

Neutralizing Antibodies Limit Cell-Associated Spread of Human Cytomegalovirus in Epithelial Cells and Fibroblasts

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Antibodies
2.3. Plaque Reduction Assay (PRA)
2.4. Statistical Analysis
3. Results
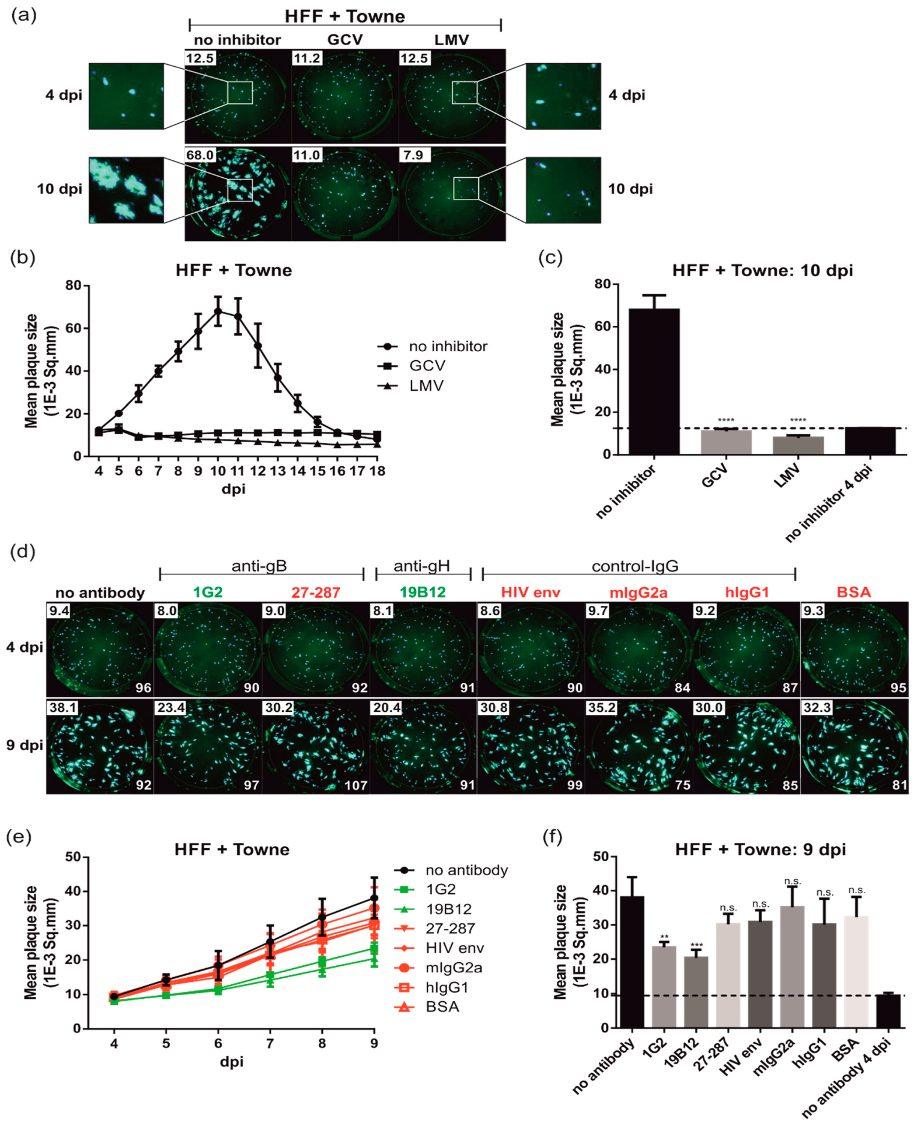
3.1. Establishment of a Live Cell Plaque Reduction Assay (PRA) for Quantitative Measurement of HCMV Spread
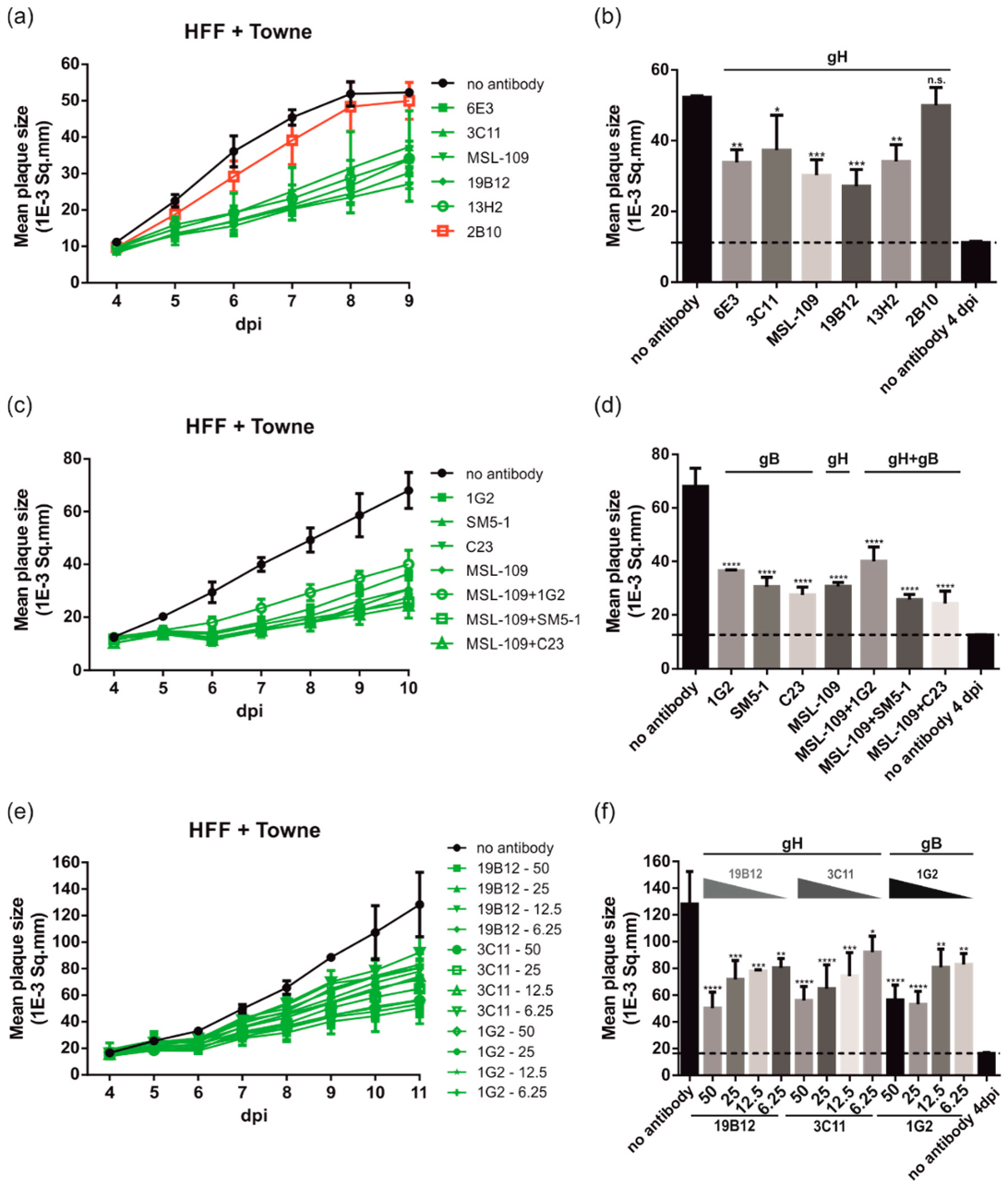
3.2. Neutralizing Antibodies to gB and gH Significantly Inhibit HCMV Spread in Fibroblasts
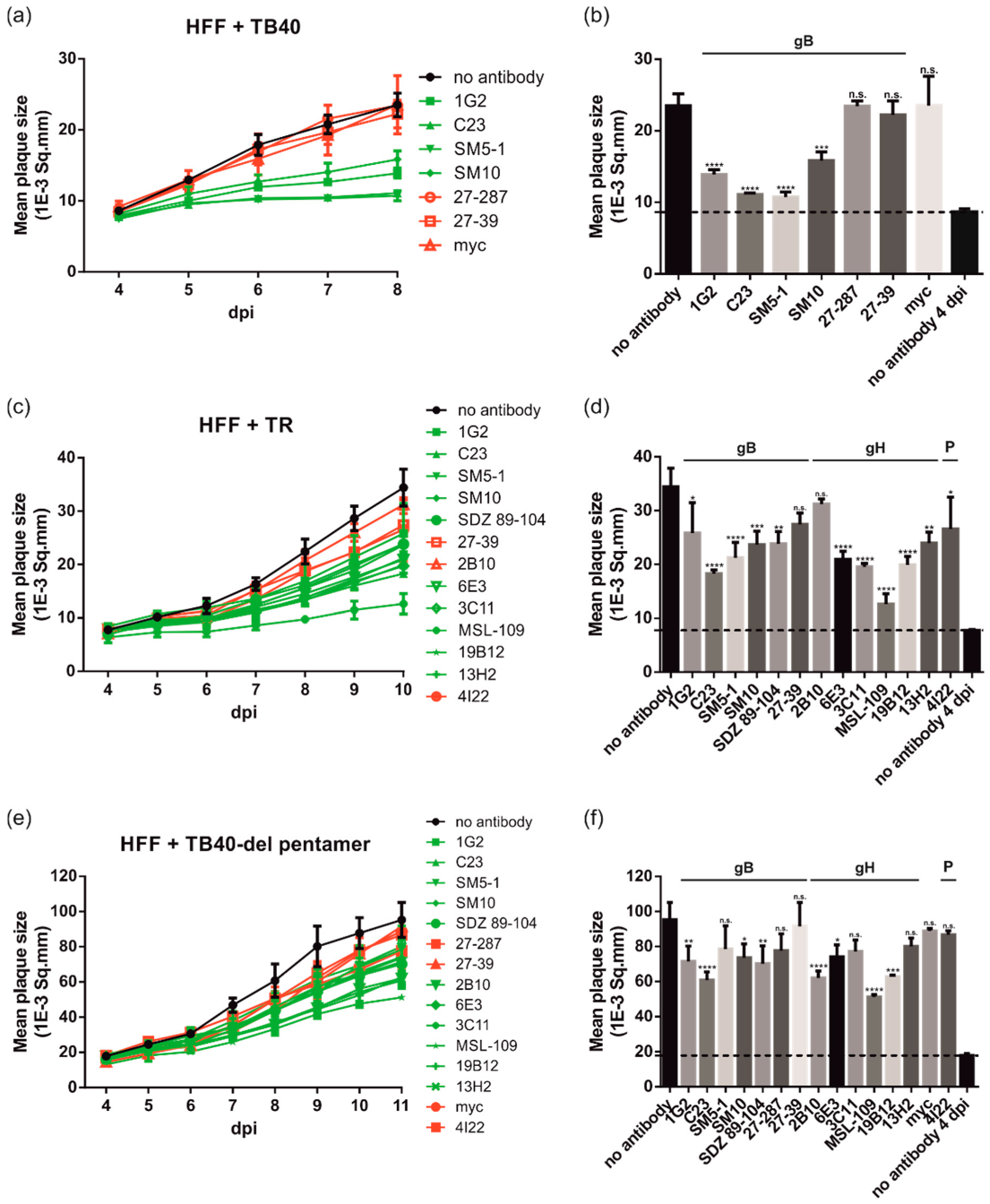
3.3. gB and gH mAbs Limit Spread of Diverse HCMV Strains in Fibroblasts
3.4. gB and gH mAbs Limit HCMV Spread Efficiently in Both Fibroblasts and Epithelial Cells
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the worldwide seroprevalence of cytomegalovirus: A systematic review and meta-analysis. Rev. Med. Virol. 2019, 29, e2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manicklal, S.; Emery, V.C.; Lazzarotto, T.; Boppana, S.B.; Gupta, R.K. The “silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 2013, 26, 86–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreher, A.M.; Arora, N.; Fowler, K.B.; Novak, Z.; Britt, W.J.; Boppana, S.B.; Ross, S.A. Spectrum of disease and outcome in children with symptomatic congenital cytomegalovirus infection. J. Pediatr. 2014, 164, 855–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanan, P.; Razonable, R.R. Cytomegalovirus infections in solid organ transplantation: A review. Infect. Chemother. 2013, 45, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Teira, P.; Battiwalla, M.; Ramanathan, M.; Barrett, A.J.; Ahn, K.W.; Chen, M.; Green, J.S.; Saad, A.; Antin, J.H.; Savani, B.N.; et al. Early cytomegalovirus reactivation remains associated with increased transplant-related mortality in the current era: A CIBMTR analysis. Blood 2016, 127, 2427–2438. [Google Scholar] [CrossRef]
- Chou, S. Approach to drug-resistant cytomegalovirus in transplant recipients. Curr. Opin. Infect. Dis. 2015, 28, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Arvin, A.M.; Fast, P.; Myers, M.; Plotkin, S.; Rabinovich, R.; National Vaccine Advisory, C. Vaccine development to prevent cytomegalovirus disease: Report from the National Vaccine Advisory Committee. Clin. Infect. Dis. 2004, 39, 233–239. [Google Scholar]
- Fowler, K.B.; Stagno, S.; Pass, R.F. Maternal immunity and prevention of congenital cytomegalovirus infection. JAMA 2003, 289, 1008–1011. [Google Scholar] [CrossRef] [Green Version]
- Schoppel, K.; Schmidt, C.; Einsele, H.; Hebart, H.; Mach, M. Kinetics of the antibody response against human cytomegalovirus-specific proteins in allogeneic bone marrow transplant recipients. J. Infect. Dis. 1998, 178, 1233–1243. [Google Scholar] [CrossRef]
- Nigro, G.; Adler, S.P.; La Torre, R.; Best, A.M.; Congenital Cytomegalovirus Collaborating, G. Passive immunization during pregnancy for congenital cytomegalovirus infection. N. Engl. J. Med. 2005, 353, 1350–1362. [Google Scholar] [CrossRef] [Green Version]
