
Lentiviral Vectors for T Cell Engineering: Clinical Applications, Bioprocessing and Future Perspectives

Abstract
:1. Introduction
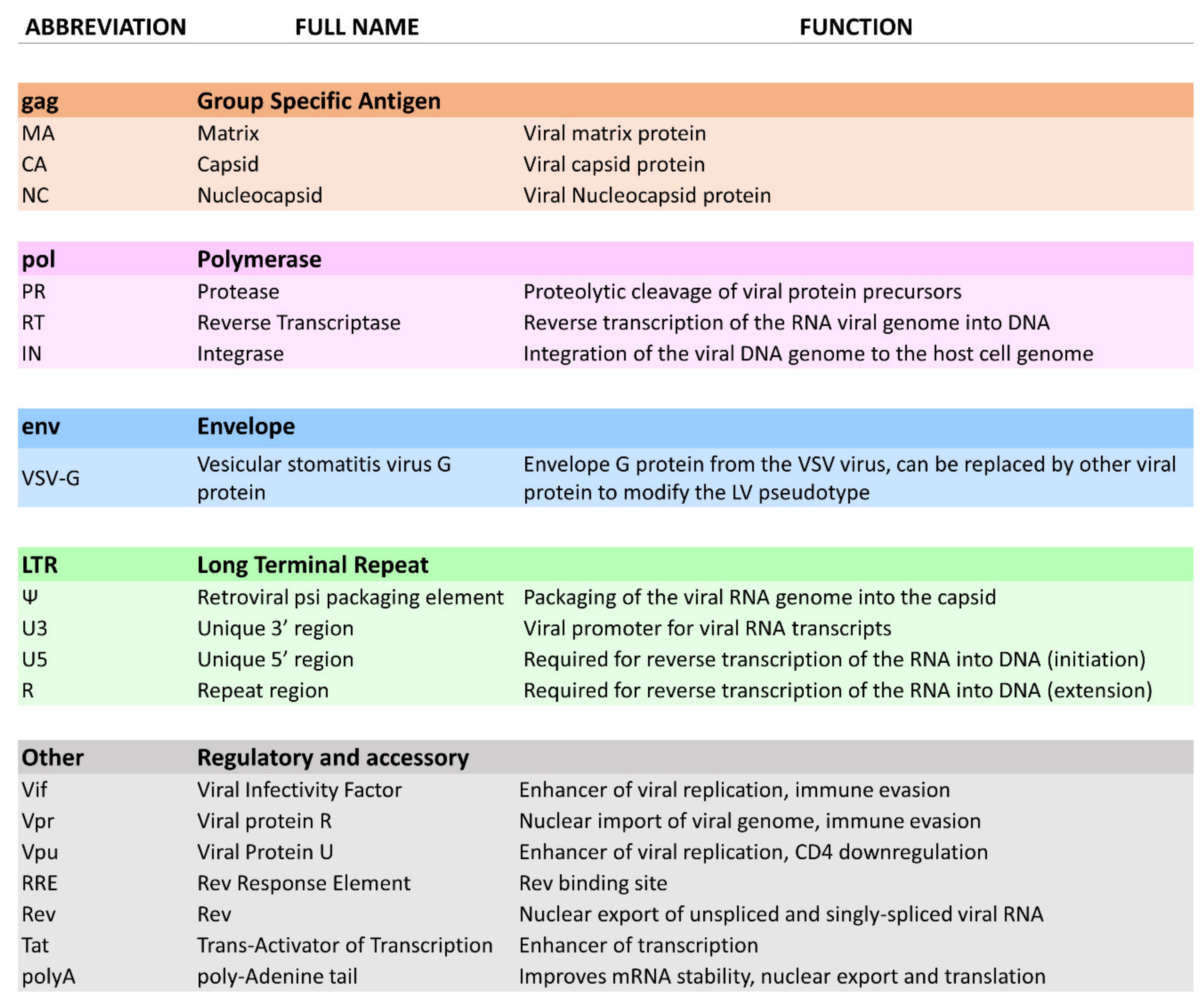
2. Lentiviral Vectors
2.1. Biological Principles
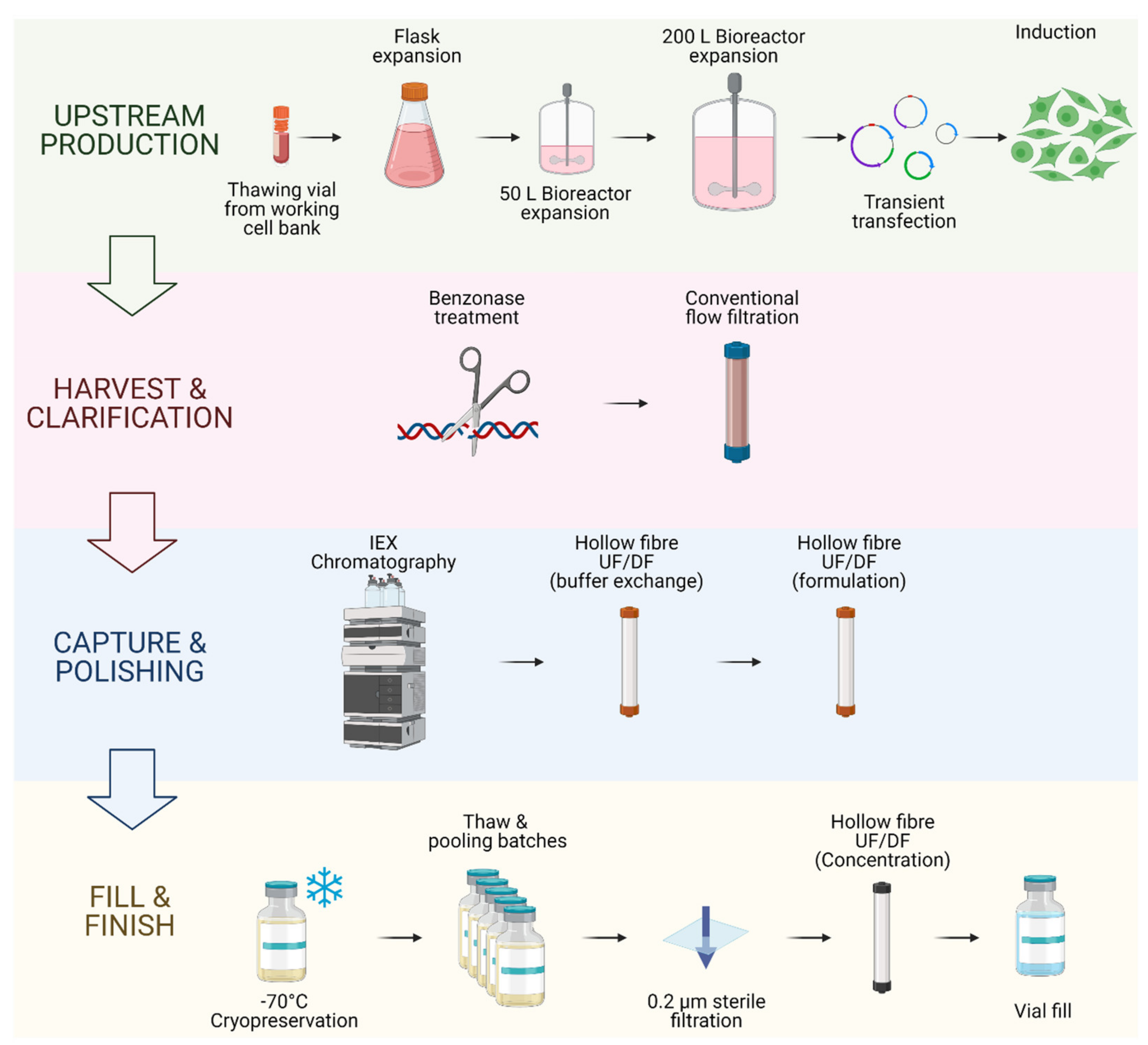
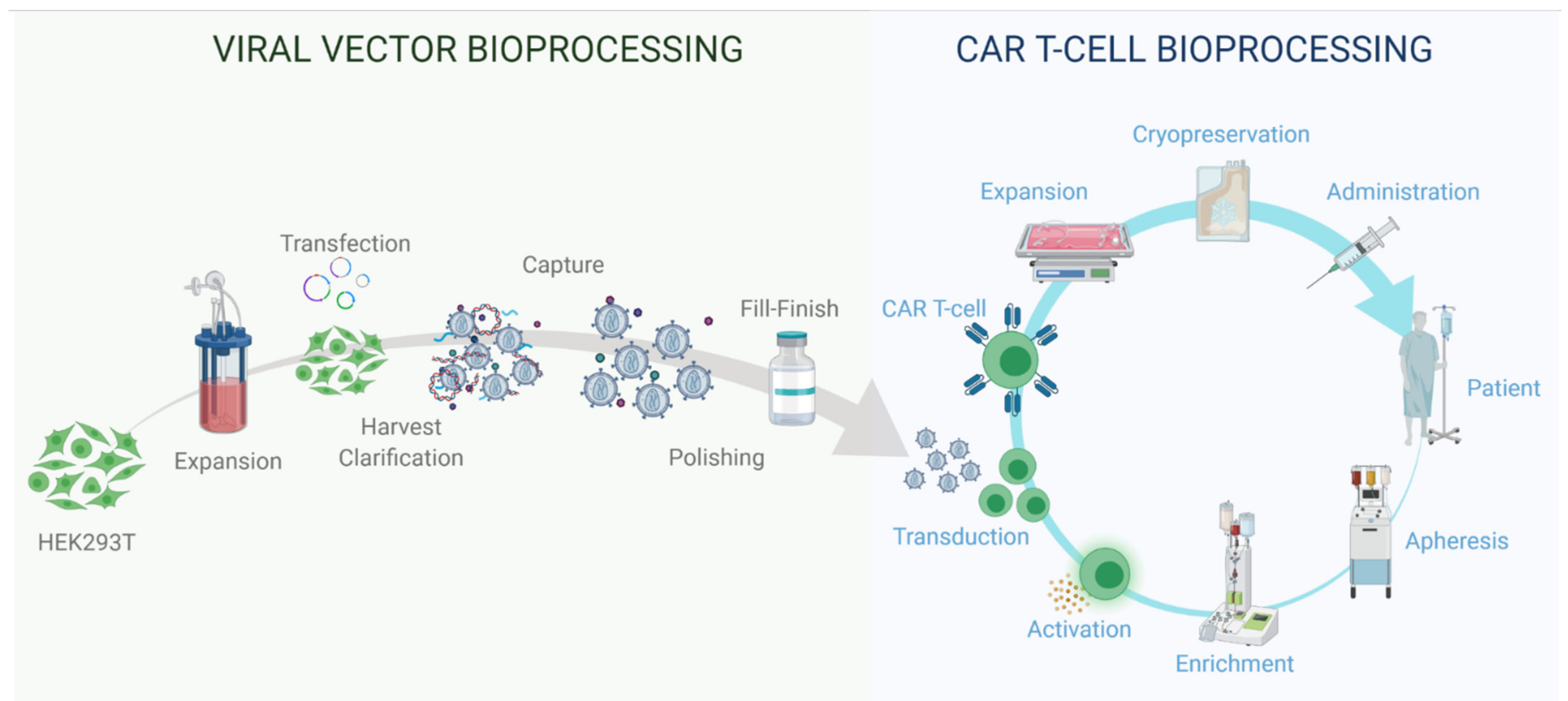
2.2. LV Bioprocessing
3. Gene-Modified T Cell Therapy
3.1. Biological Principles
3.2. T Cell Therapy Bioprocessing
4. Applications of LV Vector Technologies for T Cell Engineering
4.1. Introduction
4.2. Commercially Available Gene-Modified T Cell Therapies
4.3. Limitations, Safety and Efficacy
4.4. Development of LV-Based Tools for CAR T Cell Therapy Research
4.5. Development of Instruments Integrating LV to Current T Cell Bioprocesses
4.6. CAR T Cell Allogeneic Approach Using LV Vectors
4.7. Stable Production of LV Vectors to Address Increasing Demand in Vectors
4.8. Dedicated LV Technology for CAR T Cell Therapy Applications
4.9. Alternative Approaches to CAR T Cell Therapy Using LV Technology
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- David, R. Wessner Origin of Viruses. Nat. Educ. 2010, 3, 37. [Google Scholar]
- Milone, M.C.; O’Doherty, U. Clinical Use of Lentiviral Vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Miller, A.D. Retroviral Vectors: From Cancer Viruses to Therapeutic Tools. Hum. Gene Ther. 2014, 25, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, R.; Chowdhury, C.R.; Arega, S.; Sen, P.; Ganguly, P.; Ganguly, N. CAR T Cell Therapy: A New Era for Cancer Treatment (Review). Oncol. Rep. 2019, 42, 2183–2195. [Google Scholar] [CrossRef]
- European Medicines Agency; Committee for Medicinal Products for Human Use; Committee for Medicinal Products for Human Use (CHMP). Summary of Positive Opinion Kymriah. 2018. [Google Scholar]
- Food and Drug Administration; Center for Biologics Evaluation and Research. August 30, 2017 Approval Letter—KYMRIAH; Food and Drug Administration: Silver Spring, MD, USA, 2017. [Google Scholar]
- European Medicines Agency; Committee for Medicinal Products for Human Use; Committee for Medicinal Products for Human Use (CHMP). Summary of Positive Opinion for Yescarta. 2018. [Google Scholar]
- Food and Drug Administration; Center for Biologics Evaluation and Research. October 18, 2017 Approval Letter—YESCARTA; Food and Drug Administration: Silver Spring, MD, USA, 2017. [Google Scholar]
- European Medicines Agency; Committee for Medicinal Products for Human Use; Committee for Medicinal Products for Human Use (CHMP). Summary of Positive Opinion for Tecartus. 2020. [Google Scholar]
- Food and Drug Administration; Center for Biologics Evaluation and Research. July 24, 2020 Approval Letter—TECARTUS; Food and Drug Administration: Silver Spring, MD, USA, 2020. [Google Scholar]
- Elsner, C.; Bohne, J. The Retroviral Vector Family: Something for Everyone. Virus Genes 2017, 53, 714–722. [Google Scholar] [CrossRef]
- Cantore, A.; Ranzani, M.; Bartholomae, C.C.; Volpin, M.; Valle, P.D.; Sanvito, F.; Sergi, L.S.; Gallina, P.; Benedicenti, F.; Bellinger, D.; et al. Liver-Directed Lentiviral Gene Therapy in a Dog Model of Hemophilia B. Sci. Transl. Med. 2015, 12, 7. [Google Scholar] [CrossRef] [Green Version]
- Campochiaro, P.A.; Lauer, A.K.; Sohn, E.H.; Mir, T.A.; Naylor, S.; Anderton, M.C.; Kelleher, M.; Harrop, R.; Ellis, S.; Mitrophanous, K.A. Lentiviral Vector Gene Transfer of Endostatin/Angiostatin for Macular Degeneration (GEM) Study. Hum. Gene Ther. 2017, 28, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Munis, A.M. Gene Therapy Applications of Non-Human Lentiviral Vectors. Viruses 2020, 12, 1106. [Google Scholar] [CrossRef]
- Shaw, A.M.; Joseph, G.L.; Jasti, A.C.; Sastry-Dent, L.; Witting, S.; Cornetta, K. Differences in Vector-Genome Processing and Illegitimate Integration of Non-Integrating Lentiviral Vectors. Gene Ther. 2017, 24, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Powell, S.K.; Rivera-Soto, R.; Gray, S.J. Viral Expression Cassette Elements to Enhance Transgene Target Specificity and Expression in Gene Therapy. Discov. Med. 2015, 19, 49–57. [Google Scholar]
