
The Thiazole-5-Carboxamide GPS491 Inhibits HIV-1, Adenovirus, and Coronavirus Replication by Altering RNA Processing/Accumulation

 ,
,  , , ,
, , ,  , , , ,
, , , , 
 add
Show full author list
add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Virus Strains
2.2. GPS491 Inhibition of Virus Replication
2.3. Effect on Viral and Host Protein Expression
2.4. Toxicity Assays
2.5. RNA/DNA Analysis
2.6. Role of Proteasome in Reducing HIV-1 Tat Expression
2.7. Statistical Analysis
3. Results
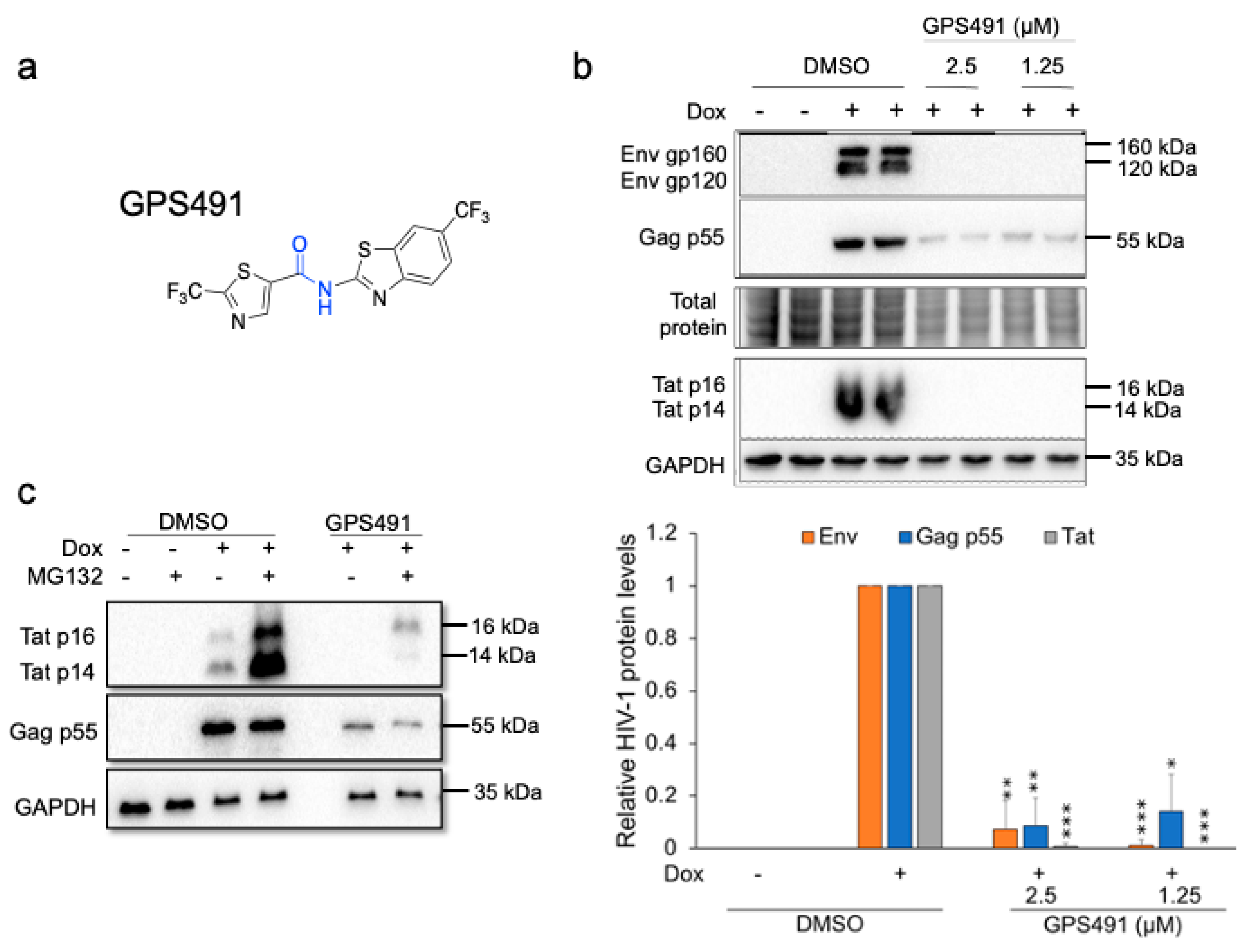
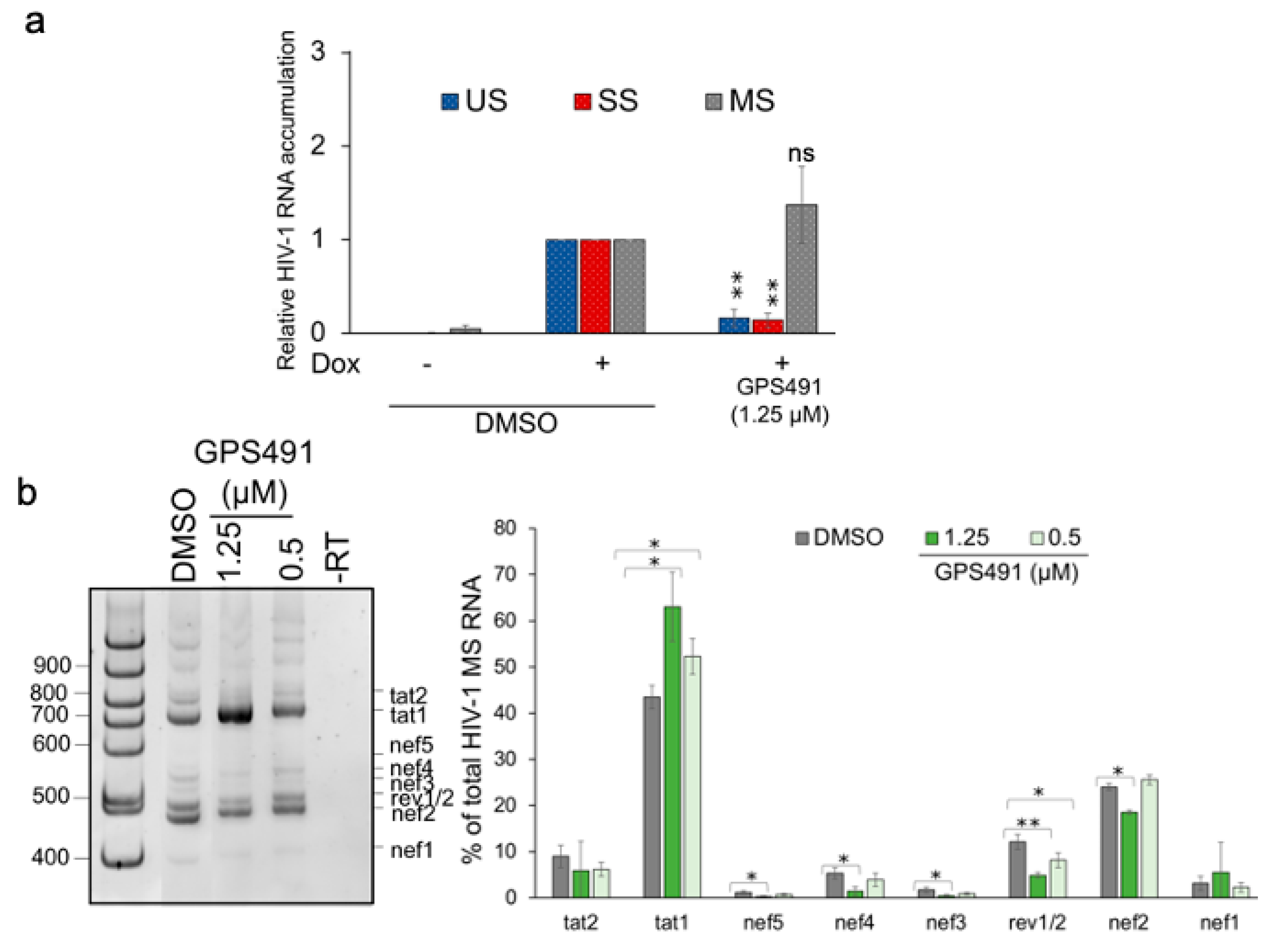
3.1. GPS491 Inhibits HIV-1 Replication by Disrupting Viral RNA Accumulation
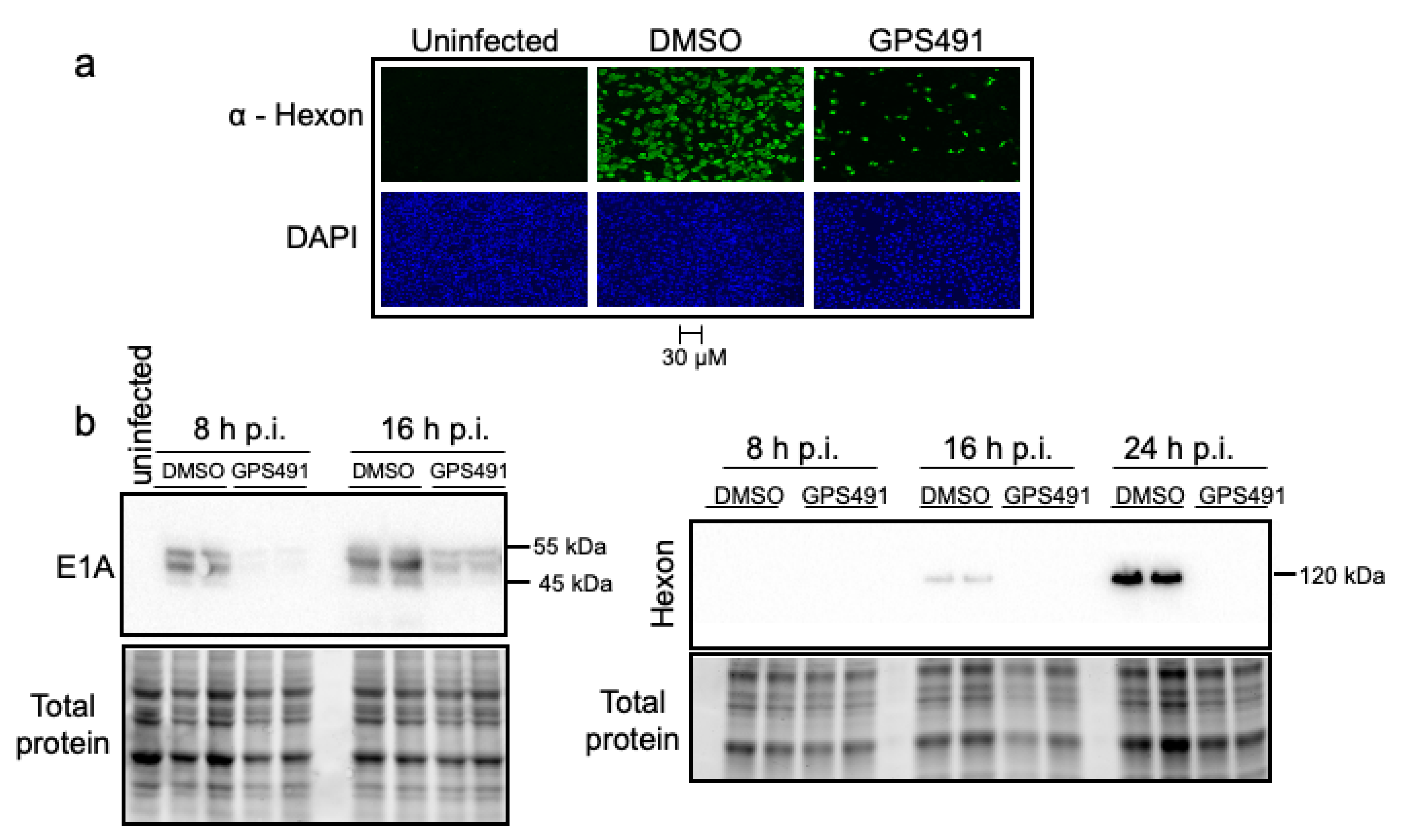
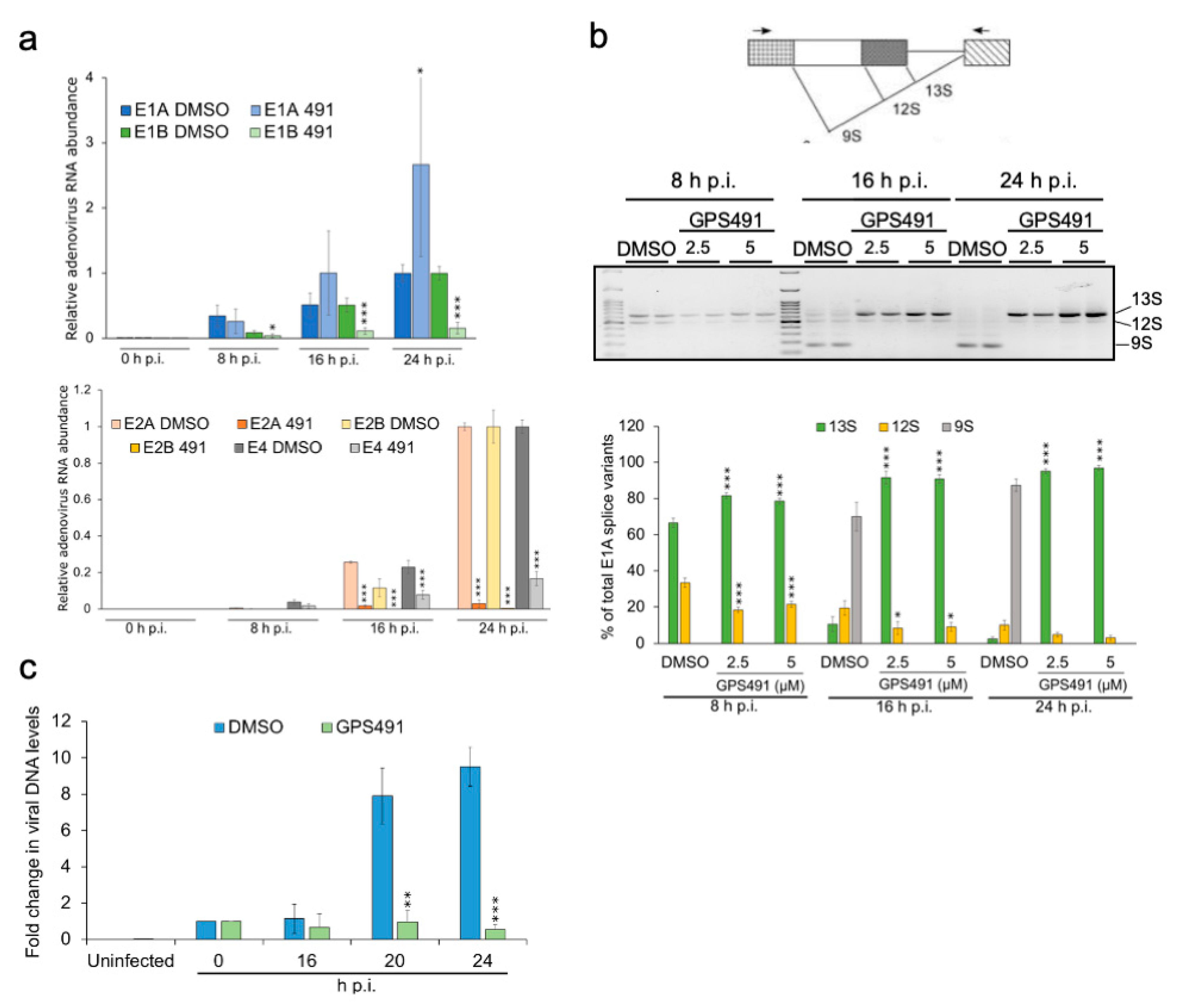
3.2. GPS491 Inhibits Adenovirus Replication
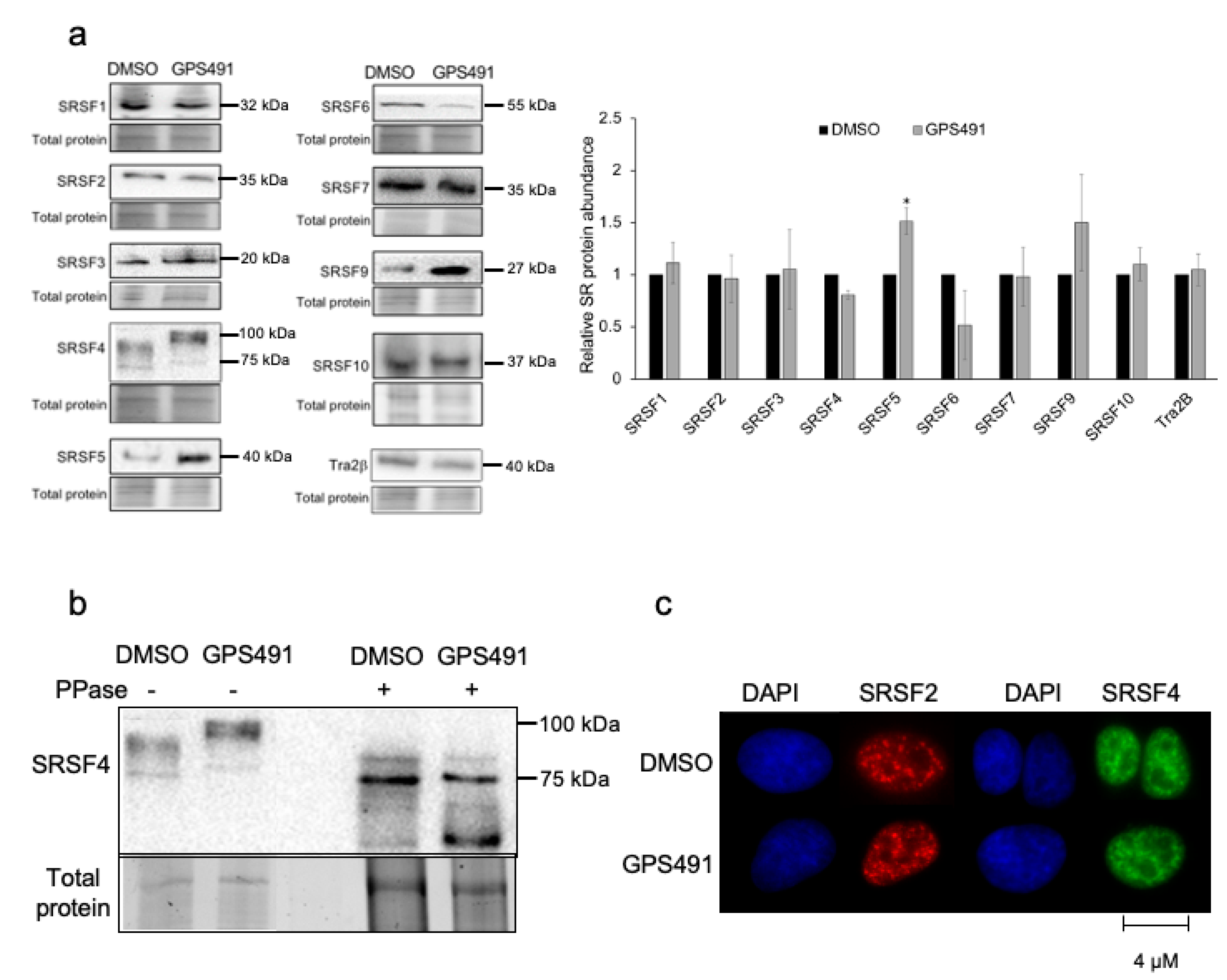
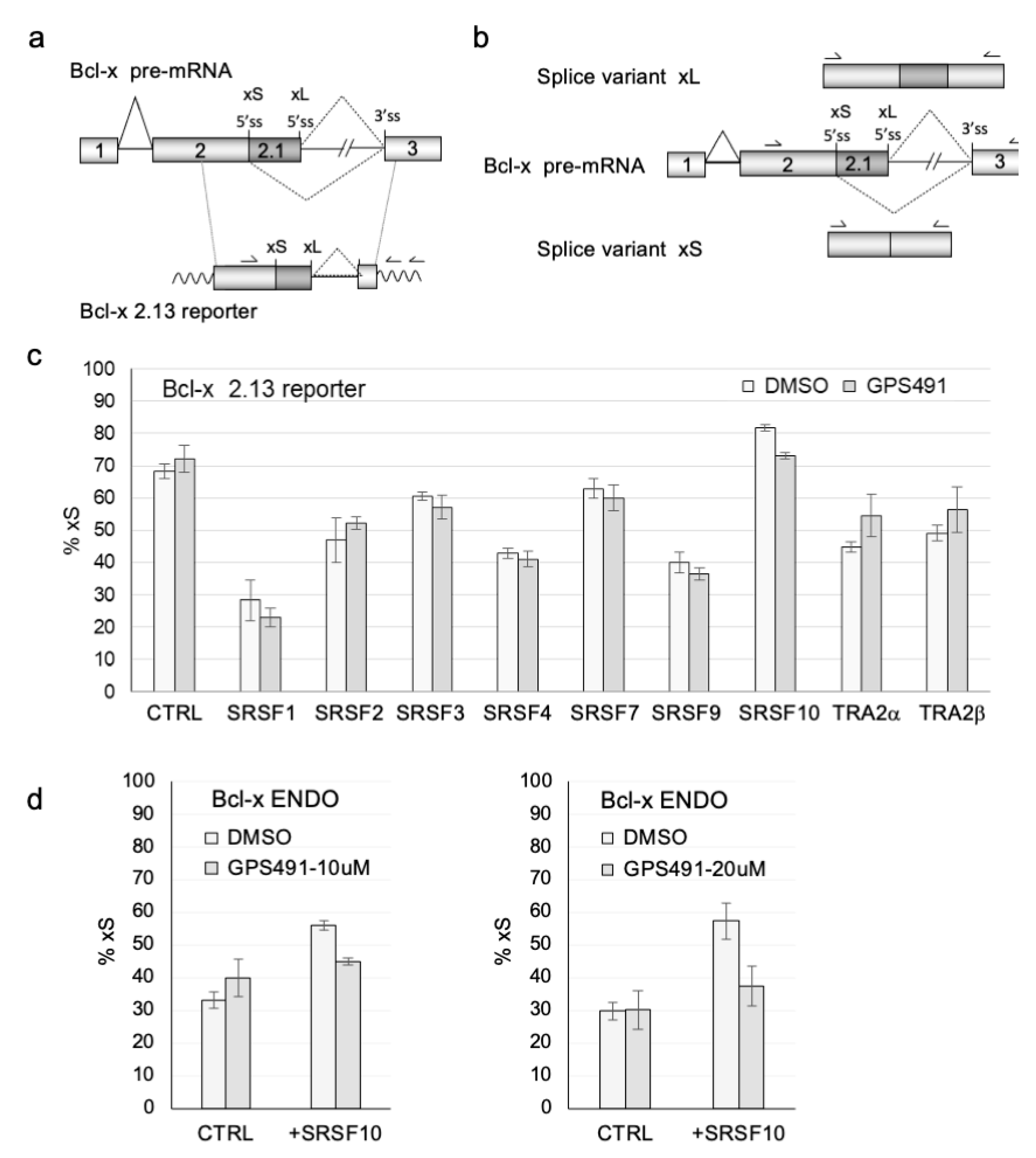
3.3. GPS491 Alters Host SR Protein Abundance, Modification, and Function
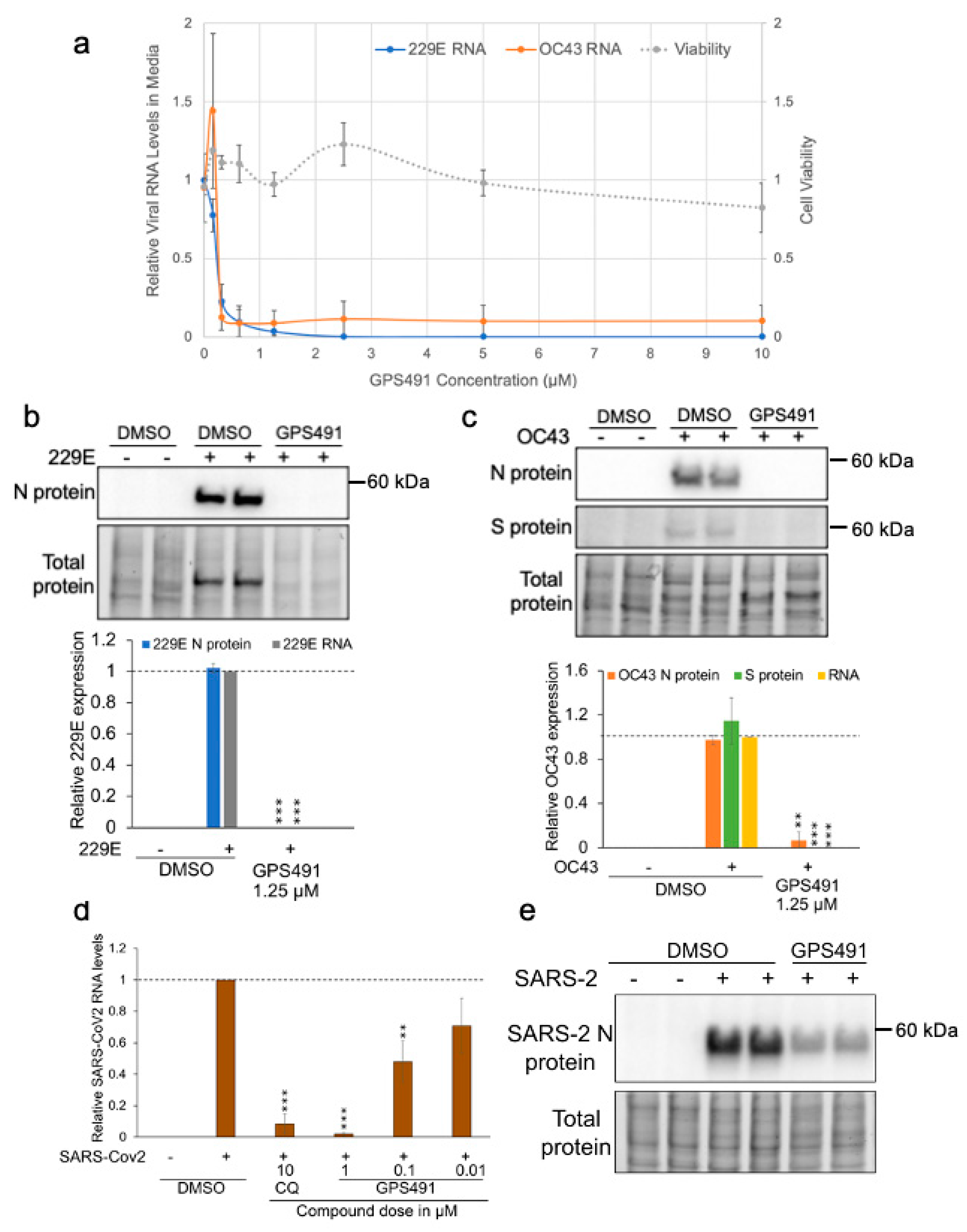
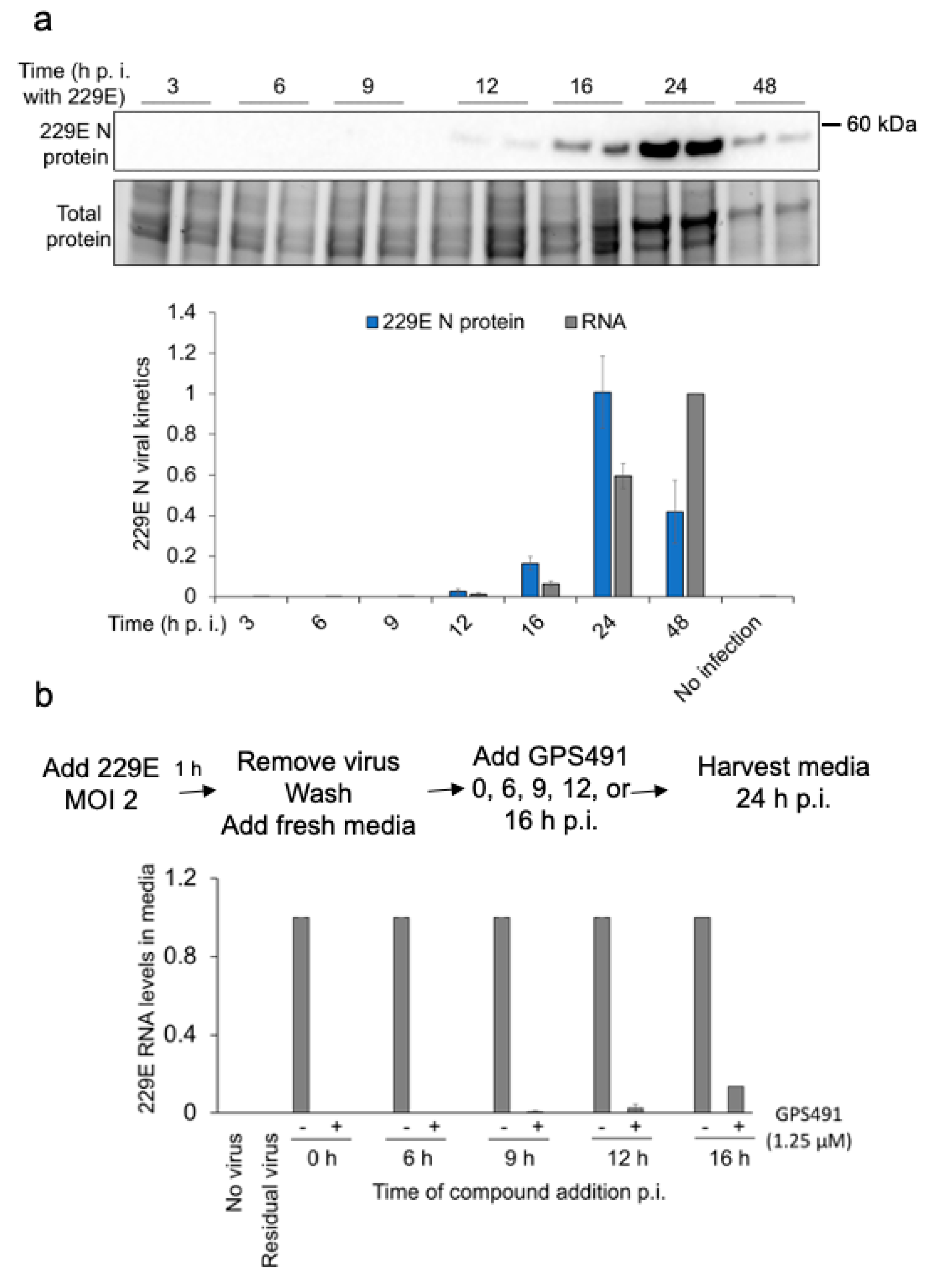
3.4. GPS491 Inhibits Coronavirus Replication
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Chaudhuri, S.; Symons, J.A.; Deval, J. Innovation and trends in the development and approval of antiviral medicines: 1987–2017 and beyond. Antivir. Res. 2018, 155, 76–88. [Google Scholar] [CrossRef]
- Kumar, N.; Sharma, S.; Kumar, R.; Tripathi, B.N.; Barua, S.; Ly, H.; Rouse, B.T. Host-Directed Antiviral Therapy. Clin. Microbiol. Rev. 2020, 33, e00168-19. [Google Scholar] [CrossRef]
- Cakir, M.; Obernier, K.; Forget, A.; Krogan, N.J. Target Discovery for Host-Directed Antiviral Therapies: Application of Proteomics Approaches. Msystems 2021, 6, e00388-21. [Google Scholar] [CrossRef]
- Keener, A.B. Host with the most: Targeting host cells instead of pathogens to fight infectious disease. Nat. Med. 2017, 23, 528–531. [Google Scholar] [CrossRef]
- Prussia, A.; Thepchatri, P.; Snyder, J.P.; Plemper, R.K. Systematic approaches towards the development of host-directed antiviral therapeutics. Int. J. Mol. Sci. 2011, 12, 4027–4052. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, A.; Ming, L.; Cochrane, A. Teetering on the Edge: The Critical Role of RNA Processing Control During HIV-1 Replication. In Retrovirus-Cell Interactions; Academic Press: Cambridge, MA, USA, 2018; pp. 229–257. [Google Scholar]
- Wong, R.W.; Balachandran, A.; Haaland, M.; Stoilov, P.; Cochrane, A. Characterization of novel inhibitors of HIV-1 replication that function via alteration of viral RNA processing and rev function. Nucleic Acids Res. 2013, 41, 9471–9483. [Google Scholar] [CrossRef] [Green Version]
- Zamiri, M.; Cheung, P.K.; Brockman, M.A.; Brumme, Z.L.; Chabot, B.; Cochrane, A.; Grierson, D.S. 2-Trifluoromethylthiazole-5-carboxamides: Analogues of a Stilbene-Based Anti-HIV Agent that Impact HIV mRNA Processing. ACS Med. Chem. Lett. 2021, 12, 1818–1823. [Google Scholar] [CrossRef]
- Wong, R.; Balachandran, A.; Mao, A.Y.; Dobson, W.; Gray-Owen, S.; Cochrane, A. Differential effect of CLK SR Kinases on HIV-1 gene expression: Potential novel targets for therapy. Retrovirology 2011, 8, 47. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Vink, M.; Berkhout, B.; Das, A.T. Modification of the Tet-On regulatory system prevents the conditional-live HIV-1 variant from losing doxycycline-control. Retrovirology 2006, 3, 82. