
In Silico Analysis and Synthesis of Nafamostat Derivatives and Evaluation of Their Anti-SARS-CoV-2 Activity

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. In Silico Analysis
2.2. Reagents
2.3. Cell Lines and Virus Preparation
2.4. Infection and MTS Assays
3. Results
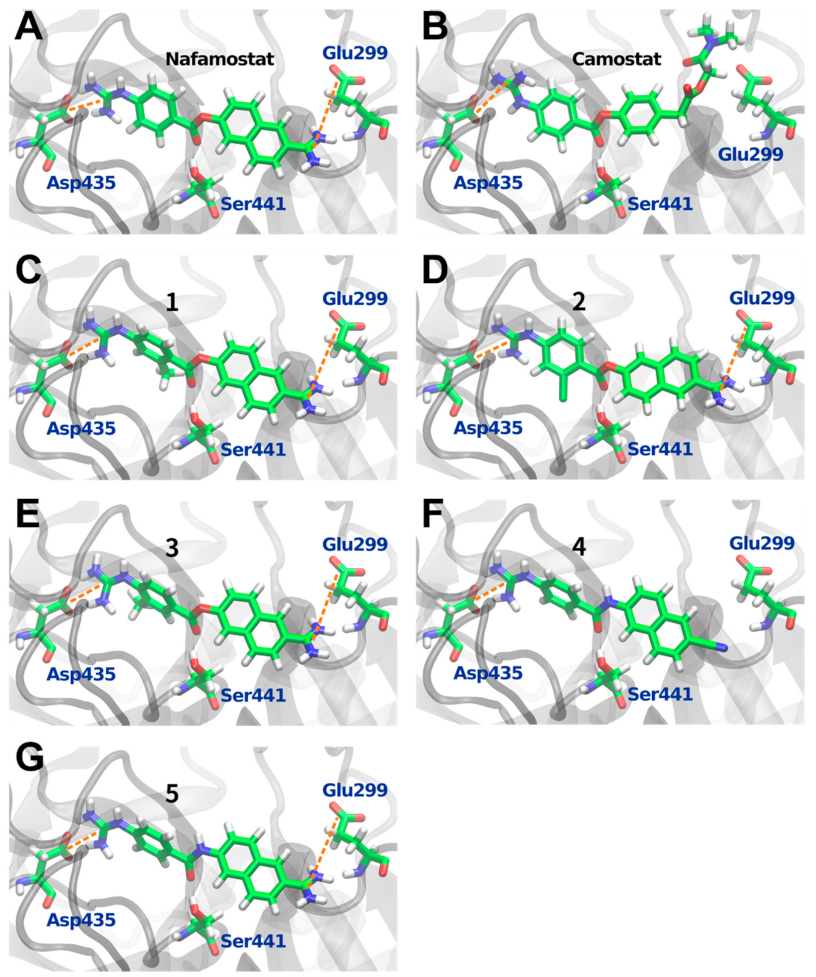
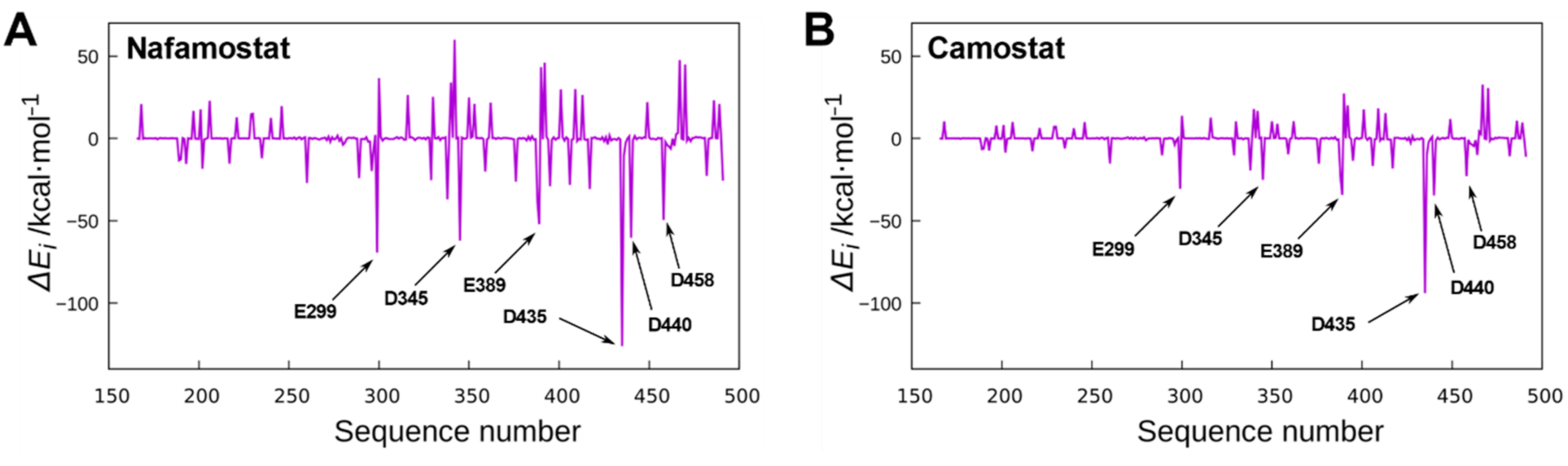
3.1. In Silico Analysis of Nafamostat Binding to TMPRSS2
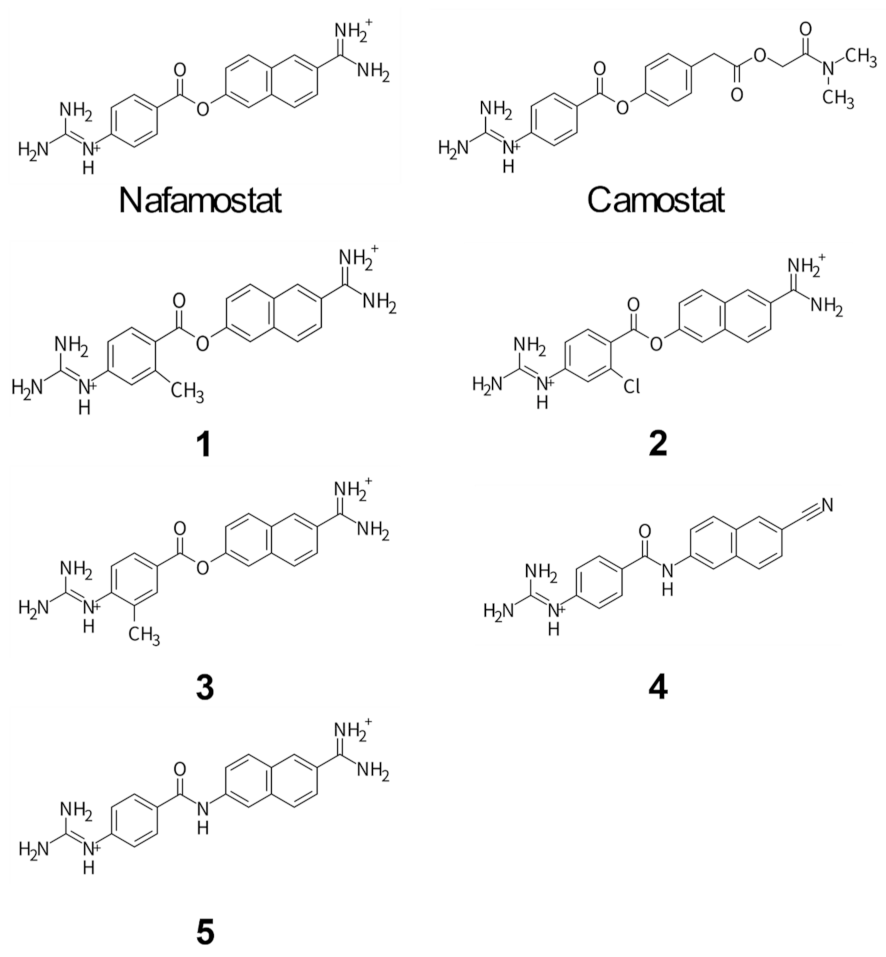