- Gándara, C.; Affleck, V.; Stoll, E.A. Manufacture of Third-Generation Lentivirus for Preclinical Use, with Process Development Considerations for Translation to Good Manufacturing Practice. Hum. Gene Ther. Methods 2018, 29, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barajas, B.C.; Tanaka, M.; Robinson, B.A.; Phuong, D.J.; Chutiraka, K.; Reed, J.C.; Lingappa, J.R. Identifying the Assembly Intermediate in Which Gag First Associates with Unspliced HIV-1 RNA Suggests a Novel Model for HIV-1 RNA Packaging. PLoS Pathog. 2018, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.H.; Huang, K.J.; Wang, C.T. HIV-1 Mutant Assembly, Processing and Infectivity Expresses Pol Independent of Gag. Viruses 2020, 12, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandres, J.C.; Wang, Q.F.; O’Leary, J.; Baleaux, F.; Amara, A.; Hoxie, J.A.; Zolla-Pazner, S.; Gorny, M.K. Human Immunodeficiency Virus (HIV) Envelope Binds to CXCR4 Independently of CD4, and Binding Can Be Enhanced by Interaction with Soluble CD4 or by HIV Envelope Deglycosylation. J. Virol. 1998, 72, 2500–2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balachandran, A.; Wong, R.; Stoilov, P.; Pan, S.; Blencowe, B.; Cheung, P.; Harrigan, P.R.; Cochrane, A. Identification of Small Molecule Modulators of HIV-1 Tat and Rev Protein Accumulation. Retrovirology 2017, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Zotova, A.; Atemasova, A.; Pichugin, A.; Filatov, A.; Mazurov, D. Distinct Requirements for HIV-1 Accessory Proteins during Cell Coculture and Cell-Free Infection. Viruses 2019, 11, 390. [Google Scholar] [CrossRef] [Green Version]
- Daniels, S.M.; Sinck, L.; Ward, N.J.; Melendez-Pe~ Na, C.E.; Scarborough, R.J.; Azar, I.; Rance, E.; Icha Daher, A.; Pang, K.-M.; Rossi, J.J.; et al. HIV-1 RRE RNA Acts as an RNA Silencing Suppressor by Competing with TRBP-Bound SiRNAs. RNA Biol. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.; Datta, G.; Geiger, J.D.; Chen, X. Apolipoprotein E Isoform Dependently Affects Tat-Mediated HIV-1 LTR Transactivation. J. Neuroinflammation 2018, 15. [Google Scholar] [CrossRef]
- Kay, M.A.; Glorioso, J.C.; Naldini, L. Viral Vectors for Gene Therapy: The Art of Turning Infectious Agents into Vehicles of Therapeutics. Nat. Med. 2001, 7, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, L.; Lai, M.; Chiuppesi, F.; Ceccherini-Nelli, L.; Pistello, M. Viral Vectors: A Look Back and Ahead on Gene Transfer Technology. New Microbiol. 2013, 36, 1–22. [Google Scholar]
- Rothe, M.; Modlich, U.; Schambach, A. Biosafety Challenges for Use of Lentiviral Vectors in Gene Therapy. Curr. Gene Ther. 2014, 13, 453–468. [Google Scholar] [CrossRef]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A Third-Generation Lentivirus Vector with a Conditional Packaging System. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, D.L.; Case, S.S.; Crooks, G.M.; Kohn, D.B. Critical Factors Influencing Stable Transduction of Human CD34+ Cells with HIV-1-Derived Lentiviral Vectors. Mol. Ther. 2000, 2, 71–80. [Google Scholar] [CrossRef]
- Vink, C.A.; Counsell, J.R.; Perocheau, D.P.; Karda, R.; Buckley, S.M.K.; Brugman, M.H.; Galla, M.; Schambach, A.; Mckay, T.R.; Waddington, S.N.; et al. Eliminating HIV-1 Packaging Sequences from Lentiviral Vector Proviruses Enhances Safety and Expedites Gene Transfer for Gene Therapy. Mol. Ther. 2017, 25, 1790–1804. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, C.; Hosiner, S.; Tichy, B.; Aldrian, S.; Hajdu, S.; Nürnberger, S. Comparison of Lentiviral Packaging Mixes and Producer Cell Lines for RNAi Applications. Mol. Biotechnol. 2015, 57, 499–505. [Google Scholar] [CrossRef]
- Valkama, A.J.; Leinonen, H.M.; Lipponen, E.M.; Turkki, V.; Malinen, J.; Heikura, T.; Ylä-Herttuala, S.; Lesch, H.P. Optimization of Lentiviral Vector Production for Scale-up in Fixed-Bed Bioreactor. Gene Ther. 2018, 25, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Strobel, B.; Zuckschwerdt, K.; Zimmermann, G.; Mayer, C.; Eytner, R.; Rechtsteiner, P.; Kreuz, S.; Lamla, T. Standardized, Scalable, and Timely Flexible Adeno-Associated Virus Vector Production Using Frozen High-Density HEK-293 Cell Stocks and CELLdiscs. Hum. Gene Ther. Methods 2019, 30, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Rout-Pitt, N.; McCarron, A.; McIntyre, C.; Parsons, D.; Donnelley, M. Large-Scale Production of Lentiviral Vectors Using Multilayer Cell Factories. J. Biol. Methods 2018, 5, 90. [Google Scholar] [CrossRef] [Green Version]
- McCarron, A.; Donnelley, M.; McIntyre, C.; Parsons, D. Transient Lentiviral Vector Production Using a Packed-Bed Bioreactor System. Hum. Gene Ther. Methods 2019, 30, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Diaconu, I.; Ballard, B.; Zhang, M.; Chen, Y.; West, J.; Dotti, G.; Savoldo, B. Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol. Ther. 2017, 25, 580–592. [Google Scholar] [CrossRef] [Green Version]