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Vink, M.; Klaver, B.; Verhoef, K.; Marzio, G.; Das, A.T.; Berkhout, B. The genetic stability of a conditional live HIV-1 variant can be improved by mutations in the Tet-On regulatory system that restrain evolution. J. Biol. Chem. 2006, 281, 17084–17091. [Google Scholar] [CrossRef] [Green Version]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockman, M.A.; Tanzi, G.O.; Walker, B.D.; Allen, T.M. Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. J. Virol. Methods 2006, 131, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Mwimanzi, P.; Tietjen, I.; Miller, S.C.; Shahid, A.; Cobarrubias, K.; Kinloch, N.N.; Baraki, B.; Richard, J.; Finzi, A.; Fedida, D.; et al. Novel Acylguanidine-Based Inhibitor of HIV-1. J. Virol. 2016, 90, 9495–9508. [Google Scholar] [CrossRef] [Green Version]
- Brown, M. Selection of nonfastidious adenovirus species in 293 cells inoculated with stool specimens containing adenovirus 40. J. Clin. Microbiol. 1985, 22, 205–209. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Nasir, J.A.; Budylowski, P.; Yip, L.; Aftanas, P.; Christie, N.; Ghalami, A.; Baid, K.; Raphenya, A.R.; Hirota, J.A.; et al. Isolation, Sequence, Infectivity, and Replication Kinetics of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. 2020, 26, 2054–2063. [Google Scholar] [CrossRef]
- Grosso, F.; Stoilov, P.; Lingwood, C.; Brown, M.; Cochrane, A. Suppression of Adenovirus Replication by Cardiotonic Steroids. J. Virol. 2017, 91, e01623-16. [Google Scholar] [CrossRef] [Green Version]
- Purcell, D.; Martin, M.A. Alternative splicing of human immunodeficiency virus type 1 mRNA modulates viral protein expression, replication, and infectivity. J. Virol. 1993, 67, 6365–6378. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, A.; Wong, R.; Stoilov, P.; Pan, S.; Blencowe, B.; Cheung, P.; Harrigan, P.R.; Cochrane, A. Identification of small molecule modulators of HIV-1 Tat and Rev protein accumulation. Retrovirology 2017, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Stoltzfus, C. Regulation of HIV-1 Alternative RNA Splicing and Its Role in Virus Replication. Adv. Virus Res. 2009, 74, 1–40. [Google Scholar]
- Ocwieja, K.E.; Sherrill-Mix, S.; Mukherjee, R.; Custers-Allen, R.; David, P.; Brown, M.; Wang, S.; Link, D.R.; Olson, J.; Travers, K.; et al. Dynamic regulation of HIV-1 mRNA populations analyzed by single-molecule enrichment and long-read sequencing. Nucleic Acids Res. 2012, 40, 10345–10355. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.; Zhou, S.; Pollom, E.; Swanstrom, R. Characterizing HIV-1 Splicing by Using Next-Generation Sequencing. J. Virol. 2017, 91, e02515–e02516. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.W.; Balachandran, A.; Cheung, P.K.; Cheng, R.; Pan, Q.; Stoilov, P.; Harrigan, P.R.; Blencowe, B.J.; Branch, D.R.; Cochrane, A. An activator of G protein-coupled receptor and MEK1/2-ERK1/2 signaling inhibits HIV-1 replication by altering viral RNA processing. PLoS Pathog. 2020, 16, e1008307. [Google Scholar] [CrossRef] [Green Version]
- Biasiotto, R.; Akusjarvi, G. Regulation of human adenovirus alternative RNA splicing by the adenoviral L4-33K and L4-22K proteins. Int. J. Mol. Sci. 2015, 16, 2893–2912. [Google Scholar] [CrossRef] [Green Version]