3.2. Design and Synthesis of Nafamostat Derivatives
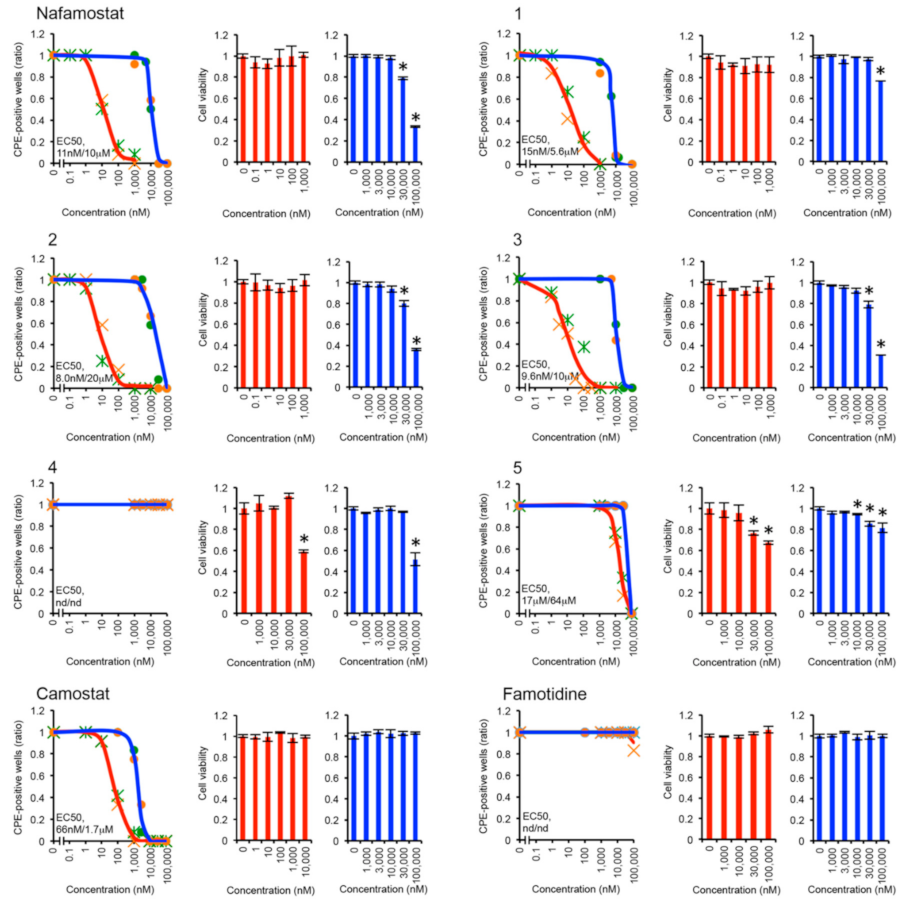
3.3. Antiviral Effects of Nafamostat Derivatives against SARS-CoV-2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; Groot, R.J.D.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. Severe acute respiratory syndrome-related coronavirus: The species and its viruses—A statement of the Coronavirus Study Group. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, M.J.; Lyke, K.E.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Raabe, V.; Bailey, R.; Swanson, K.A.; et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 2020, 586, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: An interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111. [Google Scholar] [CrossRef]
- Fujita Health University School of Medicine. Available online: https://www.fujita-hu.ac.jp/news/j93sdv000000b3zd.html (accessed on 25 August 2021).