- Nigro, G.; Adler, S.P.; Parruti, G.; Anceschi, M.M.; Coclite, E.; Pezone, I.; Di Renzo, G.C. Immunoglobulin therapy of fetal cytomegalovirus infection occurring in the first half of pregnancy-a case-control study of the outcome in children. J. Infect. Dis. 2012, 205, 215–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, K.O.; Enders, M.; Schampera, M.S.; Baeumel, E.; Hoopmann, M.; Geipel, A.; Berg, C.; Goelz, R.; De Catte, L.; Wallwiener, D.; et al. Prevention of maternal-fetal transmission of cytomegalovirus after primary maternal infection in the first trimester by biweekly hyperimmunoglobulin administration. Ultrasound Obstet. Gynecol. 2019, 53, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; Safdar, N. Polyclonal immunoglobulins and hyperimmune globulins in prevention and management of infectious diseases. Infect. Dis. Clin. N. Am. 2011, 25, 773–788. [Google Scholar] [CrossRef] [PubMed]
- Maidji, E.; Nigro, G.; Tabata, T.; McDonagh, S.; Nozawa, N.; Shiboski, S.; Muci, S.; Anceschi, M.M.; Aziz, N.; Adler, S.P.; et al. Antibody treatment promotes compensation for human cytomegalovirus-induced pathogenesis and a hypoxia-like condition in placentas with congenital infection. Am. J. Pathol. 2010, 177, 1298–1310. [Google Scholar] [CrossRef] [PubMed]
- Snydman, D.R.; Werner, B.G.; Heinze-Lacey, B.; Berardi, V.P.; Tilney, N.L.; Kirkman, R.L.; Milford, E.L.; Cho, S.I.; Bush, H.L., Jr.; Levey, A.S.; et al. Use of cytomegalovirus immune globulin to prevent cytomegalovirus disease in renal-transplant recipients. N. Engl. J. Med. 1987, 317, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Bonaros, N.; Mayer, B.; Schachner, T.; Laufer, G.; Kocher, A. CMV-hyperimmune globulin for preventing cytomegalovirus infection and disease in solid organ transplant recipients: A meta-analysis. Clin. Transplant. 2008, 22, 89–97. [Google Scholar] [CrossRef]
- Fisher, R.A.; Kistler, K.D.; Ulsh, P.; Bergman, G.E.; Morris, J. The association between cytomegalovirus immune globulin and long-term recipient and graft survival following liver transplantation. Transplant. Infect. Dis. 2012, 14, 121–131. [Google Scholar] [CrossRef]
- Klenovsek, K.; Weisel, F.; Schneider, A.; Appelt, U.; Jonjic, S.; Messerle, M.; Bradel-Tretheway, B.; Winkler, T.H.; Mach, M. Protection from CMV infection in immunodeficient hosts by adoptive transfer of memory B cells. Blood 2007, 110, 3472–3479. [Google Scholar] [CrossRef]
- Cekinovic, D.; Golemac, M.; Pugel, E.P.; Tomac, J.; Cicin-Sain, L.; Slavuljica, I.; Bradford, R.; Misch, S.; Winkler, T.H.; Mach, M.; et al. Passive immunization reduces murine cytomegalovirus-induced brain pathology in newborn mice. J. Virol. 2008, 82, 12172–12180. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.S.; Cruz, D.V.; Tran, D.; Bialas, K.M.; Stamper, L.; Wu, H.; Gilbert, M.; Blair, R.; Alvarez, X.; Itell, H.; et al. Preexisting antibodies can protect against congenital cytomegalovirus infection in monkeys. JCI Insight 2017, 2, e94002. [Google Scholar] [CrossRef] [Green Version]
- Auerbach, M.R.; Yan, D.; Vij, R.; Hongo, J.A.; Nakamura, G.; Vernes, J.M.; Meng, Y.G.; Lein, S.; Chan, P.; Ross, J.; et al. A neutralizing anti-gH/gL monoclonal antibody is protective in the guinea pig model of congenital CMV infection. PLoS Pathog. 2014, 10, e1004060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bootz, A.; Karbach, A.; Spindler, J.; Kropff, B.; Reuter, N.; Sticht, H.; Winkler, T.H.; Britt, W.J.; Mach, M. Protective capacity of neutralizing and non-neutralizing antibodies against glycoprotein B of cytomegalovirus. PLoS Pathog. 2017, 13, e1006601. [Google Scholar] [CrossRef] [PubMed]
- Sandonis, V.; Garcia-Rios, E.; McConnell, M.J.; Perez-Romero, P. Role of Neutralizing Antibodies in CMV Infection: Implications for New Therapeutic Approaches. Trends Microbiol. 2020, 28, 900–912. [Google Scholar] [CrossRef] [PubMed]