- Blessing, D.; Déglon, N.; Schneider, B.L. Scalable production and purification of adeno-associated viral vectors (AAV). In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2018; Volume 1850, pp. 259–274. [Google Scholar]
- Labisch, J.J.; Bollmann, F.; Wolff, M.W.; Pflanz, K. A New Simplified Clarification Approach for Lentiviral Vectors Using Diatomaceous Earth Improves Throughput and Safe Handling. J. Biotechnol. 2021, 326, 11–20. [Google Scholar] [CrossRef]
- Sastry, L.; Xu, Y.; Cooper, R.; Pollok, K.; Cornetta, K. Evaluation of Plasmid DNA Removal from Lentiviral Vectors by Benzonase Treatment. Hum. Gene Ther. 2004, 15, 221–226. [Google Scholar] [CrossRef]
- Merten, O.W.; Hebben, M.; Bovolenta, C. Production of Lentiviral Vectors. Mol. Ther. Methods Clin. Dev. 2016, 3, 16017. [Google Scholar] [CrossRef]
- Ruscic, J.; Perry, C.; Mukhopadhyay, T.; Takeuchi, Y.; Bracewell, D.G. Lentiviral Vector Purification Using Nanofiber Ion-Exchange Chromatography. Mol. Ther. Methods Clin. Dev. 2019, 15, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Kumru, O.S.; Wang, Y.; Gombotz, C.W.R.; Kelley-Clarke, B.; Cieplak, W.; Kim, T.; Joshi, S.B.; Volkin, D.B. Physical Characterization and Stabilization of a Lentiviral Vector Against Adsorption and Freeze-Thaw. J. Pharm. Sci. 2018, 107, 2764–2774. [Google Scholar] [CrossRef]
- Valkama, A.J.; Oruetxebarria, I.; Lipponen, E.M.; Leinonen, H.M.; Käyhty, P.; Hynynen, H.; Turkki, V.; Malinen, J.; Miinalainen, T.; Heikura, T.; et al. Development of Large-Scale Downstream Processing for Lentiviral Vectors. Mol. Ther. Methods Clin. Dev. 2020, 17, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Jones, P. Lentiviral Vector CMC Considerations for Clinical and Commercial Use. In Proceedings of the ATMP Manufacturing Community 19th Technical Meeting: Manufacturing ATMPs at Scale, Dublin, Ireland, 10 October 2019. [Google Scholar]
- Truran, R.; Buckley, R.; Radcliffe, P.; Miskin, J.; Mitrophanous, K. Virus Purification; United States Patent and Trademark Office: Washington, DC, USA, 2015. [Google Scholar]
- Newell, E.W.; Davis, M.M. Beyond Model Antigens: High-Dimensional Methods for the Analysis of Antigen-Specific T Cells. Nat. Biotechnol. 2014, 32, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Q.; Ma, K.Y.; Schonnesen, A.A.; Zhang, M.; He, C.; Sun, E.; Williams, C.M.; Jia, W.; Jiang, N. High-Throughput Determination of the Antigen Specificities of T Cell Receptors in Single Cells. Nat. Biotechnol. 2018, 36, 1156–1159. [Google Scholar] [CrossRef] [PubMed]
- Spottiswoode, C.N.; Busch, R. Vive La Difference! Self/Non-Self Recognition and the Evolution of Signatures of Identity in Arms Races with Parasites. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Eun, S.-Y.; Lee, S.-W.; Xu, Y.; Croft, M. 4-1BB Ligand Signaling to T Cells Limits T Cell Activation. J. Immunol. 2015, 194, 134–141. [Google Scholar] [CrossRef]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Lin, Q.; Zhang, Z.; Zhang, L. Therapeutic Strategies for the Costimulatory Molecule OX40 in T-Cell-Mediated Immunity. Acta Pharm. Sin. B 2020, 10, 414–433. [Google Scholar] [CrossRef] [PubMed]
- Pennock, N.D.; White, J.T.; Cross, E.W.; Cheney, E.E.; Tamburini, B.A.; Kedl, R.M. T Cell Responses: Naïve to Memory and Everything in Between. Am. J. Physiol. Adv. Physiol. Educ. 2013, 37, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clénet, M.L.; Gagnon, F.; Moratalla, A.C.; Viel, E.C.; Arbour, N. Peripheral Human CD4+CD8+ T Lymphocytes Exhibit a Memory Phenotype and Enhanced Responses to IL-2, IL-7 and IL-15. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Baliu-Piqué, M.; Verheij, M.W.; Drylewicz, J.; Ravesloot, L.; de Boer, R.J.; Koets, A.; Tesselaar, K.; Borghans, J.A.M. Short Lifespans of Memory T-Cells in Bone Marrow, Blood, and Lymph Nodes Suggest That T-Cell Memory Is Maintained by Continuous Self-Renewal of Recirculating Cells. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Germain, R.N. T-Cell Development and the CD4-CD8 Lineage Decision. Nat. Rev. Immunol. 2002, 2, 309–322. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candeias, S.M.; Gaipl, U.S. The Immune System in Cancer Prevention, Development and Therapy. Anticancer. Agents Med. Chem. 2015, 16, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Aguilera, J.V.; Palermo, B.; Markovic, S.N.; Nisticò, P.; Signore, A. Relevance of Immune Cell and Tumor Microenvironment Imaging in the New Era of Immunotherapy. J. Exp. Clin. Cancer Res. 2020, 39, 1–21. [Google Scholar] [CrossRef]
