
Viral Aggregation: The Knowns and Unknowns

 , , , and
, , , and
Abstract
:1. Introduction
2. A Brief Historical Review of Studies on Viral Aggregation
2.1. Factors Influencing Viral Aggregation
2.2. The Research Landscape of Viral Aggregation in Comparison to Their Bacterial Counterparts
3. Viral Aggregation and the Stoichiometry of MOI
3.1. The Stochasticity in Early Events of Viral Infection Often Leads to Unproductive Infection
3.2. Segmented and Multipartite Viruses Have Low Infection Probability
4. Viral Aggregation in the Context of Infectious Viral Life Cycle
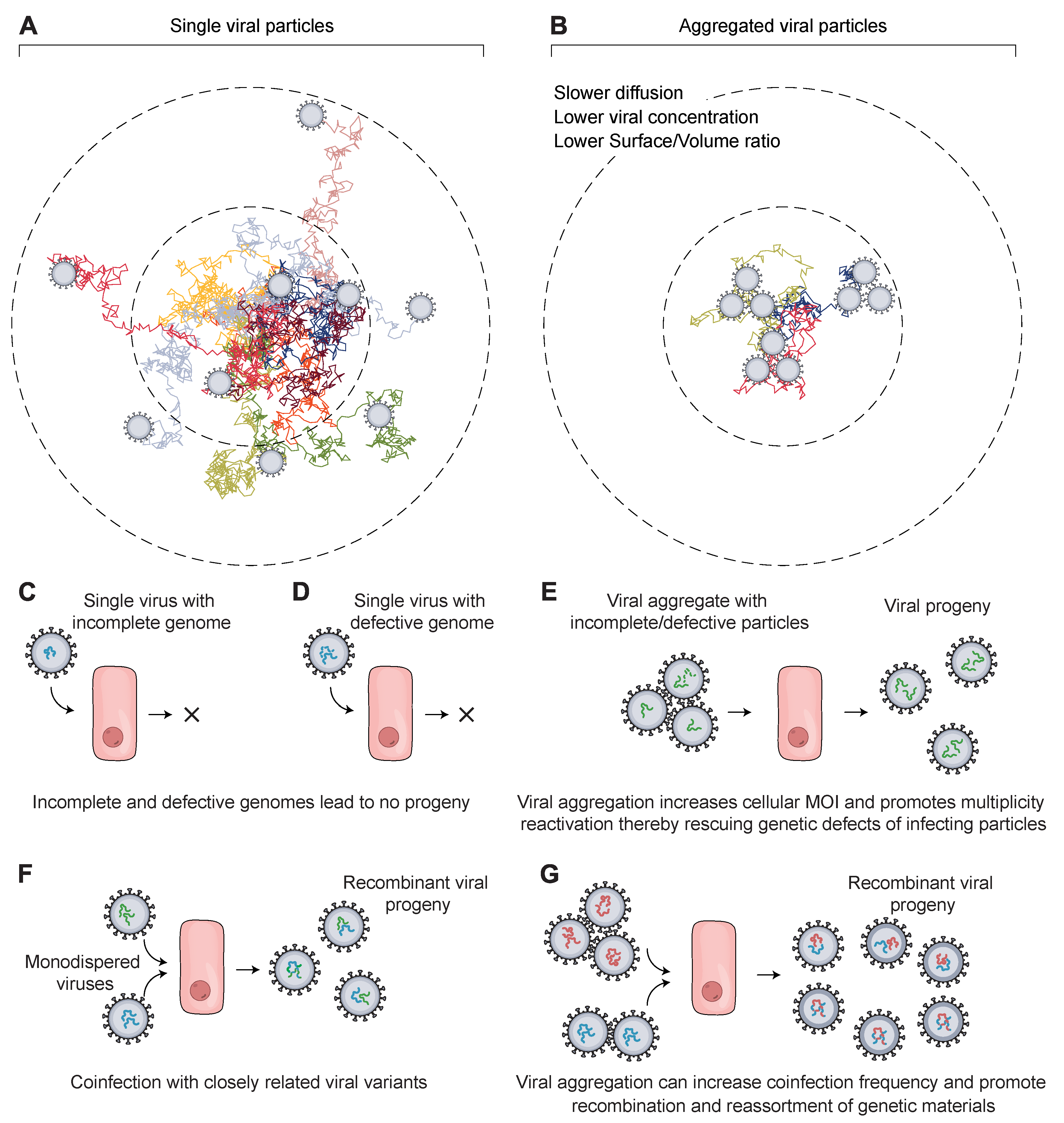
4.1. Viral Aggregation Influencing Viral Motion
4.2. Viral Aggregation Influencing Replication Inside Host Cells
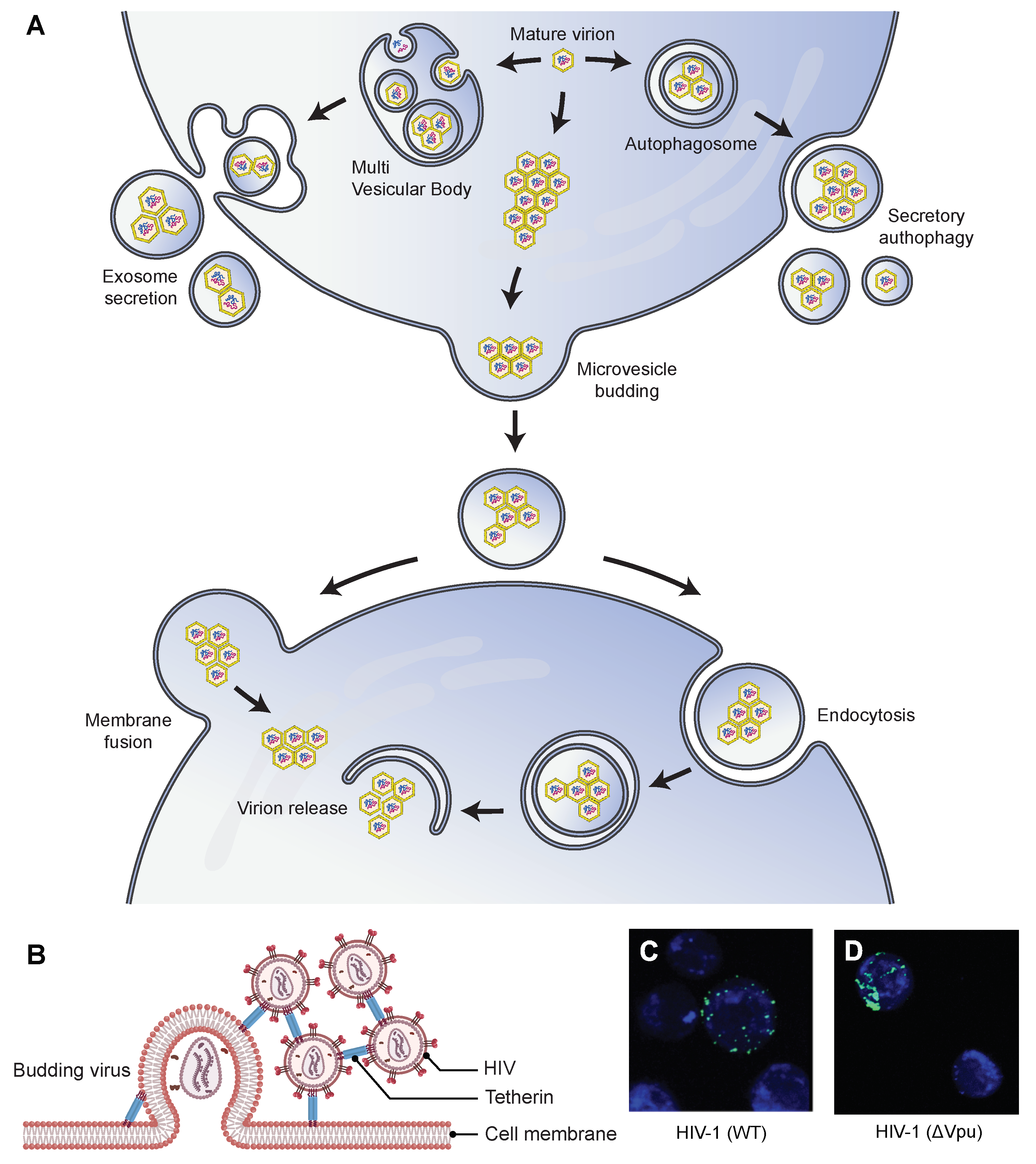
4.3. Viral Aggregation Influencing Release from Host Cells
4.3.1. Extracellular Vesicles-Mediated Release of Viral Aggregates
4.3.2. Tetherin Mediated Viral Aggregation and the Consequent Inhibition of Viral Release
5. Viral Aggregation as an Antiviral Response
6. Harnessing Viral Aggregation as a Therapeutic Tool
7. Concluding Remarks and Prospects
- How commonly do aggregates of pandemic/epidemic/endemic strains of viruses occur in different environments, such as inside a host cell versus a wastewater treatment plant?