- Chow, L.T.; Broker, T.R.; Lewis, J.B. Complex splicing patterns of RNAs from the early regions of adenovirus-2. J. Mol. Biol. 1979, 134, 265–303. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, M.; Pettersson, U. A new look at adenovirus splicing. Virology 2014, 456–457, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Akusjarvi, G. Temporal regulation of adenovirus major late alternative RNA splicing. Front. Biosci. 2008, 13, 5006–5015. [Google Scholar] [CrossRef]
- Thomas, G.P.; Mathews, M.B. DNA replication and the early to late transition in adenovirus infection. Cell 1980, 22, 523–533. [Google Scholar] [CrossRef]
- Sanford, J.R.; Ellis, J.; Caceres, J.F. Multiple roles of arginine/serine-rich splicing factors in RNA processing. Biochem. Soc. Trans. 2005, 33, 443–446. [Google Scholar] [CrossRef]
- Sanford, J.R.; Gray, N.K.; Beckmann, K.; Caceres, J.F. A novel role for shuttling SR proteins in mRNA translation. Genes. Dev. 2004, 18, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Howard, J.M.; Sanford, J.R. The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip. Rev. RNA 2015, 6, 93–110. [Google Scholar] [CrossRef]
- Ngo, J.C.; Chakrabarti, S.; Ding, J.H.; Velazquez-Dones, A.; Nolen, B.; Aubol, B.E.; Adams, J.A.; Fu, X.D.; Ghosh, G. Interplay between SRPK and Clk/Sty kinases in phosphorylation of the splicing factor ASF/SF2 is regulated by a docking motif in ASF/SF2. Mol. Cell 2005, 20, 77–89. [Google Scholar] [CrossRef]
- Prasad, J.; Colwill, K.; Pawson, T.; Manley, J.L. The protein kinase Clk/Sty directly modulates SR protein activity: Both hyper- and hypophosphorylation inhibit splicing. Mol. Cell Biol. 1999, 19, 6991–7000. [Google Scholar] [CrossRef] [Green Version]
- Colwill, K.; Pawson, T.; Andrews, B.; Prasad, J.; Manley, J.L.; Bell, J.C.; Duncan, P.I. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. EMBO J. 1996, 15, 265–275. [Google Scholar] [CrossRef]
- Duncan, P.I.; Stojdl, D.F.; Marius, R.M.; Scheit, K.H.; Bell, J.C. The Clk2 and Clk3 dual-specificity protein kinases regulate the intranuclear distribution of SR proteins and influence pre-mRNA splicing. Exp. Cell Res. 1998, 241, 300–308. [Google Scholar] [CrossRef]
- Du Toit, A. Coronavirus replication factories. Nat. Rev. Microbiol. 2020, 18, 411. [Google Scholar] [CrossRef]
- Fung, T.S.; Liu, D.X. Human Coronavirus: Host-Pathogen Interaction. Annu. Rev. Microbiol. 2019, 73, 529–557. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.L.; Guo, D.; Rottier, P.J. Coronavirus: Epidemiology, genome replication and the interactions with their hosts. Virol. Sin. 2016, 31, 1–2. [Google Scholar] [CrossRef] [Green Version]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2020, 19, 155–170. [Google Scholar] [CrossRef]
- Ajiro, M.; Zheng, Z.M. Oncogenes and RNA splicing of human tumor viruses. Emerg. Microbes Infect. 2014, 3, e63. [Google Scholar] [CrossRef]
- Dubois, J.; Terrier, O.; Rosa-Calatrava, M. Influenza viruses and mRNA splicing: Doing more with less. mBio 2014, 5, e00070-00014. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Carmichael, G.G. RNA processing in the polyoma virus life cycle. Front. Biosci. 2009, 14, 4968–4977. [Google Scholar] [CrossRef] [Green Version]
- Journo, C.; Douceron, E.; Mahieux, R. HTLV gene regulation: Because size matters, transcription is not enough. Future Microbiol. 2009, 4, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Karn, J.; Stoltzfus, C.M. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb. Perspect. Med. 2012, 2, a006916. [Google Scholar] [CrossRef]