- Seedah, E.A.; Frye, Z.P.; Maynard, J.A. Immunotherapeutic Approaches to Prevent Cytomegalovirus-Mediated Disease. Microbiol. Spectr. 2014, 2, AID-0009-2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, J.H.; Patel, A.; Mehta, A.K.; Gatault, P.; McBride, J.M.; Burgess, T.; Derby, M.A.; Snydman, D.R.; Emu, B.; Feierbach, B.; et al. Phase 2 Randomized, Double-Blind, Placebo-Controlled Trial of RG7667, a Combination Monoclonal Antibody, for Prevention of Cytomegalovirus Infection in High-Risk Kidney Transplant Recipients. Antimicrob. Agents Chemother. 2017, 61, e01794-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, R.S.; Heldwein, E.E. Herpesvirus gB: A Finely Tuned Fusion Machine. Viruses 2015, 7, 6552–6569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Wisner, T.W.; Johnson, D.C.; Heldwein, E.E. HCMV gB shares structural and functional properties with gB proteins from other herpesviruses. Virology 2013, 435, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.C.; Kamil, J.P. Pathogen at the Gates: Human Cytomegalovirus Entry and Cell Tropism. Viruses 2018, 10, 704. [Google Scholar] [CrossRef] [Green Version]
- Burke, H.G.; Heldwein, E.E. Crystal Structure of the Human Cytomegalovirus Glycoprotein B. PLoS Pathog. 2015, 11, e1005227. [Google Scholar] [CrossRef] [Green Version]
- Adler, B.; Scrivano, L.; Ruzcics, Z.; Rupp, B.; Sinzger, C.; Koszinowski, U. Role of human cytomegalovirus UL131A in cell type-specific virus entry and release. J. Gen. Virol. 2006, 87 Pt 9, 2451–2460. [Google Scholar] [CrossRef]
- Ryckman, B.J.; Jarvis, M.A.; Drummond, D.D.; Nelson, J.A.; Johnson, D.C. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 2006, 80, 710–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, G.; Revello, M.G.; Patrone, M.; Percivalle, E.; Campanini, G.; Sarasini, A.; Wagner, M.; Gallina, A.; Milanesi, G.; Koszinowski, U.; et al. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 2004, 78, 10023–10033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerna, G.; Percivalle, E.; Lilleri, D.; Lozza, L.; Fornara, C.; Hahn, G.; Baldanti, F.; Revello, M.G. Dendritic-cell infection by human cytomegalovirus is restricted to strains carrying functional UL131-128 genes and mediates efficient viral antigen presentation to CD8+ T cells. J. Gen. Virol. 2005, 86 Pt 2, 275–284. [Google Scholar] [CrossRef]
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158. [Google Scholar] [CrossRef] [Green Version]
- Mach, M.; Wiegers, A.K.; Spindler, N.; Winkler, T.; Reddehase, M.J. Protective humoral immunity. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M., Ed.; Caister Academic Press: Norfolk, UK, 2013; pp. 215–231. [Google Scholar]
- Zydek, M.; Petitt, M.; Fang-Hoover, J.; Adler, B.; Kauvar, L.M.; Pereira, L.; Tabata, T. HCMV infection of human trophoblast progenitor cells of the placenta is neutralized by a human monoclonal antibody to glycoprotein B and not by antibodies to the pentamer complex. Viruses 2014, 6, 1346–1364. [Google Scholar] [CrossRef]
- Macagno, A.; Bernasconi, N.L.; Vanzetta, F.; Dander, E.; Sarasini, A.; Revello, M.G.; Gerna, G.; Sallusto, F.; Lanzavecchia, A. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J. Virol. 2010, 84, 1005–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Freed, D.C.; Wang, D.; Qiu, P.; Li, F.; Fu, T.M.; Kauvar, L.M.; McVoy, M.A. Impact of Antibodies and Strain Polymorphisms on Cytomegalovirus Entry and Spread in Fibroblasts and Epithelial Cells. J. Virol. 2017, 91, e01650-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattentau, Q. Avoiding the void: Cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 2008, 6, 815–826. [Google Scholar] [CrossRef]
