
New Insights into Avian Infectious Bronchitis Virus in Colombia from Whole-Genome Analysis
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
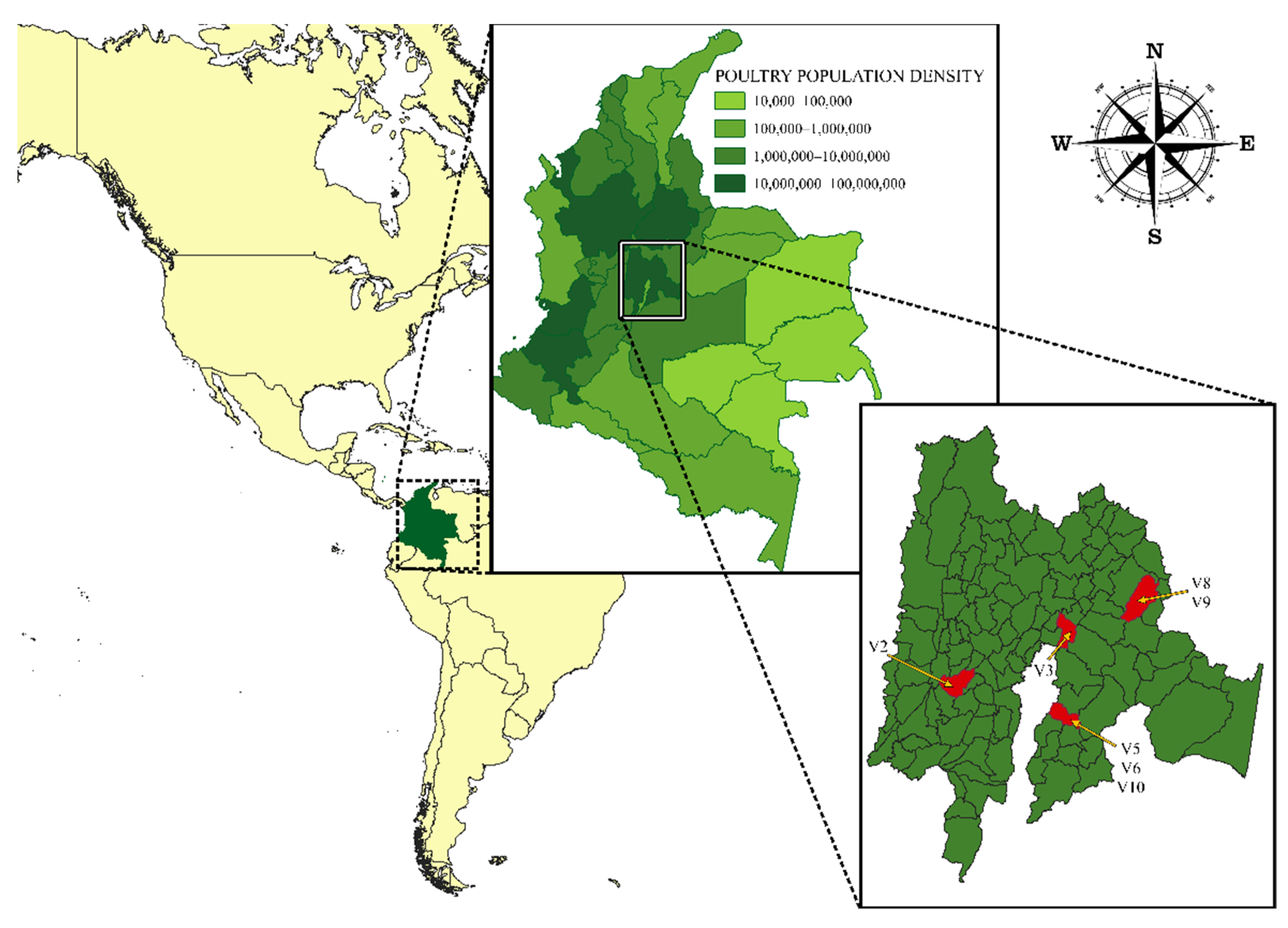
2.1. Biological Samples and Virus Isolation
2.2. RNA Extraction, cDNA Synthesis, and PCR Amplification
2.3. Illumina Sequencing and Genome Assembly
2.4. IBV Maximum Likelihood Phylogenetic Analyses
2.5. Open Reading Frames (ORFs) Prediction and Recombination Analysis
3. Results
3.1. Strains and Genome Sequencing
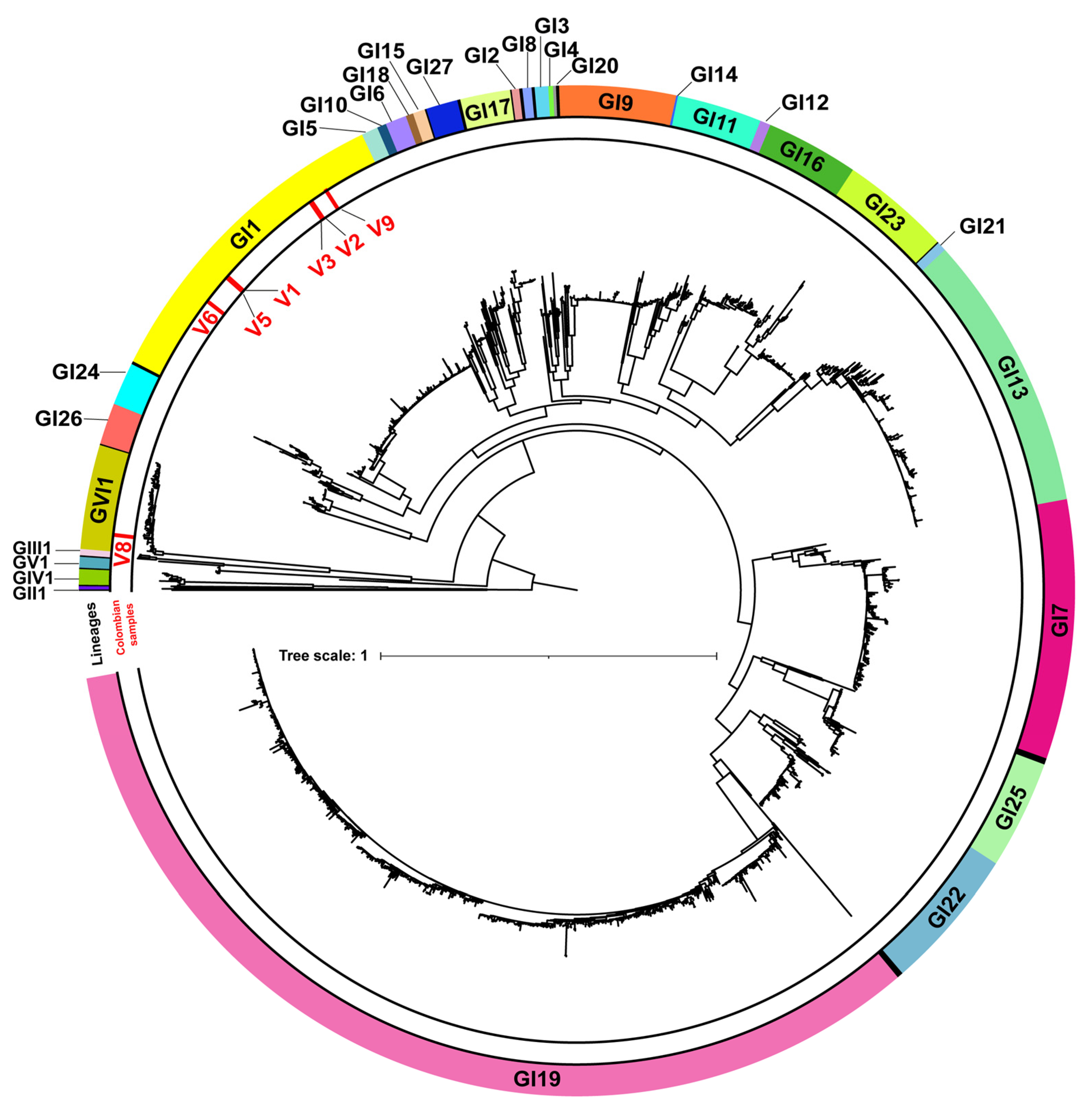
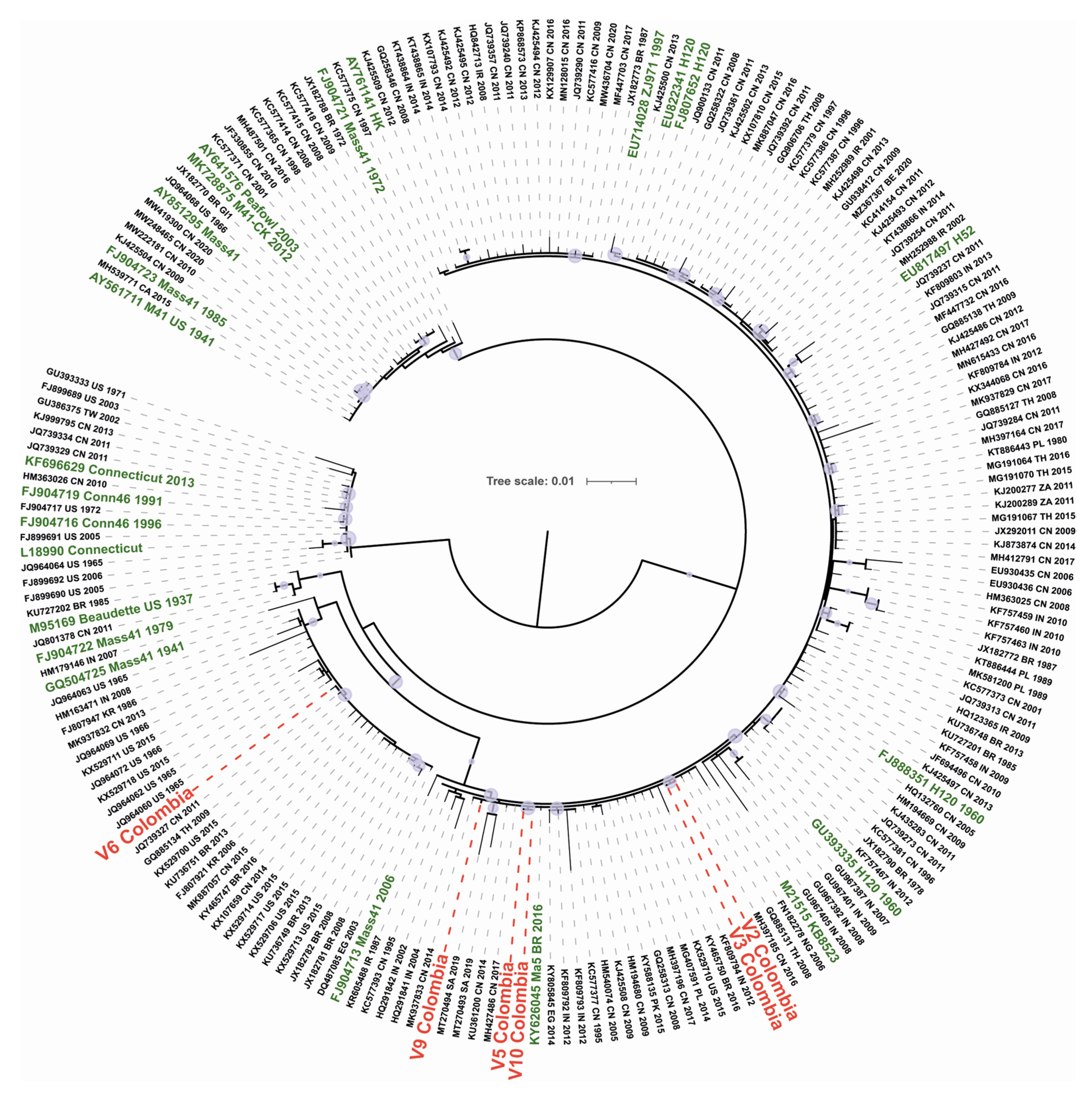
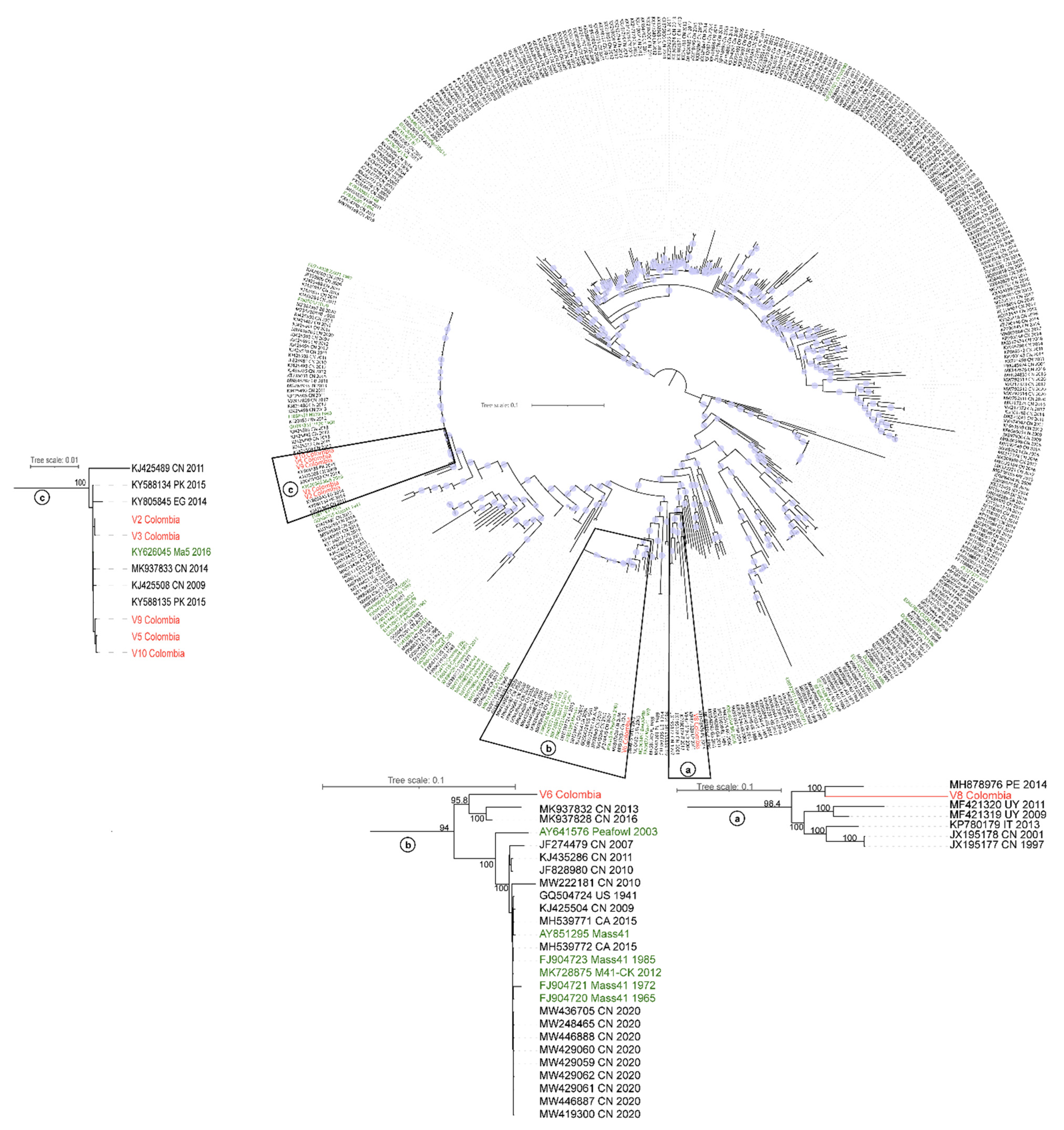
3.2. Phylogenetic Analysis of IBV
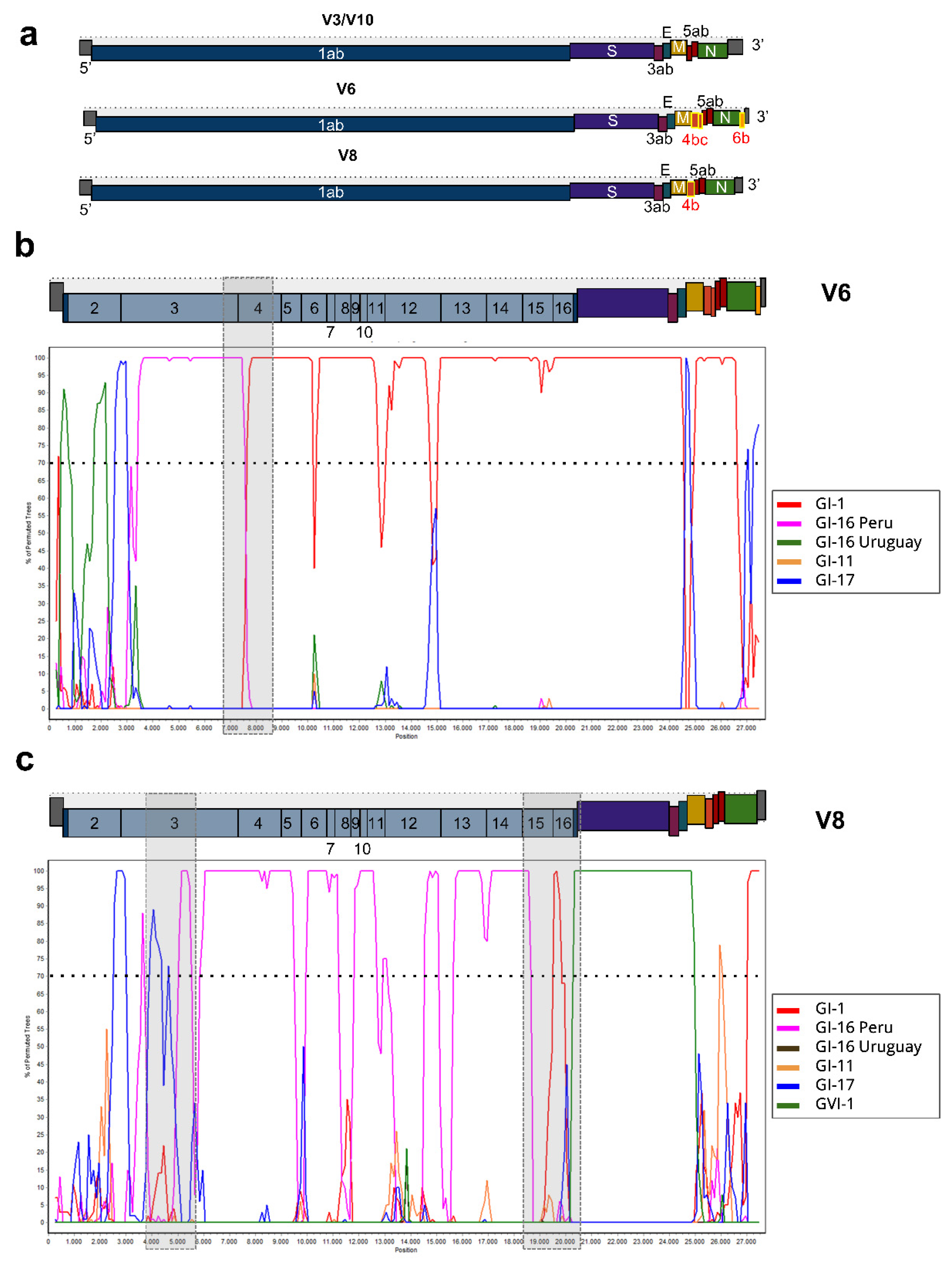
3.3. ORF Prediction and Recombination Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Cavanagh, D. Coronavirus Avian Infectious Bronchitis Virus. Vet. Res. 2007, 38, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Boursnell, M.E.; Brown, T.D.; Foulds, I.J.; Green, P.F.; Tomley, F.M.; Binns, M.M. Completion of the Sequence of the Genome of the Coronavirus Avian Infectious Bronchitis Virus. J. Gen. Virol. 1987, 68 Pt 1, 57–77. [Google Scholar] [CrossRef]
- Lin, S.Y.; Chen, H.W. Infectious Bronchitis Virus Variants: Molecular Analysis and Pathogenicity Investigation. Int. J. Mol. Sci. 2017, 18, 2030. [Google Scholar] [CrossRef] [Green Version]
- Jackwood, M.W.; de Wit, J.J. Infectious Bronchitis. In Diseases of Poultry; Swayne, D.E., Ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2019; pp. 167–188. [Google Scholar]
- Valastro, V.; Holmes, E.C.; Britton, P.; Fusaro, A.; Jackwood, M.W.; Cattoli, G.; Monne, I. S1 Gene-Based Phylogeny of Infectious Bronchitis Virus: An Attempt to Harmonize Virus Classification. Infect. Genet. Evol. 2016, 39, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, M.; Sáenz, L.; Hidalgo, H. Molecular and Antigenic Characterization of GI-13 and GI-16 Avian Infectious Bronchitis Virus Isolated in Chile from 2009 to 2017 Regarding 4/91 Vaccine Introduction. Animals 2019, 9, 656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marandino, A.; Tomás, G.; Panzera, Y.; Greif, G.; Parodi-Talice, A.; Hernández, M.; Techera, C.; Hernández, D.; Pérez, R. Whole-Genome Characterization of Uruguayan Strains of Avian Infectious Bronchitis Virus Reveals Extensive Recombination between the Two Major South American Lineages. Infect. Genet. Evol. 2017, 54, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Tataje-Lavanda, L.; Izquierdo-Lara, R.; Ormeño-Vásquez, P.; Huamán-Gutiérrez, K.; Zimic-Peralta, M.; Fernández-Díaz, M. Near-Complete Genome Sequence of Infectious Bronchitis Virus Strain VFAR-047 (GI-16 Lineage), Isolated in Peru. Microbiol. Resour. Announc. 