- Klymenko, T.; Graham, S.V. Human papillomavirus gene expression is controlled by host cell splicing factors. Biochem. Soc. Trans. 2012, 40, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Krug, L.T. Complexities of gammaherpesvirus transcription revealed by microarrays and RNAseq. Curr. Opin. Virol. 2013, 3, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, S. Papillomavirus transcripts and posttranscriptional regulation. Virology 2013, 445, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Xiong, J.; Wang, D.; Fu, X.D. Pre-mRNA splicing: Where and when in the nucleus. Trends Cell Biol. 2011, 21, 336–343. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Martinez-Contreras, R.; Cloutier, P.; Shkreta, L.; Fisette, J.F.; Revil, T.; Chabot, B. hnRNP proteins and splicing control. Adv. Exp. Med. Biol. 2007, 623, 123–147. [Google Scholar]
- Long, J.C.; Caceres, J.F. The SR protein family of splicing factors: Master regulators of gene expression. Biochem. J. 2009, 417, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Shkreta, L.; Blanchette, M.; Toutant, J.; Wilhelm, E.; Bell, B.; Story, B.A.; Balachandran, A.; Cochrane, A.; Cheung, P.K.; Harrigan, P.R.; et al. Modulation of the splicing regulatory function of SRSF10 by a novel compound that impairs HIV-1 replication. Nucleic Acids Res. 2017, 45, 4051–4067. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.W.; Balachandran, A.; Ostrowski, M.A.; Cochrane, A. Digoxin suppresses HIV-1 replication by altering viral RNA processing. PLoS Pathog. 2013, 9, e1003241. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.W.; Lingwood, C.A.; Ostrowski, M.A.; Cabral, T.; Cochrane, A. Cardiac glycoside/aglycones inhibit HIV-1 gene expression by a mechanism requiring MEK1/2-ERK1/2 signaling. Sci. Rep. 2018, 8, 850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, G.L.; Korth, M.J.; Benecke, A.G.; Katze, M.G. Systems virology: Host-directed approaches to viral pathogenesis and drug targeting. Nat. Rev. Microbiol. 2013, 11, 455–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, N.; Myburgh, R.; Garcel, A.; Vautrin, A.; Lapasset, L.; Nadal, E.S.; Mahuteau-Betzer, F.; Najman, R.; Fornarelli, P.; Tantale, K.; et al. Long lasting control of viral rebound with a new drug ABX464 targeting Rev—Mediated viral RNA biogenesis. Retrovirology 2015, 12, 30. [Google Scholar] [CrossRef] [Green Version]
- Rutsaert, S.; Steens, J.M.; Gineste, P.; Cole, B.; Kint, S.; Barrett, P.N.; Tazi, J.; Scherrer, D.; Ehrlich, H.J.; Vandekerckhove, L. Safety, tolerability and impact on viral reservoirs of the addition to antiretroviral therapy of ABX464, an investigational antiviral drug, in individuals living with HIV-1: A Phase IIa randomised controlled study. J. Virus Erad. 2019, 5, 10–22. [Google Scholar] [CrossRef]
- Steens, J.M.; Scherrer, D.; Gineste, P.; Barrett, P.N.; Khuanchai, S.; Winai, R.; Ruxrungtham, K.; Tazi, J.; Murphy, R.; Ehrlich, H. Safety, Pharmacokinetics, and Antiviral Activity of a Novel HIV Antiviral, ABX464, in Treatment-Naive HIV-Infected Subjects in a Phase 2 Randomized, Controlled Study. Antimicrob. Agents Chemother. 2017, 61, e00545-17. [Google Scholar] [CrossRef] [Green Version]
- Bakkour, N.; Lin, Y.L.; Maire, S.; Ayadi, L.; Mahuteau-Betzer, F.; Nguyen, C.H.; Mettling, C.; Portales, P.; Grierson, D.; Chabot, B.; et al. Small-molecule inhibition of HIV pre-mRNA splicing as a novel antiretroviral therapy to overcome drug resistance. PLoS Pathog. 2007, 3, 1530–1539. [Google Scholar] [CrossRef]