- Are there any genetic determinants of viral aggregation? What factors, genetic and otherwise, influence and distinguish the formation of different kinds of viral aggregates, for instance, vesicle-enclosed viral aggregates versus virus–virus binding aggregates versus aggregates formed by virus binding to other surfaces/molecules?
- Does the nature of viral aggregates determine their fate regarding immune evasion and clearance? For instance, vesicle-enclosed viral aggregates show enhanced immune evasion. In contrast, aggregates formed by antibodies are more potent immune stimuli triggering enhanced opsonization and immune clearance.
- How does viral aggregation influence different events of an infectious viral life cycle, including viral adhesion, entry, replication, assembly, and release? What molecular and cellular factors/mechanisms drive those outcomes? Is aggregation conditional on any stage of the viral life cycle?
- How does viral aggregation influence the infectivity and virulence of different viral species or even different strains of the same viral species? Are there aggregation patterns exhibited by viral strains/species that can be traced back to the similarities and differences in their structural/genetic makeup?
- How does aggregation contribute to the viral fitness, diversity, and evolution landscape?
- Can we develop model systems to study viral aggregation? Can we induce viral aggregation in vitro, in vivo, and ex vivo to modulate infectivity, virulence and neutralization?
- How does viral aggregation influence the kinetics and efficiency of viral vectors in gene therapy?