2019, 8, e01555-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Fraga, A.P.; Gräf, T.; Pereira, C.S.; Ikuta, N.; Fonseca, A.S.K.; Lunge, V.R. Phylodynamic Analysis and Molecular Diversity of the Avian Infectious Bronchitis Virus of Chickens in Brazil. Infect. Genet. Evol. 2018, 61, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Marandino, A.; Pérez, R. Genetic and Antigenic Diversity of Infectious Bronchitis Virus in South America. Avian Dis. 2021, 65, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Bande, F.; Arshad, S.S.; Omar, A.R.; Hair-Bejo, M.; Mahmuda, A.; Nair, V. Global Distributions and Strain Diversity of Avian Infectious Bronchitis Virus: A Review. Anim. Health Res. Rev. 2017, 18, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, M.W.; Boynton, T.O.; Hilt, D.A.; McKinley, E.T.; Kissinger, J.C.; Paterson, A.H.; Robertson, J.; Lemke, C.; McCall, A.W.; Williams, S.M.; et al. Emergence of a Group 3 Coronavirus through Recombination. Virology 2010, 398, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Zhang, M.; Tian, X.; Shao, H.; Qian, K.; Ye, J.; Qin, A. Identification of a Novel Recombinant Virulent Avian Infectious Bronchitis Virus. Vet. Microbiol. 2017, 199, 120–127. [Google Scholar] [CrossRef]
- Luque Forero, G.; Yepez, M.S.; Morales, A. Aislamiento Del Virus de La Bronquitis Infecciosa de Las Aves En Colombia—Dialnet. Rev. Fac. Med. Vet. Zootécnia 1963, 26, 1024–1027. [Google Scholar]
- ICA Medicamentos y Biológicos. Available online: https://www.ica.gov.co/areas/pecuaria/servicios/regulacion-y-control-de-medicamentos-veterinarios.aspx (accessed on 4 October 2020).