- Cheung, P.K.; Horhant, D.; Bandy, L.E.; Zamiri, M.; Rabea, S.M.; Karagiosov, S.K.; Matloobi, M.; McArthur, S.; Harrigan, P.R.; Chabot, B.; et al. A Parallel Synthesis Approach to the Identification of Novel Diheteroarylamide-Based Compounds Blocking HIV Replication: Potential Inhibitors of HIV-1 Pre-mRNA Alternative Splicing. J. Med. Chem. 2016, 59, 1869–1879. [Google Scholar] [CrossRef]
- Hope, T.J. The ins and outs of HIV Rev. Arch. Biochem. Biophys. 1999, 365, 186–191. [Google Scholar] [CrossRef]
- Pollard, V.; Malim, M. The HIV-1 Rev Protein. Annu. Rev. Microbiol. 1998, 52, 491–532. [Google Scholar] [CrossRef]
- Reichel, R.; Neill, S.D.; Kovesdi, I.; Simon, M.C.; Raychaudhuri, P.; Nevins, J.R. The adenovirus E4 gene, in addition to the E1A gene, is important for trans-activation of E2 transcription and for E2F activation. J. Virol. 1989, 63, 3643–3650. [Google Scholar] [CrossRef] [Green Version]
- Babiss, L.E. The cellular transcription factor E2f requires viral E1A and E4 gene products for increased DNA-binding activity and functions to stimulate adenovirus E2A gene expression. J. Virol. 1989, 63, 2709–2717. [Google Scholar] [CrossRef] [Green Version]
- Estmer Nilsson, C.; Petersen-Mahrt, S.; Durot, C.; Shtrichman, R.; Krainer, A.R.; Kleinberger, T.; Akusjarvi, G. The adenovirus E4-ORF4 splicing enhancer protein interacts with a subset of phosphorylated SR proteins. EMBO J. 2001, 20, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Kanopka, A.; Muhlemann, O.; Petersen-Mahrt, S.; Estmer, C.; Ohrmalm, C.; Akusjarvi, G. Regulation of adenovirus alternative RNA splicing by dephosphorylation of SR proteins. Nature 1998, 393, 185–187. [Google Scholar] [CrossRef]
- Chabrolles, H.; Auclair, H.; Vegna, S.; Lahlali, T.; Pons, C.; Michelet, M.; Coute, Y.; Belmudes, L.; Chadeuf, G.; Kim, Y.; et al. Hepatitis B virus Core protein nuclear interactome identifies SRSF10 as a host RNA-binding protein restricting HBV RNA production. PLoS Pathog. 2020, 16, e1008593. [Google Scholar] [CrossRef] [PubMed]
- Muraki, M.; Ohkawara, B.; Hosoya, T.; Onogi, H.; Koizumi, J.; Koizumi, T.; Sumi, K.; Yomoda, J.; Murray, M.V.; Kimura, H.; et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. J. Biol. Chem. 2004, 279, 24246–24254. [Google Scholar] [CrossRef] [Green Version]
- Jacquenet, S.; Decimo, D.; Muriaux, D.; Darlix, J.L. Dual effect of the SR proteins ASF/SF2, SC35 and 9G8 on HIV-1 RNA splicing and virion production. Retrovirology 2005, 2, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ropers, D.; Ayadi, L.; Gattoni, R.; Jacquenet, S.; Damier, L.; Branlant, C.; Stevenin, J. Differential effects of the SR proteins 9G8, SC35, ASF/SF2 and SRp40 on the utilization of the A1 to A5 splicing sites of HIV-1 RNA. J. Biol. Chem. 2004, 279, 29963–29973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jablonski, J.A.; Caputi, M. Role of cellular RNA processing factors in human immunodeficiency virus type 1 mRNA metabolism, replication, and infectivity. J. Virol. 2009, 83, 981–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, N.; Milev, M.P.; Wong, R.; Sanmuganantham, T.; Woolaway, K.; Chabot, B.; Abou Elela, S.; Mouland, A.J.; Cochrane, A. Differential effects of hnRNP D/AUF1 isoforms on HIV-1 gene expression. Nucleic Acids Res. 2012, 40, 3663–3675. [Google Scholar] [CrossRef]