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| AWOL | Autophagosome-mediated exit without lysis |
| EB | Entry blocker |
| EM | Electron microscopy |
| EMV | Extracellular microvesicles |
| EV | Extracellular vesicle |
| FGF-4 | Fibroblast growth factor-4 |
| GCXV | Guaico CuleX virus |
| HA | Hemagglutinin |
| HAV | Hepatitis A virus |
| HIV | Human immunodeficiency virus |
| IAV | Influenza A virus |
| MOI | Multiplicity of infection |
| MSD | Mean-squared displacement |
| MVB | Multivesicular body |
| NA | Neuraminidase |
| NET | Neutrophil extracellular trap |
| OBs | Occlusion bodies |
| PS | Phosphatidylserine |
| RSV | Respiratory syncytial virus |
| TMV | Tobacco mosaic virus |
| VSV | Vesicular stomatitis virus |
References
- Rohrmann, G.F. Baculovirus structural proteins. J. Gen. Virol. 1992, 73 Pt 4, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Federici, B.A.; Vlak, J.M.; Hamm, J.J. Comparative study of virion structure, protein composition and genomic DNA of three ascovirus isolates. J. Gen. Virol. 1990, 71 Pt 8, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Hirst, G.K.; Pons, M.W. Mechanism of influenza recombination: II. Virus aggregation and its effect on plaque formation by so-called noninfective virus. Virol. J. 1973, 56, 620–631. [Google Scholar] [CrossRef]
- Galasso, G.J.; Sharp, J.; Sharp, D.G. The influence of degree of aggregation and virus quality on the plaque titer of aggregated vaccinia virus. J. Immunol. 1964, 92, 870–878. [Google Scholar]
- Floyd, R.; Sharp, D.G. Aggregation of poliovirus and reovirus by dilution in water. Appl. Environ. Microbiol. 1977, 33, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Narang, H.K.; Codd, A.A. Frequency of preclumped virus in routine fecal specimens from patients with acute nonbacterial gastroenteritis. J. Clin. Microbiol. 1981, 13, 982–988. [Google Scholar] [CrossRef] [Green Version]
- Williams, F.P. Membrane-associated viral complexes observed in stools and cell culture. Appl. Environ. Microbiol. 1985, 50, 523–526. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.C. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Santiana, M.; Ghosh, S.; Ho, B.A.; Rajasekaran, V.; Du, W.L.; Mutsafi, Y.; De Jésus-Diaz, D.A.; Sosnovtsev, S.V.; Levenson, E.A.; Parra, G.I. Vesicle-cloaked virus clusters are optimal units for inter-organismal viral transmission. Cell Host Microbe 2018, 24, 208–220.e8. [Google Scholar] [CrossRef] [Green Version]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Arnaud, F.; Black, S.G.; Murphy, L.; Griffiths, D.J.; Neil, S.J.; Spencer, T.E.; Palmarini, M. Interplay between ovine bone marrow stromal cell antigen 2/tetherin and endogenous retroviruses. J. Virol. 2010, 84, 4415–4425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaletsky, R.L.; Francica, J.R.; Agrawal-Gamse, C.; Bates, P. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc. Natl. Acad. Sci. USA 2009, 106, 2886–2891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilinskaya, A.; Derse, D.; Hill, S.; Princler, G.; Heidecker, G. Cell–cell transmission allows human T-lymphotropic virus 1 to circumvent tetherin restriction. Virology 2013, 436, 201–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjuán, R. Collective infectious units in viruses. Trends Microbiol. 2017, 25, 402–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bald, J.G.; Briggs, G.E. Aggregation of Virus Particles. Nature 1937, 140, 111. [Google Scholar] [CrossRef]
- Hirst, G.K. Studies of antigenic differences among strains of influenza A by means of red cell agglutination. J. Exp. Med. 1983, 78, 407–423. [Google Scholar] [CrossRef]
- Hilleman, M.R. System for measuring and designating antigenic components of influenza viruses with analyses of recently isolated strains. Proc. Soc. Exp. Biol. Med. 1951, 78, 208–215. [Google Scholar] [CrossRef]
- Floyd, R. Viral aggregation: Mixed suspensions of poliovirus and reovirus. Appl. Environ. Microbiol. 1979, 38, 980–986. [Google Scholar] [CrossRef] [Green Version]
- Sharp, D.G.; Floyd, R.; Johnson, J.D. Nature of the surviving plaque-forming unit of reovirus in water containing bromine. Appl. Environ. Microbiol. 1975, 29, 94–101. [Google Scholar] [CrossRef]
- Galdiero, F. Adenovirus aggregation and preservation in extracellular environment. Arch. Virol. 1979, 59, 99–105. [Google Scholar] [CrossRef]
- Fiszman, M.; Bucchini, D.; Girard, M. Purification of the Sabin strain of poliovirus type I through treatment with sarkozyl. J. Virol. 1971, 7, 687–689. [Google Scholar] [CrossRef] [Green Version]
- Young, D.C.; Sharp, D.G. Poliovirus aggregates and their survival in water. Appl. Environ. Microbiol. 1977, 33, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Floyd, R.; Sharp, D.G. Viral aggregation: Effects of salts on the aggregation of poliovirus and reovirus at low pH. Appl. Environ. Microbiol. 1978, 35, 1084–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totsuka, A.; Ohtaki, K.; Tagaya, I. Aggregation of enterovirus small plaque variants and polioviruses under low ionic strength conditions. J. Gen. Virol. 1978, 38, 519–533. [Google Scholar] [CrossRef]
- Floyd, R.; Sharp, D.G. Viral aggregation: Quantitation and kinetics of the aggregation of poliovirus and reovirus. Appl. Environ. Microbiol. 1978, 35, 1079–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Zeipel, G. Neutralization of Aggregated Strains of Enterovirus 71 and Echovirus Type 4 in RD and Vero or GMK-AH1 Cells. Acta Pathol. Microbiol. Scand. B 1979, 87, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Katouzian-safadi, M.; Favre, A.; Haenni, A. Effect of freezing and thawing on the structure of turnip yellow mosaic virus. Eur. J. Biochem. 1980, 112, 479–486. [Google Scholar] [CrossRef]