- Córdoba Argoti, G.; Vera Alfonso, V.J.; Correa Jaime, J.; Ramírez Nieto, G.C. Comportamiento Del Virus de La Bronquitis Infecciosa Aviar En Aves Con Sintomatología Respiratoria Provenientes de Granjas de Producción Del Departamento de Cundinamarca. Nova 2015, 13, 47. [Google Scholar] [CrossRef] [Green Version]
- Alvarado, I.R.; Villegas, P.; Mossos, N.; Jackwood, M.W. Molecular Characterization of Avian Infectious Bronchitis Virus Strains Isolated in Colombia during 2003. Avian Dis. 2005, 49, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes-Rincón, A.; Lopes, P.D.; Sanmiguel, R.A. Genotipificación de Variantes Del Virus de Bronquitis Infecciosa Aviar En El Departamento Del Tolima, Colombia. Rev. MVZ Córdoba 2016, 21, 5500–5510. [Google Scholar] [CrossRef] [Green Version]
- Santana-Clavijo, N.F.; Brandão, P.E. Emergence of Avian Coronavirus Genotype GI-11 in Colombia. Braz. J. Microbiol. 2021, 52, 455–459. [Google Scholar] [CrossRef]
- Villalobos-Agüero, R.A.; Ramírez-Carvajal, L.; Zamora-Sanabria, R.; León, B.; Karkashian-Córdoba, J. Molecular Characterization of an Avian GA13-like Infectious Bronchitis Virus Full-Length Genome from Costa Rica. Virus Dis. 2021, 32, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Marandino, A.; Pereda, A.; Tomás, G.; Hernández, M.; Iraola, G.; Craig, M.I.; Hernaández, D.; Banda, A.; Villegas, P.; Panzera, Y.; et al. Phylodynamic Analysis of Avian Infectious Bronchitis Virus in South America. J. Gen. Virol. 2015, 96, 1340–1346. [Google Scholar] [CrossRef] [Green Version]
- Cordoba Argoti, G.M. Detección Molecular e Intento de Aislamiento del Virus de La Bronquitis Infecciosa Aviar (Coronavirus) en Explotaciones Avícolas del Departamento de Cundinamarca; Universidad Nacional de Colombia: Bogotá, Colombia, 2015. [Google Scholar]
- Williams, A.K.; Wang, L.; Sneed, L.W.; Collisson, E.W. Analysis of a Hypervariable Region in the 3’ Non-Coding End of the Infectious Bronchitis Virus Genome. Virus Res. 1993, 28, 19–27. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P. The Sequence Manipulation Suite: JavaScript Programs for Analyzing and Formatting Protein and DNA Sequences. Biotechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandão, P.E.; Taniwaki, S.A.; Berg, M.; Hora, A.S. Complete Genome of Avian Coronavirus Vaccine Strains Ma5 and BR-I. Genome Announc. 2017, 5, e00201-17. [Google Scholar] [CrossRef] [Green Version]
- Davelaar, F.G.; Kouwenhoven, B.; Burger, A.G. Occurrence and Significance of Infectious Bronchitis Virus Variant Strains in Egg and Broiler Production in the Netherlands. Vet. Q. 1984, 6, 114–120. [Google Scholar] [CrossRef]
- Cortés, V.; Sevilla-Navarro, S.; García, C.; Marín, C.; Catalá-Gregori, P. Seroprevalence and Prevalence of Infectious Bronchitis Virus in Broilers, Laying Hens and Broiler Breeders in Spain. Poult. Sci. 2022, 101, 101760. [Google Scholar] [CrossRef]
- Youn, S.-Y.; Lee, J.-Y.; Bae, Y.-C.; Kwon, Y.-K.; Kim, H.-R. Genetic and Pathogenic Characterization of QX(GI-19)-Recombinant Infectious Bronchitis Viruses in South Korea. Viruses 2021, 13, 1163. [Google Scholar] [CrossRef]
- Gallardo, R.A.; da Silva, A.P.; Gilbert, R.; Alfonso, M.; Conley, A.; Jones, K.; Stayer, P.A.; Hoerr, F.J. Testicular Atrophy and Epididymitis-Orchitis Associated with Infectious Bronchitis Virus in Broiler Breeder Roosters. Avian Dis. 2022, 66, 112–118. [Google Scholar] [CrossRef]