- Woolaway, K.; Asai, K.; Emili, A.; Cochrane, A. hnRNP E1 and E2 have distinct roles in modulating HIV-1 gene expression. Retrovirology 2007, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Erkelenz, S.; Hillebrand, F.; Widera, M.; Theiss, S.; Fayyaz, A.; Degrandi, D.; Pfeffer, K.; Schaal, H. Balanced splicing at the Tat-specific HIV-1 3′ss A3 is critical for HIV-1 replication. Retrovirology 2015, 12, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkelenz, S.; Poschmann, G.; Theiss, S.; Stefanski, A.; Hillebrand, F.; Otte, M.; Stuhler, K.; Schaal, H. Tra2-mediated recognition of HIV-1 5′ splice site D3 as a key factor in the processing of vpr mRNA. J. Virol. 2013, 87, 2721–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, C.; Calimano, M.; Nemet, J.; Bubenik, J.; Cochrane, A. Differential Effects of Tra2ss Isoforms on HIV-1 RNA Processing and Expression. PLoS ONE 2015, 10, e0125315. [Google Scholar] [CrossRef] [PubMed]
- Caputi, M.; Freund, M.; Kammler, S.; Asang, C.; Schaal, H. A bidirectional SF2/ASF- and SRp40-dependent splicing enhancer regulates human immunodeficiency virus type 1 rev, env, vpu, and nef gene expression. J. Virol. 2004, 78, 6517–6526. [Google Scholar] [CrossRef] [Green Version]
- Tranell, A.; Fenyo, E.M.; Schwartz, S. Serine- and arginine-rich proteins 55 and 75 (SRp55 and SRp75) induce production of HIV-1 vpr mRNA by inhibiting the 5′-splice site of exon 3. J. Biol. Chem. 2010, 285, 31537–31547. [Google Scholar] [CrossRef] [Green Version]
- Mahiet, C.; Swanson, C.M. Control of HIV-1 gene expression by SR proteins. Biochem. Soc. Trans. 2016, 44, 1417–1425. [Google Scholar] [CrossRef]
- Swanson, C.M.; Sherer, N.M.; Malim, M.H. SRp40 and SRp55 promote the translation of unspliced human immunodeficiency virus type 1 RNA. J. Virol. 2010, 84, 6748–6759. [Google Scholar] [CrossRef] [Green Version]
- Exline, C.M.; Feng, Z.; Stoltzfus, C.M. Negative and positive mRNA splicing elements act competitively to regulate human immunodeficiency virus type 1 vif gene expression. J. Virol. 2008, 82, 3921–3931. [Google Scholar] [CrossRef] [Green Version]
- Fukuhara, T.; Hosoya, T.; Shimizu, S.; Sumi, K.; Oshiro, T.; Yoshinaka, Y.; Suzuki, M.; Yamamoto, N.; Herzenberg, L.A.; Hagiwara, M. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc. Natl. Acad. Sci. USA 2006, 103, 11329–11333. [Google Scholar] [CrossRef] [Green Version]
- Czubaty, A.; Piekielko-Witkowska, A. Protein kinases that phosphorylate splicing factors: Roles in cancer development, progression and possible therapeutic options. Int. J. Biochem. Cell Biol. 2017, 91, 102–115. [Google Scholar] [CrossRef]
- Stamm, S. Regulation of alternative splicing by reversible protein phosphorylation. J. Biol. Chem. 2008, 283, 1223–1227. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Fu, X.D. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [CrossRef]
- Molin, M.; Akusjarvi, G. Overexpression of essential splicing factor ASF/SF2 blocks the temporal shift in adenovirus pre-mRNA splicing and reduces virus progeny formation. J. Virol. 2000, 74, 9002–9009. [Google Scholar] [CrossRef] [Green Version]
- Yomoda, J.; Muraki, M.; Kataoka, N.; Hosoya, T.; Suzuki, M.; Hagiwara, M.; Kimura, H. Combination of Clk family kinase and SRp75 modulates alternative splicing of Adenovirus E1A. Genes Cells Devoted Mol. Cell. Mech. 2008, 13, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Fu, X.D.; Ou, J.H. Suppression of hepatitis B virus replication by SRPK1 and SRPK2 via a pathway independent of the phosphorylation of the viral core protein. Virology 2005, 342, 150–158. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.C.; Peng, T.Y.; Tarn, W.Y. Functional interplay between viral and cellular SR proteins in control of post-transcriptional gene regulation. FEBS J. 2009, 276, 1517–1526. [Google Scholar] [CrossRef]