- Galasso, G.J.; Sharp, D.G. Effect of particle aggregation on the survival of irradiated vaccinia virus. J. Bacteriol. 1965, 90, 1138–1142. [Google Scholar] [CrossRef] [Green Version]
- Kahler, A.M.; Cromeans, T.L.; Metcalfe, M.G.; Humphrey, C.D.; Hill, V.R. Aggregation of adenovirus 2 in source water and impacts on disinfection by chlorine. Food Environ. Virol. 2016, 8, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Tucker, S.P.; Thornton, C.L.; Wimmer, E.; Compans, R.W. Vectorial release of poliovirus from polarized human intestinal epithelial cells. J. Virol. 1993, 67, 4274–4282. [Google Scholar] [CrossRef] [Green Version]
- Gollins, S.W.; Porterfield, J.S. Flavivirus infection enhancement in macrophages: An electron microscopic study of viral cellular entry. J. Gen. Virol. 1985, 66, 1969–1982. [Google Scholar] [CrossRef] [PubMed]
- Gassilloud, B.; Gantzer, C. Adhesion-aggregation and inactivation of poliovirus 1 in groundwater stored in a hydrophobic container. Appl. Environ. Microbiol. 2005, 71, 912–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michen, B.; Graule, T. Isoelectric points of viruses. J. Appl. Microbiol. 2010, 109, 388–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.; Mukherjee, B.; Kahler, A.M.; Zepp, R.; Molina, M. Influence of Inorganic Ions on Aggregation and Adsorption Behaviors of Human Adenovirus. Environ. Sci. Technol. 2012, 46, 11145–11153. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.K.; Kavanagh, O.V.; Estes, M.K.; Elimelech, M. Adsorption and aggregation properties of norovirus GI and GII virus-like particles demonstrate differing responses to solution chemistry. Environ. Sci. Technol. 2011, 45, 520–526. [Google Scholar] [CrossRef] [Green Version]
- Langlet, J.; Gaboriaud, F.; Gantzer, C. Effects of pH on plaque forming unit counts and aggregation of MS2 bacteriophage. J. Appl. Microbiol. 2007, 103, 1632–1638. [Google Scholar] [CrossRef]
- Zoueva, O.P.; Bailly, J.E.; Nicholls, R.; Brown, E.G. Aggregation of influenza virus ribonucleocapsids at low pH. Virus Res. 2002, 85, 141–149. [Google Scholar] [CrossRef]
- Libersou, S.; Albertini, A.A.V.; Ouldali, M.; Maury, V.; Maheu, C.; Raux, H.; de Haas, F.; Roche, S.; Gaudin, Y.; Lepault, J. Distinct structural rearrangements of the VSV glycoprotein drive membrane fusion. J. Cell Biol. 2010, 191, 199–210. [Google Scholar] [CrossRef]
- Davis, H.E.; Rosinski, M.; Morgan, J.R.; Yarmush, M.L. Charged Polymers Modulate Retrovirus Transduction via Membrane Charge Neutralization and Virus Aggregation. Biophys. J. 2004, 86, 1234–1242. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, L.; Mylon, S.E.; Nash, B.; Nguyen, T.H. Deposition and aggregation kinetics of rotavirus in divalent cation solutions. Environ. Sci. Technol. 2010, 44, 4552–4557. [Google Scholar] [CrossRef]
- Langlet, J.; Gaboriaud, F.; Jérôme, F.; Gantzer, C. Aggregation and surface properties of F-specific RNA phages: Implication for membrane filtration processes. Water Res. 2008, 42, 2769–2777. [Google Scholar] [CrossRef] [PubMed]
- Gerba, C.P.; Betancourt, W.Q. Viral Aggregation: Impact of virus behavior in the environment. Environ. Sci. Technol. 2017, 51, 7318–7325. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, J.M.; Moreno, M.D.; Sanjuán, R. Multi-virion infectious units arise from free viral particles in an enveloped virus. Nat. Microbiol. 2017, 2, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anschau, V.; Sanjuán, R. Fibrinogen Gamma Chain Promotes Aggregation of Vesicular Stomatitis Virus in Saliva. Viruses 2020, 12, 282. [Google Scholar]
- Corno, G.; Coci, M.; Giardina, M.; Plechuk, S.; Campanile, F.; Stefani, S. Antibiotics promote aggregation within aquatic bacterial communities. Front. Microbiol. 2014, 5, 297. [Google Scholar] [CrossRef]
- Blom, J.F.; Zimmermann, Y.S.; Ammann, T.; Pernthaler, J. Scent of danger: Floc formation by a freshwater bacterium is induced by supernatants from a predator-prey coculture. Appl. Environ. Microbiol. 2010, 76, 6156–6163. [Google Scholar] [CrossRef] [Green Version]
- Hahn, M.W.; Moore, E.R.B.; Höfle, M.G. Role of Microcolony Formation in the Protistan Grazing Defense of the Aquatic Bacterium Pseudomonas sp. MWH1. Microb. Ecol. 2000, 39, 175–185. [Google Scholar]
- Sherlock, O.; Schembri, M.A.; Reisner, A.; Klemm, P. Novel roles for the AIDA adhesin from diarrheagenic Escherichia coli: Cell aggregation and biofilm formation. J. Bacteriol. 2004, 186, 8058–8065. [Google Scholar] [CrossRef] [Green Version]
- O’Toole, G.A.; Kolter, R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 1998, 30, 295–304. [Google Scholar] [CrossRef]
- Abdel-Nour, M.; Duncan, C.; Prashar, A.; Rao, C.; Ginevra, C.; Jarraud, S.; Low, D.E.; Ensminger, A.W.; Terebiznik, M.R.; Guyard, C. The Legionella pneumophila collagen-like protein mediates sedimentation, autoaggregation, and pathogen-phagocyte interactions. Appl. Environ. Microbiol. 2014, 80, 1441–1454. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, M.; Ito, R.; Tanaka, Y.; Yao, M.; Matoba, K.; Saito, S.; Tanaka, I.; Ohta, T. Staphylococcus aureus surface protein SasG contributes to intercellular autoaggregation of Staphylococcus aureus. Biochem. Biophys. Res. Commun. 2008, 377, 1102–1106. [Google Scholar] [PubMed]