- Cavanagh, D.; Mawditt, K.; Britton, P.; Naylor, C.J. Longitudinal Field Studies of Infectious Bronchitis Virus and Avian Pneumovirus in Broilers Using Type-Specific Polymerase Chain Reactions. Avian Pathol. 1999, 28, 593–605. [Google Scholar] [CrossRef]
- Farsang, A.; Ros, C.; Renström, L.H.M.; Baule, C.; Soós, T.; Belák, S. Molecular Epizootiology of Infectious Bronchitis Virus in Sweden Indicating the Involvement of a Vaccine Strain. Avian Pathol. 2002, 31, 236. [Google Scholar] [CrossRef] [Green Version]
- Ghorbiani, M.; Boroomand, Z.; Mayahi, M.; Seyfi Abad Shapouri, M.R. Molecular Identification of Infectious Bronchitis Virus Isolated from Respiratory Diseases in Some Iranian Broiler Flocks. Mol. Biol. Rep. 2020, 47, 7168. [Google Scholar] [CrossRef]
- Li, L.; Xue, C.; Chen, F.; Qin, J.; Xie, Q.; Bi, Y.; Cao, Y. Isolation and Genetic Analysis Revealed No Predominant New Strains of Avian Infectious Bronchitis Virus Circulating in South China during 2004–2008. Vet. Microbiol. 2010, 143, 145–154. [Google Scholar] [CrossRef]
- Lim, T.-H.; Kim, M.-S.; Jang, J.-H.; Lee, D.-H.; Park, J.-K.; Youn, H.-N.; Lee, J.-B.; Park, S.-Y.; Choi, I.-S.; Song, C.-S. Live Attenuated Nephropathogenic Infectious Bronchitis Virus Vaccine Provides Broad Cross Protection against New Variant Strains. Poult. Sci. 2012, 91, 89–94. [Google Scholar] [CrossRef]
- Mase, M.; Hiramatsu, K.; Watanabe, S.; Iseki, H. Genetic Analysis of the Complete S1 Gene in Japanese Infectious Bronchitis Virus Strains. Viruses 2022, 14, 716. [Google Scholar] [CrossRef]
- Raja, A.; Raj, D.; Kumanan, K. Emergence of Variant Avian Infectious Bronchitis Virus in India. Iran. J. Vet. Res. 2020, 21, 33. [Google Scholar]
- Le, T.B.; Lee, H.-J.; Le, V.P.; Choi, K.-S. Multiple Genotypes of Avian Infectious Bronchitis Virus Circulating in Vietnam. Korean J. Poult. Sci. 2019, 46, 127–136. [Google Scholar] [CrossRef]
- Ren, M.; Sheng, J.; Ma, T.; Xu, L.; Han, Z.; Li, H.; Zhao, Y.; Sun, J.; Liu, S. Molecular and Biological Characteristics of the Infectious Bronchitis Virus TC07-2/GVI-1 Lineage Isolated in China. Infect. Genet. Evol. 2019, 75, 103942. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Listorti, V.; Naylor, C.J.; Lupini, C.; Laconi, A.; Felice, V.; Drigo, M.; Catelli, E.; Cecchinato, M. Molecular Investigation of a Full-Length Genome of a Q1-like IBV Strain Isolated in Italy in 2013. Virus Res. 2015, 210, 77–80. [Google Scholar] [CrossRef]
- Yu, L.; Jiang, Y.; Low, S.; Wang, Z.; Nam, S.J.; Liu, W.; Kwang, J. Characterization of Three Infectious Bronchitis Virus Isolates from China Associated with Proventriculus in Vaccinated Chickens. Avian Dis. 2001, 45, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, G.-H.; Jiang, J.-W.; Fu, J.-D.; Ren, T.; Cao, W.-S.; Xin, C.-A.; Liao, M.; Liu, W.-J. A Massachusetts Prototype like Coronavirus Isolated from Wild Peafowls Is Pathogenic to Chickens. Virus Res. 2007, 130, 121–128. [Google Scholar] [CrossRef]
- Jia, W.; Karaca, K.; Parrish, C.R.; Naqi, S.A. A Novel Variant of Avian Infectious Bronchitis Virus Resulting from Recombination among Three Different Strains. Arch. Virol. 1995, 140, 259–271. [Google Scholar] [CrossRef]
