between capsid protein and MS2 bacteriophage ssRNA. J. Phys. Chem. B 2017, 121, 6321–6330. [Google Scholar] [CrossRef]
- Eifler, J.; Podgornik, R.; Steinmetz, N.F.; French, R.H.; Parsegian, V.A.; Ching, W.Y. Charge distribution and hydrogen bonding of a collagen α2-chain in vacuum, hydrated, neutral, and charged structural models. Int. J. Quantum Chem. 2016, 116, 681–691. [Google Scholar] [CrossRef]
- Poudel, L.; Wen, A.M.; French, R.H.; Parsegian, V.A.; Podgornik, R.; Steinmetz, N.F.; Ching, W.Y. Electronic structure and partial charge distribution of doxorubicin in different molecular environments. ChemPhysChem 2015, 16, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Poudel, L.; Rulis, P.; Liang, L.; Ching, W.-Y. Electronic structure, stacking energy, partial charge, and hydrogen bonding in four periodic B-DNA models. Phys. Rev. E 2014, 90, 022705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikari, P.; Xiong, M.; Li, N.; Zhao, X.; Rulis, P.; Ching, W.-Y. Structure and electronic properties of a continuous random network model of an amorphous zeolitic imidazolate framework (a-ZIF). J. Phys. Chem. C 2016, 120, 15362–15368. [Google Scholar] [CrossRef]
- Ching, W.Y.; Yoshiya, M.; Adhikari, P.; Rulis, P.; Ikuhara, Y.; Tanaka, I. First-principles study in an inter-granular glassy film model of silicon nitride. J. Am. Ceram. Soc. 2018, 101, 2673–2688. [Google Scholar] [CrossRef]
- Ching, W.-Y.; San, S.; Brechtl, J.; Sakidja, R.; Zhang, M.; Liaw, P.K. Fundamental electronic structure and multiatomic bonding in 13 biocompatible high-entropy alloys. npj Comput. Mater. 2020, 6, 45. [Google Scholar] [CrossRef]
- Jawad, B.; Poudel, L.; Podgornik, R.; Ching, W.-Y. Thermodynamic Dissection of the Intercalation Binding Process of Doxorubicin to dsDNA with Implications of Ionic and Solvent Effects. J. Phys. Chem. B 2020, 124, 7803–7818. [Google Scholar] [CrossRef]
- Baral, K.; Li, A.; Ching, W.-Y. Ab Initio Study of Hydrolysis Effects in Single and Ion-Exchanged Alkali Aluminosilicate Glasses. J. Phys. Chem. B 2020, 124, 8418–8433. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef] [Green Version]
- Mulliken, R. Electronic population analysis on LCAO–MO molecular wave functions. II. Overlap populations, bond orders, and covalent bond energies. J. Chem. Phys. 1955, 23, 1841–1846. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | Total AABP | NN AABP | Non-Local AABP | AABP from HB | No. of NL AAs | Data Notation |
|---|---|---|---|---|---|---|
| WT P681 | 1.117 | 1.064 | 0.054 | 0.064 | 5 | P681-1.117-1.064-0.054-0.064-0 |
| WT D614 | 0.917 | 0.912 | 0.005 | 0.040 | 5 | D614-0.917-0.912-0.005-0.040-0 |
| Mutated R681 | 1.082 | 0.971 | 0.111 | 0.122 | 6 | R681-1.082-0.971-0.111-0.122-1 |
| Mutated G614 | 0.904 | 0.904 | 0.001 | 0.040 | 2 | G614-0.904-0.904-0.001-0.040-1 |
| DM G614-R681 | ||||||
| R681 | 1.023 | 0.975 | 0.047 | 0.066 | 6 | R681-1.023-0.975-0.047-0.066-1 |
| G614 | 0.901 | 0.901 | 0.001 | 0.041 | 2 | G614-0.901-0.901-0.001-0.041-1 |
| Feature Vector | Target | ||||
|---|---|---|---|---|---|
| TAABP | NN | NL | HB | NNL | MT |
| 1.117 | 1.064 | 0.054 | 0.064 | 5 | 0 |
| 0.917 | 0.912 | 0.005 | 0.040 | 5 | 0 |
| 1.082 | 0.971 | 0.111 | 0.122 | 6 | 1 |
| 0.904 | 0.904 | 0.001 | 0.040 | 2 | 1 |
| 1.023 | 0.975 | 0.047 | 0.066 | 6 | 1 |
| 0.901 | 0.901 | 0.001 | 0.041 | 2 | 1 |
| 1.163 | 1.021 | 0.143 | 0.154 | 6 | 0 |
| 1.093 | 1.009 | 0.084 | 0.106 | 6 | 1 |
| Feature Vector | Target | ||||
|---|---|---|---|---|---|
| TAABP | NN | NL | HB | NNL | MT |
| 1.064 | 1.024 | 0.041 | 0.145 | 7 | 0 |
| 1.181 | 1.098 | 0.084 | 0.089 | 4 | 1 |
| 1.068 | 0.993 | 0.075 | 0.055 | 4 | 0 |
| 1.113 | 1.057 | 0.056 | 0.119 | 5 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adhikari, P.; Jawad, B.; Rao, P.; Podgornik, R.; Ching, W.-Y. Delta Variant with P681R Critical Mutation Revealed by Ultra-Large Atomic-Scale Ab Initio Simulation: Implications for the Fundamentals of Biomolecular Interactions. Viruses 2022, 14, 465. https://doi.org/10.3390/v14030465
Adhikari P, Jawad B, Rao P, Podgornik R, Ching W-Y. Delta Variant with P681R Critical Mutation Revealed by Ultra-Large Atomic-Scale Ab Initio Simulation: Implications for the Fundamentals of Biomolecular Interactions. Viruses. 2022; 14(3):465. https://doi.org/10.3390/v14030465
Chicago/Turabian StyleAdhikari, Puja, Bahaa Jawad, Praveen Rao, Rudolf Podgornik, and Wai-Yim Ching. 2022. "Delta Variant with P681R Critical Mutation Revealed by Ultra-Large Atomic-Scale Ab Initio Simulation: Implications for the Fundamentals of Biomolecular Interactions" Viruses 14, no. 3: 465. https://doi.org/10.3390/v14030465