- Arenas, J.; Cano, S.; Nijland, R.; van Dongen, V.; Rutten, L.; van der Ende, A.; Tommassen, J. The meningococcal autotransporter AutA is implicated in autoaggregation and biofilm formation. Environ. Microbiol. 2015, 17, 1321–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trunk, T.; Khalil, H.S.; Leo, J.C. Bacterial autoaggregation. AIMS Microbiol. 2018, 4, 140. [Google Scholar] [CrossRef] [PubMed]
- Jung, A.; Maier, R.; Vartanian, J.P.; Bocharov, G.; Jung, V.; Fischer, U.; Meese, E.; Hobsor, S.W.; Meyerhans, A. Multiply infected spleen cells in HIV patients. Nature 2002, 418, 144. [Google Scholar] [CrossRef]
- Sanjuán, R.; Cuevas, J.M.; Furió, V.; Holmes, E.C.; Moya, A. Selection for Robustness in Mutagenized RNA Viruses. PLoS Genet. 2007, 3, e93. [Google Scholar] [CrossRef] [Green Version]
- Novella, I.S.; Reissig, D.D.; Wilke, C.O. Density-dependent selection in vesicular stomatitis virus. J. Virol. 2004, 78, 5799–5804. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.S.; Sharp, D.G. Electron microscopic observations on the nature of vaccinia virus particle aggregation. J. Immunol. 1966, 97, 197–202. [Google Scholar]
- Flint, S.J.; Enquist, L.W.; Racaniello, V.R.; Skalka, A.M. Principles of Virology, 3rd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Snijder, B.; Sacher, R.; Rämö, P.; Damm, E.M.; Liberali, P.; Pelkmans, L. Population context determines cell-to-cell variability in endocytosis and virus infection. Nature 2009, 461, 520–523. [Google Scholar] [CrossRef]
- Heldt, F.S.; Kupke, S.Y.; Dorl, S.; Reichl, U.; Frensing, T. Single-cell analysis and stochastic modelling unveil large cell-to-cell variability in influenza A virus infection. Nat. Commun. 2015, 6, 1–12. [Google Scholar] [CrossRef]
- Pearson, J.E.; Krapivsky, P.; Perelson, A.S. Stochastic theory of early viral infection: Continuous versus burst production of virions. PLoS Comput. Biol. 2011, 7, e1001058. [Google Scholar] [CrossRef] [Green Version]
- Patton, J.T.; Spencer, E. Genome replication and packaging of segmented double-stranded RNA viruses. J. Virol. 2000, 277, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reijnders, L. The origin of multicomponent small ribonucleoprotein viruses. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 1978; Volume 23, pp. 79–102. [Google Scholar]
- Jaspars, E.M.J. Plant viruses with a multipartite genome. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 1974; Volume 19, pp. 37–149. [Google Scholar]
- Palese, P.; Schulman, J.L. Mapping of the influenza virus genome: Identification of the hemagglutinin and the neuraminidase genes. Proc. Natl. Acad. Sci. USA 1976, 73, 2142–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palese, P.; Ritchey, M.B.; Schulman, J.L. Mapping of the influenza virus genome II. Identification of the P1, P2, and P3 genes. J. Virol. 1977, 76, 114–121. [Google Scholar] [CrossRef]
- Ritchey, M.B.; Palese, P.; Schulman, J.L. Mapping of the influenza virus genome. III. Identification of genes coding for nucleoprotein, membrane protein, and nonstructural protein. J. Virol. 1976, 20, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Brooke, C.B.; Ince, W.L.; Wrammert, J.; Ahmed, R.; Wilson, P.C.; Bennink, J.R.; Yewdell, J.W. Most influenza a virions fail to express at least one essential viral protein. J. Virol. 2013, 87, 3155–3162. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, N.T.; Onuoha, N.O.; Antia, A.; Steel, J.; Antia, R.; Lowen, A.C. Incomplete influenza A virus genomes occur frequently but are readily complemented during localized viral spread. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Varsani, A.; Lefeuvre, P.; Roumagnac, P.; Martin, D. Notes on recombination and reassortment in multipartite/segmented viruses. Curr. Opin. Virol. 2018, 33, 156–166. [Google Scholar] [CrossRef]
- Ladner, J.T.; Wiley, M.R.; Beitzel, B.; Auguste, A.J.; Dupuis, A.P., II; Lindquist, M.E.; Sibley, S.D.; Kota, K.P.; Fetterer, D.; Eastwood, G. A multicomponent animal virus isolated from mosquitoes. Cell Host Microbe 2016, 20, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Sanjuán, R. Collective properties of viral infectivity. Curr. Opin. Virol. 2018, 33, 1–6. [Google Scholar] [CrossRef]
- Andreu-Moreno, I.; Sanjuán, R. Collective infection of cells by viral aggregates promotes early viral proliferation and reveals a cellular-level Allee effect. Curr. Biol. 2018, 28, 3212–3219.e4. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casartelli, N.; Sourisseau, M.; Feldmann, J.; Guivel-Benhassine, F.; Mallet, A.; Marcelin, A.G.; Guatelli, J.; Schwartz, O. Tetherin restricts productive HIV-1 cell-to-cell transmission. PLoS Pathog. 2010, 6, e1000955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.C.; Settles, E.W.; Brandt, C.R.; Schultz-Cherry, S. Virus aggregating peptide enhances the cell-mediated response to influenza virus vaccine. Vaccine 2011, 29, 7696–7703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturman, L.S.; Ricard, C.S.; Holmes, K.V. Conformational change of the coronavirus peplomer glycoprotein at pH 8.0 and 37 degrees C correlates with virus aggregation and virus-induced cell fusion. J. Virol. 1990, 64, 3042–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavijo, G.; Williams, T.; Mun, D.; Caballero, P.; Lopez-Ferber, M. Mixed genotype transmission bodies and virions contribute to the maintenance of diversity in an insect virus. Proc. Biol. Sci. 2010, 277, 943–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajjan, D.B.; Hinchigeri, S.B. Structural Organization of Baculovirus Occlusion Bodies and Protective Role of multilayered polyhedron envelope protein. Food Environ. Virol. 2016, 8, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Booth, N.J.; Neil, S.J.D. Cell-cell spread of human immunodeficiency virus type 1 overcomes tetherin/BST-2-mediated restriction in T cells. J. Virol. 2010, 84, 12185–12199. [Google Scholar] [CrossRef] [Green Version]