- Forget, M.A.; Haymaker, C.; Hess, K.R.; Meng, Y.J.; Creasy, C.; Karpinets, T.; Fulbright, O.J.; Roszik, J.; Woodman, S.E.; Kim, Y.U.; et al. Prospective Analysis of Adoptive TIL Therapy in Patients with Metastatic Melanoma: Response, Impact of Anti-CTLA4, and Biomarkers to Predict Clinical Outcome. Clin. Cancer Res. 2018, 24, 4416–4428. [Google Scholar] [CrossRef] [Green Version]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune Evasion in Cancer: Mechanistic Basis and Therapeutic Strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Gorochov, G.; Waks, T.; Eshhar, Z. Generation of Effector T Cells Expressing Chimeric T Cell Receptor with Antibody Type-Specificity. Transplant. Proc. 1989, 21, 127–130. [Google Scholar]
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of Chimeric Receptor Composed of Immunoglobulin-Derived V Resions and T-Cell Receptor-Derived C Regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968. [Google Scholar] [CrossRef]
- Blaeschke, F.; Stenger, D.; Kaeuferle, T.; Willier, S.; Lotfi, R.; Kaiser, A.D.; Assenmacher, M.; Döring, M.; Feucht, J.; Feuchtinger, T. Induction of a Central Memory and Stem Cell Memory Phenotype in Functionally Active CD4+ and CD8+ CAR T Cells Produced in an Automated Good Manufacturing Practice System for the Treatment of CD19+ Acute Lymphoblastic Leukemia. Cancer Immunol. Immunother. 2018, 67, 1053–1066. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.A.; et al. Tethered IL-15 Augments Antitumor Activity and Promotes a Stem-Cell Memory Subset in Tumor-Specific T Cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, C.T.; Krenciute, G. Next Generation CAR T Cells for the Immunotherapy of High-Grade Glioma. Front. Oncol. 2019, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Townsend, M.H.; Bennion, K.; Robison, R.A.; O’Neill, K.L. Paving the Way towards Universal Treatment with Allogenic T Cells. Immunol. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Rivière, I. Clinical Manufacturing of CAR T Cells: Foundation of a Promising Therapy. Mol. Ther. Oncolytics 2016, 3, 16015. [Google Scholar] [CrossRef] [Green Version]
- Tyagarajan, S.; Spencer, T.; Smith, J. Optimizing CAR-T Cell Manufacturing Processes during Pivotal Clinical Trials. Mol. Ther. Methods Clin. Dev. 2020, 16, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Wiesinger, M.; März, J.; Kummer, M.; Schuler, G.; Dörrie, J.; Schuler-Thurner, B.; Schaft, N. Clinical-Scale Production of Car-t Cells for the Treatment of Melanoma Patients by Mrna Transfection of a Cspg4-Specific Car under Full Gmp Compliance. Cancers 2019, 11, 1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T Cell Expansion and Prolonged Persistence in Pediatric Patients with ALL Treated with a Low-Affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef]
- Xhangolli, I.; Dura, B.; Lee, G.H.; Kim, D.; Xiao, Y.; Fan, R. Single-Cell Analysis of CAR-T Cell Activation Reveals A Mixed TH1/TH2 Response Independent of Differentiation. Genom. Proteom. Bioinforma. 2019, 17, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Santomasso, B.; Bachier, C.; Westin, J.; Rezvani, K.; Shpall, E.J. The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. Am. Soc. Clin. Oncol. Educ. B. 2019, 39, 433–444. [Google Scholar] [CrossRef]
- Fesnak, A.D. The Challenge of Variability in Chimeric Antigen Receptor T Cell Manufacturing. Regen. Eng. Transl. Med. 2019, 1–8. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and Management in CAR T-Cell Therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, K.T.; Jadlowsky, J.K.; Hwang, W.T.; Suhoski-Davis, M.; Gonzalez, V.E.; Kulikovskaya, I.; Gupta, M.; Lacey, S.F.; Plesa, G.; Chew, A.; et al. Retroviral and Lentiviral Safety Analysis of Gene-Modified T Cell Products and Infused HIV and Oncology Patients. Mol. Ther. 2018, 26, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.; Stanton, J.; Powers, D.; Karnieli, O.; Nahum, S.; Abraham, E.; Parisse, J.S.; Oh, S. Managing Particulates in Cell Therapy: Guidance for Best Practice. Cytotherapy 2016, 18, 1063–1076. [Google Scholar] [CrossRef]
- Maziarz, R.T.; Waller, E.K.; Jaeger, U.; Fleury, I.; McGuirk, J.; Holte, H.; Jaglowski, S.; Schuster, S.J.; Bishop, M.R.; Westin, J.R.; et al. Patient-Reported Long-Term Quality of Life after Tisagenlecleucel in Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Blood Adv. 2020, 4, 629–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency Meeting Report: Workshop with Stakeholders on Support to Quality Development in Early Access Approaches (i.e., PRIME, Breakthrough Therapies). 2018.