- Jackson, J.W.; Sparer, T. There Is Always Another Way! Cytomegalovirus’ Multifaceted Dissemination Schemes. Viruses 2018, 10, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowden, R.A.; Slichter, S.J.; Sayers, M.; Weisdorf, D.; Cays, M.; Schoch, G.; Banaji, M.; Haake, R.; Welk, K.; Fisher, L.; et al. A comparison of filtered leukocyte-reduced and cytomegalovirus (CMV) seronegative blood products for the prevention of transfusion-associated CMV infection after marrow transplant. Blood 1995, 86, 3598–3603. [Google Scholar] [CrossRef] [Green Version]
- Hamprecht, K.; Steinmassl, M.; Einsele, H.; Jahn, G. Discordant detection of human cytomegalovirus DNA from peripheral blood mononuclear cells, granulocytes and plasma: Correlation to viremia and HCMV infection. J. Clin. Virol. 1998, 11, 125–136. [Google Scholar] [CrossRef]
- Sinzger, C.; Schmidt, K.; Knapp, J.; Kahl, M.; Beck, R.; Waldman, J.; Hebart, H.; Einsele, H.; Jahn, G. Modification of human cytomegalovirus tropism through propagation in vitro is associated with changes in the viral genome. J. Gen. Virol. 1999, 80 Pt 11, 2867–2877. [Google Scholar] [CrossRef]
- Yamane, Y.; Furukawa, T.; Plotkin, S.A. Supernatant virus release as a differentiating marker between low passage and vaccine strains of human cytomegalovirus. Vaccine 1983, 1, 23–25. [Google Scholar] [CrossRef]
- Dargan, D.J.; Douglas, E.; Cunningham, C.; Jamieson, F.; Stanton, R.J.; Baluchova, K.; McSharry, B.P.; Tomasec, P.; Emery, V.C.; Percivalle, E.; et al. Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J. Gen. Virol. 2010, 91 Pt 6, 1535–1546. [Google Scholar] [CrossRef]
- Murrell, I.; Bedford, C.; Ladell, K.; Miners, K.L.; Price, D.A.; Tomasec, P.; Wilkinson, G.W.G.; Stanton, R.J. The pentameric complex drives immunologically covert cell-cell transmission of wild-type human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2017, 114, 6104–6109. [Google Scholar] [CrossRef] [Green Version]
- Krmpotic, A.; Podlech, J.; Reddehase, M.J.; Britt, W.J.; Jonjic, S. Role of antibodies in confining cytomegalovirus after reactivation from latency: Three decades’ resume. Med. Microbiol. Immunol. 2019, 208, 415–429. [Google Scholar] [CrossRef]
- Wirtz, N.; Schader, S.I.; Holtappels, R.; Simon, C.O.; Lemmermann, N.A.; Reddehase, M.J.; Podlech, J. Polyclonal cytomegalovirus-specific antibodies not only prevent virus dissemination from the portal of entry but also inhibit focal virus spread within target tissues. Med. Microbiol. Immunol. 2008, 197, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Lee, R.; Adler, S.P.; McVoy, M.A. Antibody inhibition of human cytomegalovirus spread in epithelial cell cultures. J. Virol. Methods 2013, 192, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.J.; Adler, B.; Sampaio, K.L.; Digel, M.; Jahn, G.; Ettischer, N.; Stierhof, Y.D.; Scrivano, L.; Koszinowski, U.; Mach, M.; et al. UL74 of human cytomegalovirus contributes to virus release by promoting secondary envelopment of virions. J. Virol. 2008, 82, 2802–2812. [Google Scholar] [CrossRef] [Green Version]
- Sinzger, C.; Mangin, M.; Weinstock, C.; Topp, M.S.; Hebart, H.; Einsele, H.; Jahn, G. Effect of serum and CTL on focal growth of human cytomegalovirus. J. Clin. Virol. 2007, 38, 112–119. [Google Scholar] [CrossRef]