- Correia, A.M.P. Biofilm-Like Extracellular Viral Assemblies Mediate HTLV-1 (Human T Cell Leukemia Virus Type-1) Cell-to-Cell Transmission at Virological Synapses. Nat. Med. 2010, 16, 83–89. [Google Scholar] [CrossRef]
- Tecle, T.; White, M.R.; Gantz, D.; Crouch, E.C.; Hartshorn, K.L. Human neutrophil defensins increase neutrophil uptake of influenza A virus and bacteria and modify virus-induced respiratory burst responses. J. Immunol. 2007, 178, 8046–8052. [Google Scholar] [CrossRef]
- Doss, M.; White, M.R.; Tecle, T.; Gantz, D.; Crouch, E.C.; Jung, G.; Ruchala, P.; Waring, A.J.; Lehrer, R.I.; Hartshorn, K.L. Interactions of α-, β-, and θ-defensins with influenza A virus and surfactant protein D. J. Immunol. 2009, 182, 7878–7887. [Google Scholar] [CrossRef] [Green Version]
- Hoeksema, M.; Tripathi, S.; White, M.; Qi, L.; Taubenberger, J.; van Eijk, M.; Haagsman, H.; Hartshorn, K.L. Arginine-rich histones have strong antiviral activity for influenza A viruses. Innate Immun. 2015, 21, 736–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilera, E.R.; Erickson, A.K.; Jesudhasan, P.R.; Robinson, C.M.; Pfeiffer, J.K. Plaques Formed by Mutagenized Viral Populations Have Elevated Coinfection Frequencies. mBio 2017, 8, e02020-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, S.W.; Maynard, N.D.; Covert, M.W.; Kirkegaard, K. Nonlytic viral spread enhanced by autophagy components. Proc. Natl. Acad. Sci. USA 2014, 111, 13081–13086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreadis, S.; Lavery, T.; Davis, H.E.; Le Doux, J.M.; Yarmush, M.L.; Morgan, J.R. Toward a more accurate quantification of the activity of recombinant retroviruses: Alternatives to titer and multiplicity of infection. J. Virol. 2000, 74, 3431–3439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vahey, M.D.; Fletcher, D.A. Influenza A virus surface proteins are organized to help penetrate host mucus. Elife 2019, 8, e43764. [Google Scholar] [CrossRef] [PubMed]
- Baccam, P.; Beauchemin, C.; Macken, C.A.; Hayden, F.G.; Perelson, A.S. Kinetics of influenza A virus infection in humans. J. Virol. 2006, 80, 7590–7599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuck, A.S.; Clarke, M.F.; Palsson, B.O. Retroviral infection is limited by Brownian motion. Hum. Gene Ther. 1996, 7, 1527–1534. [Google Scholar] [CrossRef]
- Marsh, M.; Helenius, A. Virus entry: Open sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Au, S.; Pante, N. How viruses access the nucleus. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1634–1645. [Google Scholar] [CrossRef] [Green Version]
- Altan-Bonnet, N.; Perales, C.; Domingo, E. Extracellular vesicles: Vehicles of en bloc viral transmission. Virus Res. 2019, 265, 143–149. [Google Scholar] [CrossRef]
- Brandenburg, B.; Zhuang, X. Virus trafficking–learning from single-virus tracking. Nat. Rev. Microbiol. 2007, 5, 197–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, S.M.; Tsueng, G.; Sin, J.; Mangale, V.; Rahawi, S.; McIntyre, L.L.; Williams, W.; Kha, N.; Cruz, C.; Hancock, B.M. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014, 10, e1004045. [Google Scholar] [CrossRef] [PubMed]
- Kerviel, A.; Zhang, M.; Bonnet, N.A. A new infectious unit: Extracellular vesicles carrying virus populations. Annu. Rev. Cell Dev. Biol. 2021, 37, 171–197. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R. Subversion of Cellular Autophagosomal Machinery by RNA Viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [Green Version]
- Jouvenet, N.; Neil, S.J.D.; Zhadina, M.; Zang, T.; Kratovac, Z.; Lee, Y.; McNatt, M.; Hatziioannou, T.; Bieniasz, P.D. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 2009, 83, 1837–1844. [Google Scholar] [CrossRef] [Green Version]
- Pardieu, C.; Vigan, R.; Wilson, S.J.; Calvi, A.; Zang, T.; Bieniasz, P.; Kellam, P.; Towers, G.J.; Neil, S.J.D. The RING-CH Ligase K5 Antagonizes Restriction of KSHV and HIV-1 Particle Release by Mediating Ubiquitin-Dependent Endosomal Degradation of Tetherin. PLoS Pathog. 2010, 6, e1000843. [Google Scholar] [CrossRef]
- Weidner, J.M.; Jiang, D.; Pan, X.B.; Chang, J.; Block, T.M.; Guo, J.T. Interferon-Induced Cell Membrane Proteins, IFITM3 and Tetherin, Inhibit Vesicular Stomatitis Virus Infection via Distinct Mechanisms. J. Virol. 2010, 84, 12646–12657. [Google Scholar] [CrossRef] [Green Version]
- Miyagi, E.; Andrew, A.J.; Kao, S.; Strebel, K. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc. Natl. Acad. Sci. USA 2009, 106, 2868–2873. [Google Scholar] [CrossRef] [Green Version]
- Kuhl, B.D.; Sloan, R.D.; Donahue, D.A.; Bar-Magen, T.; Liang, C.; Wainberg, M.A. Tetherin restricts direct cell-to-cell infection of HIV-1. Retrovirology 2010, 7, 115. [Google Scholar] [CrossRef] [Green Version]
- Rudnicka, D.; Feldmann, J.; Porrot, F.; Wietgrefe, S.; Guadagnini, S.; Prévost, M.C.; Estaquier, J.; Haase, A.T.; Sol-Foulon, N.; Schwartz, O. Simultaneous cell-to-cell transmission of human immunodeficiency virus to multiple targets through polysynapses. J. Virol. 2009, 83, 6234–6246. [Google Scholar] [CrossRef] [Green Version]