- Stirrups, R. CAR T-Cell Therapy for Relapsed or Refractory Mantle-Cell Lymphoma. Lancet. Oncol. 2020, 21, e239. [Google Scholar] [CrossRef]
- Papadouli, I.; Mueller-Berghaus, J.; Beuneu, C.; Ali, S.; Hofner, B.; Petavy, F.; Tzogani, K.; Miermont, A.; Norga, K.; Kholmanskikh, O.; et al. EMA Review of Axicabtagene Ciloleucel (Yescarta) for the Treatment of Diffuse Large B-Cell Lymphoma. Oncologist 2020, 25, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, T.W.; Myers, G.D.; Baruchel, A.; Dietz, A.C.; Pulsipher, M.A.; Bittencourt, H.; Buechner, J.; De Moerloose, B.; Davis, K.L.; Nemecek, E.; et al. Patient-Reported Quality of Life after Tisagenlecleucel Infusion in Children and Young Adults with Relapsed or Refractory B-Cell Acute Lymphoblastic Leukaemia: A Global, Single-Arm, Phase 2 Trial. Lancet Oncol. 2019, 20, 1710–1718. [Google Scholar] [CrossRef]
- Detela, G.; Lodge, A. EU Regulatory Pathways for ATMPs: Standard, Accelerated and Adaptive Pathways to Marketing Authorisation. Mol. Ther. Methods Clin. Dev. 2019, 13, 205–232. [Google Scholar] [CrossRef] [Green Version]
- Sena-Esteves, M.; Gao, G. Titration of Lentivirus Vectors. Cold Spring Harb. Protoc. 2018, 2018, 281–285. [Google Scholar] [CrossRef]
- Lana, M.G.; Strauss, B.E. Production of Lentivirus for the Establishment of CAR-T Cells. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2020; Volume 2086, pp. 61–67. [Google Scholar]
- Yu, S.; Yi, M.; Qin, S.; Wu, K. Next Generation Chimeric Antigen Receptor T Cells: Safety Strategies to Overcome Toxicity. Mol. Cancer 2019, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, K.; Al-Obaidi, M.; Di Stasi, A. Generation of suicide gene-modified chimeric antigen receptor-redirected T-cells for cancer immunotherapy. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2019; Volume 1895, pp. 57–73. [Google Scholar]
- ClinicalTrials.gov. 3rd Generation GD-2 Chimeric Antigen Receptor and ICaspase Suicide Safety Switch. Available online: https://clinicaltrials.gov/ct2/show/NCT01822652?term=CAR+suicide&draw=2&rank=1 (accessed on 17 June 2021).
- ClinicalTrials.gov. A Phase I Trial of T Cells Expressing an Anti-GD2 Chimeric Antigen Receptor in Children and Young Adults With GD2+ Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT02107963?term=CAR+suicide&draw=2&rank=5 (accessed on 17 June 2021).
- Amatya, C.; Pegues, M.A.; Lam, N.; Vanasse, D.; Geldres, C.; Choi, S.; Hewitt, S.M.; Feldman, S.A.; Kochenderfer, J.N. Development of CAR T Cells Expressing a Suicide Gene Plus a Chimeric Antigen Receptor Targeting Signaling Lymphocytic-Activation Molecule F7. Mol. Ther. 2021, 29, 702–717. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I.; et al. Induction of Resistance to Chimeric Antigen Receptor T Cell Therapy by Transduction of a Single Leukemic B Cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.M.; Braun, A.H.; Scheib, L.; Agarwal, S.; Schneider, I.C.; Fusil, F.; Perian, S.; Sahin, U.; Thalheimer, F.B.; Verhoeyen, E.; et al. Combining T-Cell-Specific Activation and in Vivo Gene Delivery through CD3-Targeted Lentiviral Vectors. Blood Adv. 2020, 4, 5702–5715. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Sun, Q.; Liang, X.; Chen, Z.; Zhang, X.; Zhou, X.; Li, M.; Tu, H.; Liu, Y.; Tu, S.; et al. Mechanisms of Relapse After CD19 CAR T-Cell Therapy for Acute Lymphoblastic Leukemia and Its Prevention and Treatment Strategies. Front. Immunol. 2019, 10, 2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Wang, X.Q.; Zhang, R.L.; Liu, F.; Wang, Y.; Yan, Z.L.; Song, Y.P.; Yang, T.; Li, P.; Wang, Z.; et al. Donor-Derived CD19 CAR-T Cell Therapy of Relapse of CD19-Positive B-ALL Post Allotransplant. Leukemia 2021, 35, 1563–1570. [Google Scholar] [CrossRef]
- Tong, C.; Zhang, Y.; Liu, Y.; Ji, X.; Zhang, W.; Guo, Y.; Han, X.; Ti, D.; Dai, H.; Wang, C.; et al. Optimized Tandem CD19/CD20 CAR-Engineered T Cells in Refractory/Relapsed B-Cell Lymphoma. Blood 2020, 136, 1632–1644. [Google Scholar] [CrossRef]
- Fousek, K.; Watanabe, J.; Joseph, S.K.; George, A.; An, X.; Byrd, T.T.; Morris, J.S.; Luong, A.; Martínez-Paniagua, M.A.; Sanber, K.; et al. CAR T-Cells That Target Acute B-Lineage Leukemia Irrespective of CD19 Expression. Leukemia 2020. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Fujiwara, K.; Tsunei, A.; Kusabuka, H.; Ogaki, E.; Tachibana, M.; Okada, N. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells 2020, 9, 1182. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. Driving T-Cell Immunotherapy to Solid Tumors. Nat. Biotechnol. 2018, 36, 215–219. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef]
- Ang, W.X.; Ng, Y.Y.; Xiao, L.; Chen, C.; Li, Z.; Chi, Z.; Tay, J.C.K.; Tan, W.K.; Zeng, J.; Toh, H.C.; et al. Electroporation of NKG2D RNA CAR Improves Vγ9Vδ2 T Cell Responses against Human Solid Tumor Xenografts. Mol. Ther. Oncolytics 2020, 17, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, K.; Rengstl, B.; Oehm, P.; Michel, K.; Billmeier, A.; Hayduk, N.; Klein, O.; Kuna, K.; Ouchan, Y.; Wöll, S.; et al. An RNA Vaccine Drives Expansion and Efficacy of Claudin-CAR-T Cells against Solid Tumors. Science 2020, 367, 446–453. [Google Scholar] [CrossRef]
- Andersch, L.; Radke, J.; Klaus, A.; Schwiebert, S.; Winkler, A.; Schumann, E.; Grunewald, L.; Zirngibl, F.; Flemmig, C.; Jensen, M.C.; et al. CD171- and GD2-Specific CAR-T Cells Potently Target Retinoblastoma Cells in Preclinical in Vitro Testing. BMC Cancer 2019, 19. [Google Scholar] [CrossRef] [Green Version]
- Marcinkowski, B.; Stevanović, S.; Helman, S.R.; Norberg, S.M.; Serna, C.; Jin, B.; Gkitsas, N.; Kadakia, T.; Warner, A.; Davis, J.L.; et al. Cancer Targeting by TCR Gene-Engineered T Cells Directed against Kita-Kyushu Lung Cancer Antigen-1. J. Immunother. Cancer 2019, 7, 229. [Google Scholar] [CrossRef]