- Scrivano, L.; Sinzger, C.; Nitschko, H.; Koszinowski, U.H.; Adler, B. HCMV spread and cell tropism are determined by distinct virus populations. PLoS Pathog. 2011, 7, e1001256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiuppesi, F.; Wussow, F.; Johnson, E.; Bian, C.; Zhuo, M.; Rajakumar, A.; Barry, P.A.; Britt, W.J.; Chakraborty, R.; Diamond, D.J. Vaccine-Derived Neutralizing Antibodies to the Human Cytomegalovirus gH/gL Pentamer Potently Block Primary Cytotrophoblast Infection. J. Virol. 2015, 89, 11884–11898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilleri, D.; Kabanova, A.; Revello, M.G.; Percivalle, E.; Sarasini, A.; Genini, E.; Sallusto, F.; Lanzavecchia, A.; Corti, D.; Gerna, G. Fetal human cytomegalovirus transmission correlates with delayed maternal antibodies to gH/gL/pUL128-130-131 complex during primary infection. PLoS ONE 2013, 8, e59863. [Google Scholar]
- Jacob, C.L.; Lamorte, L.; Sepulveda, E.; Lorenz, I.C.; Gauthier, A.; Franti, M. Neutralizing antibodies are unable to inhibit direct viral cell-to-cell spread of human cytomegalovirus. Virology 2013, 444, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Navarro, D.; Paz, P.; Tugizov, S.; Topp, K.; La Vail, J.; Pereira, L. Glycoprotein B of human cytomegalovirus promotes virion penetration into cells, transmission of infection from cell to cell, and fusion of infected cells. Virology 1993, 197, 143–158. [Google Scholar] [CrossRef]
- Frenzel, K.; Ganepola, S.; Michel, D.; Thiel, E.; Kruger, D.H.; Uharek, L.; Hofmann, J. Antiviral function and efficacy of polyvalent immunoglobulin products against CMV isolates in different human cell lines. Med. Microbiol. Immunol. 2012, 201, 277–286. [Google Scholar] [CrossRef]
- Gerna, G.; Sarasini, A.; Patrone, M.; Percivalle, E.; Fiorina, L.; Campanini, G.; Gallina, A.; Baldanti, F.; Revello, M.G. Human cytomegalovirus serum neutralizing antibodies block virus infection of endothelial/epithelial cells, but not fibroblasts, early during primary infection. J. Gen. Virol. 2008, 89 Pt 4, 853–865. [Google Scholar] [CrossRef]
- Marchini, A.; Liu, H.Q.; Zhu, H. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J. Virol. 2001, 75, 1870–1878. [Google Scholar] [CrossRef] [Green Version]
- Sampaio, K.L.; Weyell, A.; Subramanian, N.; Wu, Z.G.; Sinzger, C. A TB40/E-derived human cytomegalovirus genome with an intact US-gene region and a self-excisable BAC cassette for immunological research. Biotechniques 2017, 63, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Collins-McMillen, D.; Caposio, P.; Yurochko, A.D. Viral binding-induced signaling drives a unique and extended intracellular trafficking pattern during infection of primary monocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 8819–8824. [Google Scholar] [CrossRef] [Green Version]
- Meyer, H.; Masuho, Y.; Mach, M. The gp116 of the gp58/116 complex of human cytomegalovirus represents the amino-terminal part of the precursor molecule and contains a neutralizing epitope. J. Gen. Virol. 1990, 71 Pt 10, 2443–2450. [Google Scholar] [CrossRef] [PubMed]