- Sherer, N.M.; Lehmann, M.J.; Jimenez-Soto, L.F.; Horensavitz, C.; Pypaert, M.; Mothes, W. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat. Cell Biol. 2007, 9, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sowinski, S.; Jolly, C.; Berninghausen, O.; Purbhoo, M.A.; Chauveau, A.; Köhler, K.; Oddos, S.; Eissmann, P.; Brodsky, F.M.; Hopkins, C. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 2008, 10, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Braciale, T.J.; Hahn, Y.S. Immunity to viruses. Immunol. Rev. 2013, 255, 5–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.N.; Rouse, B.T. Immune responses to viruses. Clin. Immunol. 2008, 2008, 421. [Google Scholar]
- Rouse, B.T.; Sehrawat, S. Immunity and immunopathology to viruses: What decides the outcome? Nat. Rev. Immunol. 2010, 10, 514–526. [Google Scholar] [CrossRef]
- Wallis, C.; Melnick, J.L. Virus aggregation as the cause of the non-neutralizable persistent fraction. J. Virol. 1967, 1, 478–488. [Google Scholar] [CrossRef] [Green Version]
- Jayasekera, J.P.; Moseman, E.A.; Carroll, M.C. Natural antibody and complement mediate neutralization of influenza virus in the absence of prior immunity. J. Virol. 2007, 81, 3487–3494. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Kash, J.C.; Dugan, V.G.; Jagger, B.W.; Lau, Y.F.; Sheng, Z.M.; Crouch, E.C.; Hartshorn, K.L.; Taubenberger, J.K. The ability of pandemic influenza virus hemagglutinins to induce lower respiratory pathology is associated with decreased surfactant protein D binding. Virology 2011, 412, 426–434. [Google Scholar] [CrossRef] [Green Version]
- Doss, M.; Ruchala, P.; Tecle, T.; Gantz, D.; Verma, A.; Hartshorn, A.; Crouch, E.C.; Luong, H.; Micewicz, E.D.; Lehrer, R.I.; et al. Hapivirins and diprovirins: Novel θ-defensin analogs with potent activity against influenza A virus. J. Immunol. 2012, 188, 2759–2768. [Google Scholar] [CrossRef] [Green Version]
- Hartshorn, K.L.; White, M.R.; Crouch, E.C. Contributions of the N-and C-terminal domains of surfactant protein d to the binding, aggregation, and phagocytic uptake of bacteria. Infect. Immun. 2002, 70, 6129–6139. [Google Scholar] [CrossRef] [Green Version]
- Hartshorn, K.L.; White, M.R.; Tecle, T.; Holmskov, U.; Crouch, E.C. Innate defense against influenza A virus: Activity of human neutrophil defensins and interactions of defensins with surfactant protein D. J. Immunol. 2006, 176, 6962–6972. [Google Scholar] [CrossRef] [PubMed]
- Hartshorn, K.L.; Webby, R.; White, M.R.; Tecle, T.; Pan, C.; Boucher, S.; Moreland, R.J.; Crouch, E.C.; Scheule, R.K. Role of viral hemagglutinin glycosylation in anti-influenza activities of recombinant surfactant protein D. Respir. Res. 2008, 9, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.A.; Gui, L.; Hom, N.; Mileant, A.; Lee, K.K. Dissection of epitope-specific mechanisms of neutralization of influenza virus by intact IgG and Fab fragments. J. Virol. 2017, 92, e02006-17. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.C.; Settles, E.W.; Brandt, C.R.; Schultz-Cherry, S. Identification of the Minimal Active Sequence of an Anti-Influenza Virus Peptide. Antimicrob. Agents Chemother. 2011, 55, 1810–1813. [Google Scholar] [CrossRef] [Green Version]
- White, M.R.; Kandel, R.; Hsieh, I.N.; De Luna, X.; Hartshorn, K.L. Critical role of C-terminal residues of the Alzheimer’s associated β-amyloid protein in mediating antiviral activity and modulating viral and bacterial interactions with neutrophils. PLoS ONE 2018, 13, e0194001. [Google Scholar] [CrossRef] [Green Version]
- Outlaw, M.C.; Dimmock, N.J. Mechanisms of neutralization of influenza virus on mouse tracheal epithelial cells by mouse monoclonal polymeric IgA and polyclonal IgM directed against the viral haemagglutinin. J. Gen. Virol. 1990, 71, 69–76. [Google Scholar] [CrossRef]
- Jones, J.C.; Turpin, E.A.; Bultmann, H.; Brandt, C.R.; Schultz-Cherry, S. Inhibition of influenza virus infection by a novel antiviral peptide that targets viral attachment to cells. J. Virol. 2006, 80, 11960–11967. [Google Scholar] [CrossRef] [Green Version]
- Kwon, P.S.; Ren, S.; Kwon, S.J.; Kizer, M.E.; Kuo, L.; Xie, M.; Zhu, D.; Zhou, F.; Zhang, F.; Kim, D. Designer DNA architecture offers precise and multivalent spatial pattern-recognition for viral sensing and inhibition. Nat. Chem. 2020, 12, 26–35. [Google Scholar] [CrossRef]
- Gao, G.; Vandenberghe, L.H.; Alvira, M.R.; Lu, Y.; Calcedo, R.; Zhou, X.; Wilson, J.M. Clades of Adeno-associated viruses are widely disseminated in human tissues. J. Virol. 2004, 78, 6381–6388. [Google Scholar] [CrossRef] [Green Version]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: Implications for gene therapy using AAV vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef]
- Zincarelli, C.; Soltys, S.; Rengo, G.; Rabinowitz, J.E. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008, 16, 1073–1080. [Google Scholar] [CrossRef]
- Halbert, C.L.; Allen, J.M.; Miller, A.D. Efficient mouse airway transduction following recombination between AAV vectors carrying parts of a larger gene. Nat. Biotechnol. 2002, 20, 697–701. [Google Scholar] [CrossRef]
- Lai, Y.; Yue, Y.; Liu, M.; Ghosh, A.; Engelhardt, J.F.; Chamberlain, J.S.; Duan, D. Efficient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat. Biotechnol. 2005, 23, 1435–1439. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Virus | Family | Source | Microenvironment of Virus and Aggregating Condition | Ref. | Image |
|---|---|---|---|---|---|
| Vaccinia | Poxiviridae | Virus propagated in Earle’s L cells in vitro | Purified virus particles were resuspended in PBS. | [28] |  |
| Human adenovirus 2 | Adenoviridae | Virus propagated in A549 cells in vitro | Cell associated virus (CAV) particles were resuspended in chlorine demand-free (CDF) grade water. | [29] |  |