- Xu, G.; Liu, X.Y.; Zhao, Y.; Chen, Y.; Zhao, J.; Zhang, G.Z. Characterization and Analysis of an Infectious Bronchitis Virus Strain Isolated from Southern China in 2013. Virol. J. 2016, 13, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Xu, Y.; Collisson, E.W. Experimental Confirmation of Recombination Upstream of the S1 Hypervariable Region of Infectious Bronchitis Virus. Virus Res. 1997, 49, 139–145. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | HA Titer | Tissue | Productive Unit | Vaccine Strain | Age at Last Vaccination | Time between Isolation and Last Vaccination | Clinical Signs |
|---|---|---|---|---|---|---|---|
| V2 | 1/2048 | Trachea | Breeder | Ma5/H120 | 26 w | 7 w | Respiratory signs and trachea lesions. |
| V3 | 1/2048 | Trachea | Broiler | M48/Ma5 | 16 d | 7 d | Ocular discharge, respiratory signs, and trachea and lung lesions. |
| V5 V6 V10 | 1/4096 1/4096 1/4096 | Tracheal swab | Broiler | Ma5 | 5 d | 15 d | Nasal-ocular discharge, swollen head, and trachea lesions. |
| V8 | 1/256 | Trachea | Broiler | Ma5 | 14 d | 15 d | Ocular discharge and kidney lesions. |
| V9 | 1/4096 | Kidney |
| GenBank Accession Number | Strain Name | Country of Origin | Genotype and Lineage |
|---|---|---|---|
| MH878976 | VFAR-047 | Peru | GI-16 |
| MF421319 | UY/09/CA/01 | Uruguay | GI-16 |
| MF421320 | UY/11/CA/18 | Uruguay | GI-11 |
| MK957245 | PR01 | Brazil | GI-11 |
| MK953937 | Brazil/SP55 | Brazil | GI-11 |
| MK957244 | PR05 | Brazil | GI-11 |
| KX258195 | 23/2013 | Brazil | GI-11 |
| MN757859 | CK/CR/1160/16 | Costa Rica | GI-17 |
| AY85129 | Mass 41 | USA | GI-1 |
| FJ807652 | H120 | USA | GI-1 |
| MK574042 | ck/CH/LHB/110615 | China | GVI-1 |
| Virus | S1 Subunit | Whole Genome | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Nearest Sequence | Genotype-Lineage | Percentage of Identity | Query Cover | Country | Nearest Sequence | Genotype-Lineage | Percentage of Identity | Query Cover | Country | |
| V2 | MH427486 | GI-1 | 99.94 | 100% | China | KY626045 | GI-1 | 99.98 | 100% | Brazil |
| V3 | MH427486 | GI-1 | 99.94 | 100% | China | KY626045 | GI-1 | 99.97 | 99% | Brazil |
| V5 | MH427486 | GI-1 | 99.94 | 100% | China | KY626045 | GI-1 | 99.98 | 100% | Brazil |
| V6 | MK937828 | GI-1 | 99.88 | 100% | China | OM912698 | GI-1 | 96.96 | 99% | Mexico |
| V8 | KC692307 | GVI-1 | 94.51 | 100% | China | MH878976 | GI-11 | 91.53 | 99% | Peru |
| V9 | MH427486 | GI-1 | 99.94 | 100% | China | KY626045 | GI-1 | 99.97 | 100% | Brazil |
| V10 | MH427486 | GI-1 | 99.88 | 100% | China | KY626045 | GI-1 | 99.99 | 99% | Brazil |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramirez-Nieto, G.; Mir, D.; Almansa-Villa, D.; Cordoba-Argotti, G.; Beltran-Leon, M.; Rodriguez-Osorio, N.; Garai, J.; Zabaleta, J.; Gomez, A.P. New Insights into Avian Infectious Bronchitis Virus in Colombia from Whole-Genome Analysis. Viruses 2022, 14, 2562. https://doi.org/10.3390/v14112562
Ramirez-Nieto G, Mir D, Almansa-Villa D, Cordoba-Argotti G, Beltran-Leon M, Rodriguez-Osorio N, Garai J, Zabaleta J, Gomez AP. New Insights into Avian Infectious Bronchitis Virus in Colombia from Whole-Genome Analysis. Viruses. 2022; 14(11):2562. https://doi.org/10.3390/v14112562
Chicago/Turabian StyleRamirez-Nieto, Gloria, Daiana Mir, Diego Almansa-Villa, Geovanna Cordoba-Argotti, Magda Beltran-Leon, Nelida Rodriguez-Osorio, Jone Garai, Jovanny Zabaleta, and Arlen P. Gomez. 2022. "New Insights into Avian Infectious Bronchitis Virus in Colombia from Whole-Genome Analysis" Viruses 14, no. 11: 2562. https://doi.org/10.3390/v14112562