- Siegler, E.L.; Wang, P. Preclinical Models in Chimeric Antigen Receptor-Engineered T-Cell Therapy. Hum. Gene Ther. 2018, 29, 534–546. [Google Scholar] [CrossRef]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [Green Version]
- Davila, M.L.; Kloss, C.C.; Gunset, G.; Sadelain, M. CD19 CAR-Targeted T Cells Induce Long-Term Remission and B Cell Aplasia in an Immunocompetent Mouse Model of B Cell Acute Lymphoblastic Leukemia. PLoS ONE 2013, 8, e61338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.H.; Xia, J.; Rafiq, S.; Huang, X.; Hu, Z.; Zhou, X.; Brentjens, R.J.; Yang, Y.G. Modeling Anti-CD19 CAR T Cell Therapy in Humanized Mice with Human Immunity and Autologous Leukemia. EBioMedicine 2019, 39, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seimetz, D.; Heller, K.; Richter, J. Approval of First CAR-Ts: Have We Solved All Hurdles for ATMPs? Cell Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, R.P.; Zylberberg, E.; Ellison, S.; Levine, B.L. Chimeric Antigen Receptor–T Cell Therapy Manufacturing: Modelling the Effect of Offshore Production on Aggregate Cost of Goods. Cytotherapy 2019, 21, 224–233. [Google Scholar] [CrossRef]
- Fernández, L.; Fernández, A.; Mirones, I.; Escudero, A.; Cardoso, L.; Vela, M.; Lanzarot, D.; de Paz, R.; Leivas, A.; Gallardo, M.; et al. GMP-Compliant Manufacturing of NKG2D CAR Memory T Cells Using CliniMACS Prodigy. Front. Immunol. 2019, 10, 2361. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Jordan, K.R.; Schulte, B.; Purev, E. Characterization of Clinical Grade CD19 Chimeric Antigen Receptor T Cells Produced Using Automated CliniMACS Prodigy System. Drug Des. Devel. Ther. 2018, 12, 3343. [Google Scholar] [CrossRef] [Green Version]
- Marín Morales, J.M.; Münch, N.; Peter, K.; Freund, D.; Oelschlägel, U.; Hölig, K.; Böhm, T.; Flach, A.C.; Keßler, J.; Bonifacio, E.; et al. Automated Clinical Grade Expansion of Regulatory T Cells in a Fully Closed System. Front. Immunol. 2019, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Ran, T.; Eichmüller, S.B.; Schmidt, P.; Schlander, M. Cost of Decentralized CAR T-cell Production in an Academic Nonprofit Setting. Int. J. Cancer 2020, 147, 3438–3445. [Google Scholar] [CrossRef]
- Zhu, F.; Shah, N.N.; Schneider, D.; Xu, H.; Chaney, K.; Luib, L.; Keever-Taylor, C.; Dropulic, B.; Orentas, R.; Hari, P.; et al. Automated Manufacturing of CD20.19 Bi-Specific Chimeric Antigen Receptor T (CAR-T) Cells at an Academic Center for a Phase I Clinical Trial in Relapsed, Refractory NHL. Biol. Blood Marrow Transplant. 2019, 25, S62. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Smith, M.; James, S.E.; Davila, M.L.; Velardi, E.; Argyropoulos, K.V.; Gunset, G.; Perna, F.; Kreines, F.M.; Levy, E.R.; et al. Donor CD19 CAR T Cells Exert Potent Graft-versus-Lymphoma Activity with Diminished Graft-versus-Host Activity. Nat. Med. 2017, 23, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Sicard, A.; Lamarche, C.; Speck, M.; Wong, M.; Rosado-Sánchez, I.; Blois, M.; Glaichenhaus, N.; Mojibian, M.; Levings, M.K. Donor-specific Chimeric Antigen Receptor Tregs Limit Rejection in Naive but Not Sensitized Allograft Recipients. Am. J. Transplant. 2020, 20, 1562–1573. [Google Scholar] [CrossRef]
- Zhang, J.P.; Zhang, R.; Tsao, S.T.; Liu, Y.C.; Chen, X.; Lu, D.P.; Castillo, P.; Chang, L.J. Sequential Allogeneic and Autologous CAR-T-Cell Therapy to Treat an Immune-Compromised Leukemic Patient. Blood Adv. 2018, 2, 1691–1695. [Google Scholar] [CrossRef] [Green Version]
- Fesnak, A.D. The Challenge of Variability in Chimeric Antigen Receptor T Cell Manufacturing. Regen. Eng. Transl. Med. 2020, 6, 322–329. [Google Scholar] [CrossRef]
- Smith, S.D.; Reddy, P.; Sokolova, A.; Chow, V.A.; Lynch, R.C.; Shadman, M.A.; Till, B.G.; Shustov, A.R.; Warren, E.H.; Ujjani, C.S.; et al. Eligibility for CAR T-cell Therapy: An Analysis of Selection Criteria and Survival Outcomes in Chemorefractory DLBCL. Am. J. Hematol. 2019, 94, E117–E116. [Google Scholar] [CrossRef] [Green Version]
- Herrera, L.; Santos, S.; Vesga, M.A.; Anguita, J.; Martin-Ruiz, I.; Carrascosa, T.; Juan, M.; Eguizabal, C. Adult Peripheral Blood and Umbilical Cord Blood NK Cells Are Good Sources for Effective CAR Therapy against CD19 Positive Leukemic Cells. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular Remission of Infant B-ALL after Infusion of Universal TALEN Gene-Edited CAR T Cells. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Karasiewicz, K.; He, S.; Ng, M.; Tess, K.; Ling, W.; Kaufmann, G.F.; Zeldis, J.B.; Ji, H.; Hariri, R.; Zhang, X. Preclinical Evaluation of Human Placental-Derived Allogeneic CD19 CAR-T Cells Against B Cell Malignancies. Blood 2019, 134, 3222. [Google Scholar] [CrossRef]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-Man Clinical Trial of CAR NK-92 Cells: Safety Test of CD33-CAR NK-92 Cells in Patients with Relapsed and Refractory Acute Myeloid Leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar]
- Nianias, A.; Themeli, M. Induced Pluripotent Stem Cell (IPSC)–Derived Lymphocytes for Adoptive Cell Immunotherapy: Recent Advances and Challenges. Curr. Hematol. Malig. Rep. 2019, 14, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N.; et al. Genome-Edited, Donor-Derived Allogeneic Anti-CD19 Chimeric Antigen Receptor T Cells in Paediatric and Adult B-Cell Acute Lymphoblastic Leukaemia: Results of Two Phase 1 Studies. Lancet 2020, 396, 1885–1894. [Google Scholar] [CrossRef]
- Clinical Trial: Safety and Efficacy of ALLO-501 Anti-CD19 Allogeneic CAR T Cells in Adults with Relapsed/Refractory Large B Cell or Follicular Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03939026 (accessed on 23 January 2021).