- Wiegers, A.K.; Sticht, H.; Winkler, T.H.; Britt, W.J.; Mach, M. Identification of a neutralizing epitope within antigenic domain 5 of glycoprotein B of human cytomegalovirus. J. Virol. 2015, 89, 361–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potzsch, S.; Spindler, N.; Wiegers, A.K.; Fisch, T.; Rucker, P.; Sticht, H.; Grieb, N.; Baroti, T.; Weisel, F.; Stamminger, T.; et al. B cell repertoire analysis identifies new antigenic domains on glycoprotein B of human cytomegalovirus which are target of neutralizing antibodies. PLoS Pathog. 2011, 7, e1002172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, B.; Kropff, B.; Kalbacher, H.; Britt, W.; Sundqvist, V.A.; Ostberg, L.; Mach, M. A continuous sequence of more than 70 amino acids is essential for antibody binding to the dominant antigenic site of glycoprotein gp58 of human cytomegalovirus. J. Virol. 1992, 66, 5290–5297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britt, W.J.; Vugler, L.G. Oligomerization of the human cytomegalovirus major envelope glycoprotein complex gB (gp55-116). J. Virol. 1992, 66, 6747–6754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoppel, K.; Hassfurther, E.; Britt, W.; Ohlin, M.; Borrebaeck, C.A.; Mach, M. Antibodies specific for the antigenic domain 1 of glycoprotein B (gpUL55) of human cytomegalovirus bind to different substructures. Virology 1996, 216, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Nokta, M.; Tolpin, M.D.; Nadler, P.I.; Pollard, R.B. Human monoclonal anti-cytomegalovirus (CMV) antibody (MSL 109): Enhancement of in vitro foscarnet- and ganciclovir-induced inhibition of CMV replication. Antiviral Res. 1994, 24, 17–26. [Google Scholar] [CrossRef]
- Thomas, M.; Kropff, B.; Schneider, A.; Winkler, T.H.; Gorzer, I.; Sticht, H.; Britt, W.J.; Mach, M.; Reuter, N. A novel strain-specific neutralizing epitope on glycoprotein H of Human Cytomegalovirus. J. Virol. 2021, JVI0065721. [Google Scholar] [CrossRef]
- Spindler, N.; Diestel, U.; Stump, J.D.; Wiegers, A.K.; Winkler, T.H.; Sticht, H.; Mach, M.; Muller, Y.A. Structural basis for the recognition of human cytomegalovirus glycoprotein B by a neutralizing human antibody. PLoS Pathog. 2014, 10, e1004377. [Google Scholar] [CrossRef]
- Spindler, N.; Rucker, P.; Potzsch, S.; Diestel, U.; Sticht, H.; Martin-Parras, L.; Winkler, T.H.; Mach, M. Characterization of a discontinuous neutralizing epitope on glycoprotein B of human cytomegalovirus. J. Virol. 2013, 87, 8927–8939. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.C.; Schroer, J.; Shenk, T. Human cytomegalovirus cell-to-cell spread in the absence of an essential assembly protein. Proc. Natl. Acad. Sci. USA 2005, 102, 2081–2086. [Google Scholar] [CrossRef] [Green Version]
- Sinzger, C.; Knapp, J.; Plachter, B.; Schmidt, K.; Jahn, G. Quantification of replication of clinical cytomegalovirus isolates in cultured endothelial cells and fibroblasts by a focus expansion assay. J. Virol. Methods 1997, 63, 103–112. [Google Scholar] [CrossRef]
- Murphy, E.; Yu, D.; Grimwood, J.; Schmutz, J.; Dickson, M.; Jarvis, M.A.; Hahn, G.; Nelson, J.A.; Myers, R.M.; Shenk, T.E. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2003, 100, 14976–14981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.H.; Adler, S.P.; Schleiss, M.R.; Arav-Boger, R.; Harrison, G.J.D.; Mcvoy, M.A. Cytomegalovirus Virions Shed in Urine Have a Reversible Block to Epithelial Cell Entry and Are Highly Resistant to Antibody Neutralization. Clin. Vaccine Immunol. 2017, 24, e00024-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, E.P.; Lanchy, J.M.; Day, L.Z.; Yu, Q.; Peterson, C.; Preece, J.; Ryckman, B.J. Specialization for Cell-Free or Cell-to-Cell Spread of BAC-Cloned Human Cytomegalovirus Strains Is Determined by Factors beyond the UL128-131 and RL13 Loci. J. Virol. 