- Flynn, R.A.; Belk, J.A.; Qi, Y.; Yasumoto, Y.; Wei, J.; Alfajaro, M.M.; Shi, Q.; Mumbach, M.R.; Limaye, A.; DeWeirdt, P.C.; et al. Discovery and functional interrogation of SARS-CoV-2 RNA-host protein interactions. Cell 2021, 184, 2394–2411.e16. [Google Scholar] [CrossRef] [PubMed]
- Kamel, W.; Noerenberg, M.; Cerikan, B.; Chen, H.; Jarvelin, A.I.; Kammoun, M.; Lee, J.Y.; Shuai, N.; Garcia-Moreno, M.; Andrejeva, A.; et al. Global analysis of protein-RNA interactions in SARS-CoV-2-infected cells reveals key regulators of infection. Mol. Cell 2021, 81, 2851–2867.e7. [Google Scholar] [CrossRef] [PubMed]
- Karakama, Y.; Sakamoto, N.; Itsui, Y.; Nakagawa, M.; Tasaka-Fujita, M.; Nishimura-Sakurai, Y.; Kakinuma, S.; Oooka, M.; Azuma, S.; Tsuchiya, K.; et al. Inhibition of hepatitis C virus replication by a specific inhibitor of serine-arginine-rich protein kinase. Antimicrob. Agents Chemother. 2010, 54, 3179–3186. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.M.; Daijogo, S.; Semler, B.L. A nucleo-cytoplasmic SR protein functions in viral IRES-mediated translation initiation. EMBO J. 2007, 26, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Yaron, T.M.; Heaton, B.E.; Levy, T.M.; Johnson, J.L.; Jordan, T.X.; Cohen, B.M.; Kerelsky, A.; Lin, T.Y.; Liberatore, K.M.; Bulaon, D.K.; et al. The FDA-approved drug Alectinib compromises SARS-CoV-2 nucleocapsid phosphorylation and inhibits viral infection in vitro. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gui, J.F.; Lane, W.S.; Fu, X.D. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature 1994, 369, 678–682. [Google Scholar] [CrossRef]
- van Der Houven Van Oordt, W.; Newton, K.; Screaton, G.R.; Caceres, J.F. Role of SR protein modular domains in alternative splicing specificity in vivo. Nucleic Acids Res. 2000, 28, 4822–4831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, S.H.; Manley, J.L. Phosphorylation of the ASF/SF2 RS domain affects both protein-protein and protein-RNA interactions and is necessary for splicing. Genes Dev. 1997, 11, 334–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeakley, J.M.; Tronchere, H.; Olesen, J.; Dyck, J.A.; Wang, H.Y.; Fu, X.D. Phosphorylation regulates in vivo interaction and molecular targeting of serine/arginine-rich pre-mRNA splicing factors. J. Cell Biol. 1999, 145, 447–455. [Google Scholar] [CrossRef] [Green Version]
- Zahler, A.M.; Lane, W.S.; Stolk, J.A.; Roth, M.B. SR proteins: A conserved family of pre-mRNA splicing factors. Genes. Dev. 1992, 6, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Mattox, W. Activation and repression functions of an SR splicing regulator depend on exonic versus intronic-binding position. Nucleic Acids Res. 2012, 40, 428–437. [Google Scholar] [CrossRef] [Green Version]
- Maciolek, N.L.; McNally, M.T. Serine/arginine-rich proteins contribute to negative regulator of splicing element-stimulated polyadenylation in rous sarcoma virus. J. Virol. 2007, 81, 11208–11217. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Gattoni, R.; Stevenin, J.; Steitz, J.A. SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol. Cell 2003, 11, 837–843. [Google Scholar] [CrossRef]