| Adenovirus | Adenoviridae | Virus present in fecal specimens of patients with gastroenteritis. | Fecal samples with virus particles were diluted in PBS. | [6] |  100 nm |
| Rotavirus | Reoviridae | Virus present in fecal specimens of patients with gastroenteritis. | Fecal samples with virus diluted in water. Image shows aggregates of Rotavirus inside membranes. | [7] |  200 nm |
| Parvovirus | Parvoviridae | Virus present in fecal specimens of patients with gastroenteritis. | Fecal samples with virus diluted in water. Image shows aggregates of Parvovirus inside membranes. | [7] |  100 nm |
| Norwalk virus | Caliciviridae | Virus present in fecal specimens of patients with gastroenteritis. | Fecal samples with virus diluted in water. Image shows three Norwalk virus particles associated with a fuzzy membranous element. | [7] |  100 nm |
| Poliovirus | Picornaviridae | Virus-infected Caco-2 cells (MOI = 1) | Arrowhead shows aggregate of poliovirus within an intracellular vesicle of infected Caco-2 cells observed at 16 hpi. | [30] |  200 nm |
| Reovirus | Reoviridae | Virus propagated in L cells in vitro | Purified virus particles were diluted in buffers of different pH. Aggregation was observed in buffer with low pH which was reversible when returned to neutral pH. | [5] |  |
| West Nile Virus | Picornaviridae | Virus propagated in Vero cells in vitro. | Aggregate of WNV observed after binding with P388D1 cells for 2 h at 0 °C. | [31] |  100 nm |
| Virus | Genetic Material | Envelope | Family | Size (nm) | Effect of Aggregation on Infection Cycle | Reference |
|---|---|---|---|---|---|---|
| Baculovirus | DNA | Enveloped | Baculoviridae | 200–450 | Co-transmission of multiple viral genomes leading to maintenance of genetic diversity [78], enhanced viral protection [79] | [78,79] |
| Coronavirus | RNA | Enveloped | Coronaviridae | 80–120 | Correlated with loss of viral infectivity although not determined as the only cause | [77] |
| Echovirus type 4 | RNA | Non-enveloped | Picornaviridae | 30 | Enhanced protection against neutralizing antibodies | [26] |
| Enterovirus | RNA | Non-enveloped | Picornaviridae | 30 | Enhanced protection against neutralizing antibodies [8,26], enhanced infectivity [8] | [8,26] |
| Hepatitis A Virus | RNA | Non-enveloped | Picornaviridae | 27 | Viral aggregates inside host-derived membranes showed enhanced infectivity and resistance against antibodies | [74] |
| Human Immunodeficiency Virus | RNA | Enveloped | Retroviridae | 120 | Tetherin-induced viral aggregates showed reduced infectivity due to impairment of their fusion capabilities [75], enhanced cell-to-cell transfer either by mediating the accumulation of virions on the cell surface or by regulating the integrity of the virological synapse [80] | [75,80] |
| Human T-lymphotropic Virus | RNA | Enveloped | Retroviridae | 120 | Facilitated attachment of virus to target cell surface | [81] |
| Influenza A Virus | RNA | Enveloped | Orthomyxoviridae | 80–120 | Enhanced infective capacity when aggregated by nucleohistones [3], enhanced opsonization and uptake by neutrophils when aggregated by collectins, defensins, or antiviral peptides [76,82,83], decrease in viral uptake and replication by host cells [84] | [3,76,82,83,84] |
| Poliovirus | RNA | Non-enveloped | Picornaviridae | 30 | Aggregates formed in low pH showed decrease in infectious viral titer [32,85] and promoted coinfection that correlated with the mutation frequency and rescue of heavily mutagenized viruses [85]. Vesicle-enclosed viral aggregates showed non-lytic release, enhanced viral spread in vitro and pathogenicity in vivo [86] | [32,85,86] |
| Vaccinia virus | DNA | Enveloped | Poxvirus | 250–360 | Enhanced viral survival via increase in cellular MOI | [28,57] |
| Rotavirus | RNA | Non-enveloped | Reoviridae | 55–70 | Vesicle-enclosed aggregates showed enhanced infectivity in vitro and in vivo by overcoming replication barriers associated with low MOI | [9] |
| Vesicular Somatitis Virus | RNA | Enveloped | Rhabdoviridae | 70 | Co-transmission of multiple viral genomes to same cells [43], saliva-induced viral aggregates showed enhanced viral fitness via increase in per capita progeny production [73] | [43,73] |
| West Nile Virus | RNA | Enveloped | Flaviviridae | 40–65 | Slower uptake and phagocytosis by macrophage-like cells | [31] |
| IAV Strain | Aggregating Factor and Conditions | Ref. | Image |
|---|---|---|---|
| H3N2 A/Philippines/2/82 | Arginine-rich histone protein (H4) | [84] |  |
| H3N2 A/Philippines/2/82 | -amyloid peptide (A22-42), which is a 19-amino acid long fragment of the Alzheimer-associated -amyloid transmembrane precursor protein | [118] |  |
| H3N2 A/X-31 | IgG antibodies | [116] |  |
| H1N1 A/PR/8/34 | Mouse serum with complement proteins and virus-specific antibodies | [110] |  |
| H1N1 A/PR/8/34 | EB (Entry Blocker) antiviral peptide derived from fibroblast growth factor 4 | [117] |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pradhan, S.; Varsani, A.; Leff, C.; Swanson, C.J.; Hariadi, R.F. Viral Aggregation: The Knowns and Unknowns. Viruses 2022, 14, 438. https://doi.org/10.3390/v14020438
Pradhan S, Varsani A, Leff C, Swanson CJ, Hariadi RF. Viral Aggregation: The Knowns and Unknowns. Viruses. 2022; 14(2):438. https://doi.org/10.3390/v14020438
Chicago/Turabian StylePradhan, Swechchha, Arvind Varsani, Chloe Leff, Carter J. Swanson, and Rizal F. Hariadi. 2022. "Viral Aggregation: The Knowns and Unknowns" Viruses 14, no. 2: 438. https://doi.org/10.3390/v14020438