- Neelapu, S.S.; Munoz, J.; Locke, F.L.; Miklos, D.B.; Brown, R.; McDevitt, J.T.; Mardiros, A.; Demirhan, E.; Konto, C.; Tees, M.T. First-in-Human Data of ALLO-501 and ALLO-647 in Relapsed/Refractory Large B-Cell or Follicular Lymphoma (R/R LBCL/FL): ALPHA Study. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Martínez-Molina, E.; Chocarro-Wrona, C.; Martínez-Moreno, D.; Marchal, J.A.; Boulaiz, H. Large-Scale Production of Lentiviral Vectors: Current Perspectives and Challenges. Pharmaceutics 2020, 12, 1051. [Google Scholar] [CrossRef]
- Karda, R.; Counsell, J.R.; Karbowniczek, K.; Caproni, L.J.; Tite, J.P.; Waddington, S.N. Production of Lentiviral Vectors Using Novel, Enzymatically Produced, Linear DNA. Gene Ther. 2019, 26, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Goto, Y.; Yonemitsu, Y.; Miyazaki, M.; Sakamoto, T.; Ishibashi, T.; Tabata, T.; Ueda, Y.; Hasegawa, M.; Tobimatsu, S.; et al. Simian Immunodeficiency Virus-Based Lentivirus Vector for Retinal Gene Transfer: A Preclinical Safety Study in Adult Rats. Gene Ther. 2003, 10, 1161–1169. [Google Scholar] [CrossRef] [Green Version]
- Throm, R.E.; Ouma, A.A.; Zhou, S.; Chandrasekaran, A.; Lockey, T.; Greene, M.; De Ravin, S.S.; Moayeri, M.; Malech, H.L.; Sorrentino, B.P.; et al. Efficient Construction of Producer Cell Lines for a SIN Lentiviral Vector for SCID-X1 Gene Therapy by Concatemeric Array Transfection. Blood 2009, 113, 5104–5110. [Google Scholar] [CrossRef] [Green Version]
- Stornaiuolo, A.; Piovani, B.M.; Bossi, S.; Zucchelli, E.; Corna, S.; Salvatori, F.; Mavilio, F.; Bordignon, C.; Rizzardi, G.P.; Bovolenta, C. RD2-Molpack-Chim3, a Packaging Cell Line for Stable Production of Lentiviral Vectors for Anti-HIV Gene Therapy. Hum. Gene Ther. Methods 2013, 24, 228–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanber, K.S.; Knight, S.B.; Stephen, S.L.; Bailey, R.; Escors, D.; Minshull, J.; Santilli, G.; Thrasher, A.J.; Collins, M.K.; Takeuchi, Y. Construction of Stable Packaging Cell Lines for Clinical Lentiviral Vector Production. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef]
- Manceur, A.P.; Kim, H.; Misic, V.; Andreev, N.; Dorion-Thibaudeau, J.; Lanthier, S.; Bernier, A.; Tremblay, S.; Gélinas, A.M.; Broussau, S.; et al. Scalable Lentiviral Vector Production Using Stable HEK293SF Producer Cell Lines. Hum. Gene Ther. Methods 2017, 28, 330–339. [Google Scholar] [CrossRef]
- Jiang, W.; Hua, R.; Wei, M.; Li, C.; Qiu, Z.; Yang, X.; Zhang, C. An Optimized Method for High-Titer Lentivirus Preparations without Ultracentrifugation. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bauler, M.; Roberts, J.K.; Wu, C.-C.; Fan, B.; Ferrara, F.; Yip, B.H.; Diao, S.; Kim, Y.-I.; Moore, J.; Zhou, S.; et al. Production of Lentiviral Vectors Using Suspension Cells Grown in Serum-Free Media. Mol. Ther. Methods Clin. Dev. 2020, 17, 58–68. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Pallant, C.; Sampson, C.J.; Boiti, A.; Johnson, S.; Brazauskas, P.; Hardwicke, P.; Marongiu, M.; Marinova, V.M.; Carmo, M.; et al. Rapid Lentiviral Vector Producer Cell Line Generation Using a Single DNA Construct. Mol. Ther. Methods Clin. Dev. 2020, 19, 47–57. [Google Scholar] [CrossRef]
- Johnson, S.; Pallant, C.; Vamva, E.; Vink, C. Stable Cell Lines for Retroviral Production; World Intellectual Property Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Verhoeyen, E.; Costa, C.; Cosset, F.L. Lentiviral Vector Gene Transfer into Human T Cells. Methods Mol. Biol. 2009, 506, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Wheeler, J.X.; Thorpe, R.; Collins, M.; Takeuchi, Y.; Zhao, Y. Mass Spectrometry Analysis Reveals Differences in the Host Cell Protein Species Found in Pseudotyped Lentiviral Vectors. Biologicals 2018, 52, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Mekkaoui, L.; Parekh, F.; Kotsopoulou, E.; Darling, D.; Dickson, G.; Cheung, G.W.; Chan, L.; MacLellan-Gibson, K.; Mattiuzzo, G.; Farzaneh, F.; et al. Lentiviral Vector Purification Using Genetically Encoded Biotin Mimic in Packaging Cell. Mol. Ther. Methods Clin. Dev. 2018, 11, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pule, M.; Mekkaoui, L. Retroviral and Lentiviral Vectors; World Intellectual Property Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Rath, J.A.; Arber, C. Engineering Strategies to Enhance TCR-Based Adoptive T Cell Therapy. Cells 2020, 9, 1485. [Google Scholar] [CrossRef]
- Zhou, J.; Jin, L.; Wang, F.; Zhang, Y.; Liu, B.; Zhao, T. Chimeric Antigen Receptor T (CAR-T) Cells Expanded with IL-7/IL-15 Mediate Superior Antitumor Effects. Protein Cell 2019, 10, 764–769. [Google Scholar] [CrossRef] [Green Version]
- Stadtmauer, E.A.; Faitg, T.H.; Lowther, D.E.; Badros, A.Z.; Chagin, K.; Dengel, K.; Iyengar, M.; Melchiori, L.; Navenot, J.M.; Norry, E.; et al. Long-term Safety and Activity of NY-ESO-1 SPEAR T Cells after Autologous Stem Cell Transplant for Myeloma. Blood Adv. 2019, 3, 2022–2034. [Google Scholar] [CrossRef]
- Clinical Trial: Redirected Auto T Cells for Advanced Myeloma. Available online: https://clinicaltrials.gov/ct2/show/NCT01352286 (accessed on 27 January 2021).
- Sanderson, J.P.; Crowley, D.J.; Wiedermann, G.E.; Quinn, L.L.; Crossland, K.L.; Tunbridge, H.M.; Cornforth, T.V.; Barnes, C.S.; Ahmed, T.; Howe, K.; et al. Preclinical Evaluation of an Affinity-Enhanced MAGE-A4-Specific T-Cell Receptor for Adoptive T-Cell Therapy. Oncoimmunology 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Abken, H. TRUCKS, the Fourth-generation CAR T Cells: Current Developments and Clinical Translation. Adv. Cell Gene Ther. 2020, 3, e84. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. EGFR-IL12-CART Cells for Patients With Metastatic Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03542799 (accessed on 20 June 2021).