2020, 94, e00034-20. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.P.; Grover, J.R.; Tolbert, W.D.; Prevost, J.; Richard, J.; Ding, S.; Baril, S.; Medjahed, H.; Evans, D.T.; Pazgier, M.; et al. Antibody-Induced Internalization of HIV-1 Env Proteins Limits Surface Expression of the Closed Conformation of Env. J. Virol. 2019, 93, e00293-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvis, M.A.; Fish, K.N.; Soderberg-Naucler, C.; Streblow, D.N.; Meyers, H.L.; Thomas, G.; Nelson, J.A. Retrieval of human cytomegalovirus glycoprotein B from cell surface is not required for virus envelopment in astrocytoma cells. J. Virol. 2002, 76, 5147–5155. [Google Scholar] [CrossRef] [Green Version]
- Leemans, A.; De Schryver, M.; Van der Gucht, W.; Heykers, A.; Pintelon, I.; Hotard, A.L.; Moore, M.L.; Melero, J.A.; McLellan, J.S.; Graham, B.S.; et al. Antibody-Induced Internalization of the Human Respiratory Syncytial Virus Fusion Protein. J. Virol. 2017, 91, e00184-17. [Google Scholar] [CrossRef] [Green Version]
- Maertens, J.; Logan, A.C.; Jang, J.; Long, G.; Tang, J.L.; Hwang, W.Y.K.; Koh, L.P.; Chemaly, R.; Gerbitz, A.; Winkler, J.; et al. Phase 2 Study of Anti-Human Cytomegalovirus Monoclonal Antibodies for Prophylaxis in Hematopoietic Cell Transplantation. Antimicrob. Agents Chemother. 2020, 64, e02467-19. [Google Scholar] [CrossRef] [Green Version]
- Pass, R.F.; Zhang, C.; Evans, A.; Simpson, T.; Andrews, W.; Huang, M.L.; Corey, L.; Hill, J.; Davis, E.; Flanigan, C.; et al. Vaccine prevention of maternal cytomegalovirus infection. N. Engl. J. Med. 2009, 360, 1191–1199. [Google Scholar] [CrossRef]
- Griffiths, P.D.; Stanton, A.; McCarrell, E.; Smith, C.; Osman, M.; Harber, M.; Davenport, A.; Jones, G.; Wheeler, D.C.; O’Beirne, J.; et al. Cytomegalovirus glycoprotein-B vaccine with MF59 adjuvant in transplant recipients: A phase 2 randomised placebo-controlled trial. Lancet 2011, 377, 1256–1263. [Google Scholar] [CrossRef] [Green Version]
- Reuter, N.; Kropff, B.; Schneiderbanger, J.K.; Alt, M.; Krawczyk, A.; Sinzger, C.; Winkler, T.H.; Britt, W.J.; Mach, M.; Thomas, M. Cell Fusion Induced by a Fusion-Active Form of Human Cytomegalovirus Glycoprotein B (gB) Is Inhibited by Antibodies Directed at Antigenic Domain 5 in the Ectodomain of gB. J. Virol. 2020, 94, e01276-20. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Meza, B.P.; Adler, S.P.; McVoy, M.A. Cytomegalovirus vaccines fail to induce epithelial entry neutralizing antibodies comparable to natural infection. Vaccine 2008, 26, 5760–5766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, C.S.; Huffman, T.; Jenks, J.A.; Cisneros de la Rosa, E.; Xie, G.; Vandergrift, N.; Pass, R.F.; Pollara, J.; Permar, S.R. HCMV glycoprotein B subunit vaccine efficacy mediated by nonneutralizing antibody effector functions. Proc. Natl. Acad. Sci. USA 2018, 115, 6267–6272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baraniak, I.; Kropff, B.; Ambrose, L.; McIntosh, M.; McLean, G.R.; Pichon, S.; Atkinson, C.; Milne, R.S.B.; Mach, M.; Griffiths, P.D.; et al. Protection from cytomegalovirus viremia following glycoprotein B vaccination is not dependent on neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2018, 115, 6273–6278. [Google Scholar] [CrossRef] [Green Version]