- Pegu, A.; O’Connell, S.E.; Schmidt, S.D.; O’Dell, S.; Talana, C.A.; Lai, L.; Albert, J.; Anderson, E.; Bennett, H.; Corbett, K.S.; et al. Durability of mRNA-1273 vaccine–induced antibodies against SARS-CoV-2 variants. Science 2021, 373, 1372–1377. [Google Scholar] [CrossRef]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.-X.; et al. Compassionate Use of Remdesivir for Patients with Severe COVID-19. N. Engl. J. Med. 2020, 382, 2327–2336. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; Elmahi, E.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar]
- Noor, R. Antiviral drugs against severe acute respiratory syndrome coronavirus 2 infection triggering the coronavirus disease-19 pandemic. Tzu Chi Med. J. 2021, 33, 7–12. [Google Scholar] [CrossRef]
- Sikandar, Y.B.; Kheya, I.S.; Noor, R. Remdesivir and Dexamethasone: The Two Eligible Candidate Drugs Against Severe Acute Respiratory Syndrome-Coronavirus 2 (SARS-CoV-2) Infection. Biomed. Res. J. 2020, 7, 29–33. [Google Scholar]
- Rosas, I.O.; Bräu, N.; Waters, M.; Go, R.C.; Hunter, B.D.; Bhagani, S.; Skiest, D.; Aziz, M.S.; Cooper, N.; Douglas, I.S.; et al. Tocilizumab in Hospitalized Patients with Severe COVID-19 Pneumonia. N. Engl. J. Med. 2021, 384, 1503–1516. [Google Scholar] [CrossRef] [PubMed]
- Fung, K.-L.; Chan, P.-L. Comment on: COVID-19: A recommendation to examine the effect of hydroxychloroquine in preventing infection and progression. J. Antimicrob. Chemother. 2020, 75, 2016–2017. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortolá, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kiso, M.; Sakai-Tagawa, Y.; Iwatsuki-Horimoto, K.; Imai, M.; Takeda, M.; Kinoshita, N.; Ohmagari, N.; Gohda, J.; Semba, K.; et al. The Anticoagulant Nafamostat Potently Inhibits SARS-CoV-2 S Protein-Mediated Fusion in a Cell Fusion Assay System and Viral Infection In Vitro in a Cell-Type-Dependent Manner. Viruses 2020, 12, 629. [Google Scholar] [CrossRef]
- Fraser, B.J.; Beldar, S.; Seitova, A.; Hutchinson, A.; Mannar, D.; Li, Y.; Kwon, D.; Tan, R.; Wilson, R.P.; Leopold, K.; et al. Structure, activity and inhibition of human TMPRSS2, a protease implicated in SARS-CoV-2 activation. bioRxiv 2021. [CrossRef]
- Spraggon, G.; Hornsby, M.; Shipway, A.; Tully, D.C.; Bursulaya, B.; Danahay, H.; Harris, J.L.; Lesley, S.A. Active site conformational changes of prostasin provide a new mechanism of protease regulation by divalent cations. Protein Sci. 2009, 18, 1081–1094. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Zhang, X.; Cummings, M.D.; Albarazanji, K.; Wu, J.; Wang, M.; Alexander, R.; Zhu, B.; Zhang, Y.; Leonard, J.; et al. Targeting Enteropeptidase with Reversible Covalent Inhibitors To Achieve Metabolic Benefits. J. Pharmacol. Exp. Ther. 2020, 375, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Sui, Y.; Zhou, Y.; Ya, J.; Yuan, C.; Jiang, L.; Huang, M. Structural Basis of Covalent Inhibitory Mechanism of TMPRSS2-Related Serine Proteases by Camostat. J. Virol. 2021, 95, e00861–e00821. [Google Scholar] [CrossRef] [PubMed]
- Hempel, T.; Raich, L.; Olsson, S.; Azouz, N.P.; Klingler, A.M.; Hoffmann, M.; Pöhlmann, S.; Rothenberg, M.E.; Noé, F. Molecular mechanism of inhibiting the SARS-CoV-2 cell entry facilitator TMPRSS2 with camostat and nafamostat. Chem. Sci. 2021, 12, 983–992. [Google Scholar] [CrossRef]
- Fujimoto, K.J.; Nema, D.; Ninomiya, M.; Koketsu, M.; Sadanari, H.; Takemoto, M.; Daikoku, T.; Murayama, T. An in silico-designed flavone derivative, 6-fluoro-4′-hydroxy-3′,5′-dimethoxyflavone, prevents replication of human cytomegalovirus in infected cells stronger than ganciclovir. Antivir. Res. 2018, 154, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Mardirossian, N.; Wang, Y.; Pearlman, D.A.; Chan, G.K.-L.; Shiozaki, T. Novel algorithms and high-performance cloud computing enable efficient fully quantum mechanical protein-ligand scoring. arXiv 2020, arXiv:2004.08725. [Google Scholar]