- Zimmermann, K.; Kuehle, J.; Dragon, A.C.; Galla, M.; Kloth, C.; Rudek, L.S.; Sandalcioglu, I.E.; Neyazi, B.; Moritz, T.; Meyer, J.; et al. Design and Characterization of an “All-in-One” Lentiviral Vector System Combining Constitutive Anti-Gd2 Car Expression and Inducible Cytokines. Cancers 2020, 12, 375. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Burga, R.A.; Powell, A.B.; Chorvinsky, E.A.; Hoq, N.; McCormack, S.E.; Van Pelt, S.N.; Hanley, P.J.; Cruz, C.R.Y. Beyond CAR T Cells: Other Cell-Based Immunotherapeutic Strategies against Cancer. Front. Oncol. 2019, 9, 196. [Google Scholar] [CrossRef] [Green Version]
- Heczey, A.; Liu, D.; Tian, G.; Courtney, A.N.; Wei, J.; Marinova, E.; Gao, X.; Guo, L.; Yvon, E.; Hicks, J.; et al. Invariant NKT Cells with Chimeric Antigen Receptor Provide a Novel Platform for Safe and Effective Cancer Immunotherapy. Blood 2014, 124, 2824–2833. [Google Scholar] [CrossRef] [Green Version]
- Vantourout, P.; Hayday, A. Six-of-the-Best: Unique Contributions of Γδ T Cells to Immunology. Nat. Rev. Immunol. 2013, 13, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Minculescu, L.; Marquart, H.V.; Ryder, L.P.; Andersen, N.S.; Schjoedt, I.; Friis, L.S.; Kornblit, B.T.; Petersen, S.L.; Haastrup, E.; Fischer-Nielsen, A.; et al. Improved Overall Survival, Relapse-Free-Survival, and Less Graft-vs.-Host-Disease in Patients with High Immune Reconstitution of TCR Gamma Delta Cells 2 Months after Allogeneic Stem Cell Transplantation. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.N.; Wen, Q.; He, W.T.; Yang, J.H.; Zhou, C.Y.; Xiong, W.J.; Ma, L. Optimized Protocols for Γδ T Cell Expansion and Lentiviral Transduction. Mol. Med. Rep. 2019, 19, 1471–1480. [Google Scholar] [CrossRef] [Green Version]
- Sutton, K.; Dasgupta, A.; David, M.; Doering, C.; Spencer, H.T. 402. Bioengineering of Peripheral Blood Derived Gamma Delta T Cells in a Serum-Free Expansion Medium. Mol. Ther. 2016, 24, S159. [Google Scholar] [CrossRef]
- Charles David, P.; Haishan, L.; Lahusen, T.; Liou, M. Methods and Compositions for the Activation of Gamma-Delta T-Cells; World Intellectual Property Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Clinical Trial: Novel Gamma-Delta (Γδ) T Cell Therapy for Treatment of Patients With Newly Diagnosed Glioblastoma. Available online: https://clinicaltrials.gov/ct2/show/study/NCT04165941 (accessed on 22 January 2021).
- Altman, J.B.; Benavides, A.D.; Das, R.; Bassiri, H. Antitumor Responses of Invariant Natural Killer T Cells. J. Immunol. Res. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Heczey, A.; Courtney, A.N.; Montalbano, A.; Robinson, S.; Liu, K.; Li, M.; Ghatwai, N.; Dakhova, O.; Liu, B.; Raveh-Sadka, T.; et al. Anti-GD2 CAR-NKT Cells in Patients with Relapsed or Refractory Neuroblastoma: An Interim Analysis. Nat. Med. 2020, 26, 1686–1690. [Google Scholar] [CrossRef]
- Xu, X.; Huang, W.; Heczey, A.; Liu, D.; Guo, L.; Wood, M.; Jin, J.; Courtney, A.N.; Liu, B.; Di Pierro, E.J.; et al. NKT Cells Coexpressing a GD2-Specific Chimeric Antigen Receptor and IL15 Show Enhanced in Vivo Persistence and Antitumor Activity against Neuroblastoma. Clin. Cancer Res. 2019, 25, 7126–7138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.gov. GD2 Specific CAR and Interleukin-15 Expressing Autologous NKT Cells to Treat Children With Neuroblastoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03294954?cond=CAR+NKT&draw=2&rank=1 (accessed on 20 June 2021).
- ClinicalTrials.gov. Clinical Study of CAR-INKT Cells in the Treatment of Relapsed/Refractory/High-Risk B-Cell Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT04814004?cond=iNKT&draw=2&rank=2 (accessed on 20 June 2021).
- Zhao, H.; Liao, X.; Kang, Y. Tregs: Where We Are and What Comes Next? Front. Immunol. 2017, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lu, W.; Liang, C.-L.; Chen, Y.; Liu, H.; Qiu, F.; Dai, Z. Chimeric Antigen Receptor (CAR) Treg: A Promising Approach to Inducing Immunological Tolerance. Front. Immunol. 2018, 9, 2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsche, E.; Volk, H.D.; Reinke, P.; Abou-El-Enein, M. Toward an Optimized Process for Clinical Manufacturing of CAR-Treg Cell Therapy. Trends Biotechnol. 2020, 38, 1099–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency Clinical Trials Register. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2019-001730-34/NL (accessed on 23 January 2021).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| INN 1 (Commercial Name) | Manufacturer | Application(s) | Therapy Type | Market Approval | Price per Dose (USD 4) | Reference | ||
|---|---|---|---|---|---|---|---|---|
| EMA 2 | FDA 3 | |||||||
| brexucabtagene autoleucel (Tecartus®) | Kite Pharma Inc. (Gilead) | Mantle cell lymphoma | CAR T cellGRV vector | 2020 | 2020 | $ | 373,000 | [9,10] |
| tisagenlecleucel (Kymriah®) | Novartis AG | Acute B-cell lymphoblastic leukaemia | CAR T cellLV vector | 2018 | 2017 | $ | 475,000 | [5,6] |
| axicabtagene ciloleucel (Yescarta®) | Kite Pharma Inc. (Gilead) | B cell lymphoma | CAR T cellGRV vector | 2018 | 2017 | $ | 373,000 | [7,8] |
| Product | Activation Method | Antibody Scaffold |
|---|---|---|
| Dynabeads (Gibco) | CD3/CD28 Antibody-mediated | Magnetic beads |
| TransAct (Miltenyi Biotec) | CD3/CD28 Antibody-mediated | Polymer beads |
| Cloudz (Bio-Techne) | CD3/CD28 Antibody-mediated | Dissolvable beads |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Labbé, R.P.; Vessillier, S.; Rafiq, Q.A. Lentiviral Vectors for T Cell Engineering: Clinical Applications, Bioprocessing and Future Perspectives. Viruses 2021, 13, 1528. https://doi.org/10.3390/v13081528
Labbé RP, Vessillier S, Rafiq QA. Lentiviral Vectors for T Cell Engineering: Clinical Applications, Bioprocessing and Future Perspectives. Viruses. 2021; 13(8):1528. https://doi.org/10.3390/v13081528
Chicago/Turabian StyleLabbé, Roman P., Sandrine Vessillier, and Qasim A. Rafiq. 2021. "Lentiviral Vectors for T Cell Engineering: Clinical Applications, Bioprocessing and Future Perspectives" Viruses 13, no. 8: 1528. https://doi.org/10.3390/v13081528