- Huang, Y.; Yario, T.A.; Steitz, J.A. A molecular link between SR protein dephosphorylation and mRNA export. Proc. Natl. Acad. Sci. USA 2004, 101, 9666–9670. [Google Scholar] [CrossRef] [Green Version]
- Hartenian, E.; Nandakumar, D.; Lari, A.; Ly, M.; Tucker, J.M.; Glaunsinger, B.A. The molecular virology of coronaviruses. J. Biol. Chem. 2020, 295, 12910–12934. [Google Scholar] [CrossRef]
- Schneider, W.M.; Luna, J.M.; Hoffmann, H.H.; Sanchez-Rivera, F.J.; Leal, A.A.; Ashbrook, A.W.; Le Pen, J.; Ricardo-Lax, I.; Michailidis, E.; Peace, A.; et al. Genome-Scale Identification of SARS-CoV-2 and Pan-coronavirus Host Factor Networks. Cell 2021, 184, 120–132.e14. [Google Scholar] [CrossRef]
- Nikolakaki, E.; Giannakouros, T. SR/RS Motifs as Critical Determinants of Coronavirus Life Cycle. Front. Mol. Biosci. 2020, 7, 219. [Google Scholar] [CrossRef]
- Peng, T.Y.; Lee, K.R.; Tarn, W.Y. Phosphorylation of the arginine/serine dipeptide-rich motif of the severe acute respiratory syndrome coronavirus nucleocapsid protein modulates its multimerization, translation inhibitory activity and cellular localization. FEBS J. 2008, 275, 4152–4163. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.H.; Yeh, S.H.; Tsay, Y.G.; Shieh, Y.H.; Kao, C.L.; Chen, Y.S.; Wang, S.H.; Kuo, T.J.; Chen, D.S.; Chen, P.J. Glycogen synthase kinase-3 regulates the phosphorylation of severe acute respiratory syndrome coronavirus nucleocapsid protein and viral replication. J. Biol. Chem. 2009, 284, 5229–5239. [Google Scholar] [CrossRef] [Green Version]
- Bojkova, D.; Klann, K.; Koch, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.; Munch, C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Strain | EC50 | CC50 |
|---|---|---|---|
| HIV-1 Assayed in CEM-GXR cell line | Clade A | 176 nM | 12,052 nM |
| Clade B | 248 nM | 12,052 nM | |
| NL43 E0043 RTI | 235 nM | 12,052 nM | |
| NL43 2918 PI | 230 nM | 12,052 nM | |
| NL43 11845 INI | 203 nM | 12,052 nM | |
| IIIB MVC res | 216 nM | 12,052 nM | |
| HIV-1 Assayed in PBMCs | HIV-1 Bal (average of 3 donors) | 248 nM | 2,509 nM |
| HIV-1 IIIB (average of 3 donors) | 738 nM | 2,509 nM | |
| Adenovirus Assayed in A549 cell line | HAdV-C5 | 1000 nM | >40,000 nM |
| Coronavirus Assayed in Huh7 cell line | 229E | 250 nM | >10,000 nM |
| OC43 | 250 nM | >10,000 nM | |
| SARS-CoV2 | 100 nM | >10,000 nM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahal, S.; Cheng, R.; Cheung, P.K.; Been, T.; Malty, R.; Geng, M.; Manianis, S.; Shkreta, L.; Jahanshahi, S.; Toutant, J.; et al. The Thiazole-5-Carboxamide GPS491 Inhibits HIV-1, Adenovirus, and Coronavirus Replication by Altering RNA Processing/Accumulation. Viruses 2022, 14, 60. https://doi.org/10.3390/v14010060
Dahal S, Cheng R, Cheung PK, Been T, Malty R, Geng M, Manianis S, Shkreta L, Jahanshahi S, Toutant J, et al. The Thiazole-5-Carboxamide GPS491 Inhibits HIV-1, Adenovirus, and Coronavirus Replication by Altering RNA Processing/Accumulation. Viruses. 2022; 14(1):60. https://doi.org/10.3390/v14010060
Chicago/Turabian StyleDahal, Subha, Ran Cheng, Peter K. Cheung, Terek Been, Ramy Malty, Melissa Geng, Sarah Manianis, Lulzim Shkreta, Shahrazad Jahanshahi, Johanne Toutant, and et al. 2022. "The Thiazole-5-Carboxamide GPS491 Inhibits HIV-1, Adenovirus, and Coronavirus Replication by Altering RNA Processing/Accumulation" Viruses 14, no. 1: 60. https://doi.org/10.3390/v14010060