- Murrell, I.; Tomasec, P.; Wilkie, G.S.; Dargan, D.J.; Davison, A.J.; Stanton, R.J. Impact of sequence variation in the UL128 locus on production of human cytomegalovirus in fibroblast and epithelial cells. J. Virol. 2013, 87, 10489–10500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cudini, J.; Roy, S.; Houldcroft, C.J.; Bryant, J.M.; Depledge, D.P.; Tutill, H.; Veys, P.; Williams, R.; Worth, A.J.J.; Tamuri, A.U.; et al. Human cytomegalovirus haplotype reconstruction reveals high diversity due to superinfection and evidence of within-host recombination. Proc. Natl. Acad. Sci. USA 2019, 116, 5693–5698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassalle, F.; Depledge, D.P.; Reeves, M.B.; Brown, A.C.; Christiansen, M.T.; Tutill, H.J.; Williams, R.J.; Einer-Jensen, K.; Holdstock, J.; Atkinson, C.; et al. Islands of linkage in an ocean of pervasive recombination reveals two-speed evolution of human cytomegalovirus genomes. Virus Evol. 2016, 2, vew017. [Google Scholar] [CrossRef]
- Renzette, N.; Gibson, L.; Bhattacharjee, B.; Fisher, D.; Schleiss, M.R.; Jensen, J.D.; Kowalik, T.F. Rapid intrahost evolution of human cytomegalovirus is shaped by demography and positive selection. PLoS Genet. 2013, 9, e1003735. [Google Scholar] [CrossRef] [Green Version]
- Suarez, N.M.; Wilkie, G.S.; Hage, E.; Camiolo, S.; Holton, M.; Hughes, J.; Maabar, M.; Vattipally, S.B.; Dhingra, A.; Gompels, U.A.; et al. Human Cytomegalovirus Genomes Sequenced Directly From Clinical Material: Variation, Multiple-Strain Infection, Recombination, and Gene Loss. J. Infect. Dis. 2019, 220, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Galitska, G.; Biolatti, M.; De Andrea, M.; Leone, A.; Coscia, A.; Bertolotti, L.; Ala, U.; Bertino, E.; Dell’Oste, V.; Landolfo, S. Biological relevance of Cytomegalovirus genetic variability in congenitally and postnatally infected children. J. Clin. Virol. 2018, 108, 132–140. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Target | Immunogen | Species | Reference |
|---|---|---|---|---|
| Neutralizing | ||||
| 1G2 | gB/AD-5 | Natural infection | Human | [63,64] |
| SM5-1 | gB/AD-4 | Natural infection | Human | [64,70,71] |
| SM10 | gB/AD-5 | Natural infection | Human | [63,64] |
| C23 | gB/AD-2 | Natural infection | Human | [62] |
| SDZ 89-104 | gB/AD-1 | Natural infection | Human | [65] |
| 2B10 | gH | AD169 | Mouse | [69] |
| 6E3 | gH | AD169 | Mouse | [69] |
| 3C11 | gH | AD169 | Mouse | [69] |
| 19B12 | gH | TB40/E | Mouse | [unpub.] |
| 13H2 | gH | TB40/E | Mouse | [unpub.] |
| MSL-109 | gH | Natural infection | Human | [68] |
| 4I22 | pUL128/130/131 | Natural infection | Human | [37] |
| Non-neutralizing | ||||
| 27-287 | gB/AD-1 | AD169 | Mouse | [67] |
| 27-39 | gB/AD-1 | AD169 | Mouse | [66] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reuter, N.; Kropff, B.; Britt, W.J.; Mach, M.; Thomas, M. Neutralizing Antibodies Limit Cell-Associated Spread of Human Cytomegalovirus in Epithelial Cells and Fibroblasts. Viruses 2022, 14, 284. https://doi.org/10.3390/v14020284
Reuter N, Kropff B, Britt WJ, Mach M, Thomas M. Neutralizing Antibodies Limit Cell-Associated Spread of Human Cytomegalovirus in Epithelial Cells and Fibroblasts. Viruses. 2022; 14(2):284. https://doi.org/10.3390/v14020284
Chicago/Turabian StyleReuter, Nina, Barbara Kropff, William J. Britt, Michael Mach, and Marco Thomas. 2022. "Neutralizing Antibodies Limit Cell-Associated Spread of Human Cytomegalovirus in Epithelial Cells and Fibroblasts" Viruses 14, no. 2: 284. https://doi.org/10.3390/v14020284