- Wang, Y.; Murlidaran, S.; Pearlman, D.A. Quantum simulations of SARS-CoV-2 main protease Mpro enable high-quality scoring of diverse ligands. J. Comput. Aided Mol. Des. 2021, 35, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Uehara, S.; Fujimoto, K.J.; Tanaka, S. Protein-ligand docking using fitness learning-based artificial bee colony with proximity stimuli. Phys. Chem. Chem. Phys. 2015, 17, 16412–16417. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar]
- Schrödinger Release 2021–3. Glide; Schrödinger: New York, NY, USA, 2021. [Google Scholar]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian16; Rev. C.01; Gaussian Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Murata, T.; Komoto, S.; Iwahori, S.; Sasaki, J.; Nishitsuji, H.; Hasebe, T.; Hoshinaga, K.; Yuzawa, Y. Reduction of severe acute respiratory syndrome coronavirus-2 infectivity by admissible concentration of ozone gas and water. Microbiol. Immunol. 2021, 65, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; Shirato, K.; van der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous Treatment of Human Bronchial Epithelial Cells with Serine and Cysteine Protease Inhibitors Prevents Severe Acute Respiratory Syndrome Coronavirus Entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, P.W.; Murakami, K.; Inagami, T. Specificity and Mechanism of Clostripain Catalysis. Biochemistry 1971, 10, 4246–4252. [Google Scholar] [CrossRef] [PubMed]
- Pacaud, M. Protease II from Escherichia coli. Substrate Specificity and Kinetic Properties. Eur. J. Biochem. 1978, 82, 439–451. [Google Scholar]
- Sethia, R.; Prasad, M.; Mahapatra, S.J.; Nischal, N.; Soneja, M.; Garg, P.; Shalimar. Efficacy of Famotidine for COVID-19: A Systematic Review and Meta-analysis. medRxiv 2020. [Google Scholar] [CrossRef]
- Nichi-Iko Pharmaceutical Co.Ltd. Pharmaceutical Interview Form for FUTHAN 10 INJ., FUTHAN 50 INJ. 2020. Available online: https://www.nichiiko.co.jp/medicine/file/31050/interview/31050_interview.pdf (accessed on 10 January 2022).
- Loffredo, M.; Lucero, H.; Chen, D.Y.; O’Connell, A.; Bergqvist, S.; Munawar, A.; Bandara, A.; Graef, S.D.; Weeks, S.D.; Douam, F.; et al. The in-vitro effect of famotidine on SARS-CoV-2 proteases and virus replication. Sci. Rep. 2021, 11, 5433. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | EBind1 |
|---|---|
| Nafamostat | –178.22 |
| Camostat | –128.84 |
| 1 | –172.38 |
| 2 | –174.16 |
| 3 | –178.94 |
| 4 | –127.92 |
| 5 | –179.65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujimoto, K.J.; Hobbs, D.C.F.; Umeda, M.; Nagata, A.; Yamaguchi, R.; Sato, Y.; Sato, A.; Ohmatsu, K.; Ooi, T.; Yanai, T.; et al. In Silico Analysis and Synthesis of Nafamostat Derivatives and Evaluation of Their Anti-SARS-CoV-2 Activity. Viruses 2022, 14, 389. https://doi.org/10.3390/v14020389
Fujimoto KJ, Hobbs DCF, Umeda M, Nagata A, Yamaguchi R, Sato Y, Sato A, Ohmatsu K, Ooi T, Yanai T, et al. In Silico Analysis and Synthesis of Nafamostat Derivatives and Evaluation of Their Anti-SARS-CoV-2 Activity. Viruses. 2022; 14(2):389. https://doi.org/10.3390/v14020389
Chicago/Turabian StyleFujimoto, Kazuhiro J., Daniel C. F. Hobbs, Miki Umeda, Akihiro Nagata, Rie Yamaguchi, Yoshitaka Sato, Ayato Sato, Kohsuke Ohmatsu, Takashi Ooi, Takeshi Yanai, and et al. 2022. "In Silico Analysis and Synthesis of Nafamostat Derivatives and Evaluation of Their Anti-SARS-CoV-2 Activity" Viruses 14, no. 2: 389. https://doi.org/10.3390/v14020389