
Inhibiting ACSL1-Related Ferroptosis Restrains Murine Coronavirus Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Primary Macrophages
2.3. Murine Hepatitis Virus Culture
2.4. Reagents and Drugs
2.5. Virus Infection in Cells
2.6. RNA Isolation and RT-qPCR
2.7. Bulk RNA-Seq
2.8. PI Staining
2.9. Cell Imaging
2.10. Cytokine Measurement and LDH Assay
2.11. Plaque Assay
2.12. Animal Model
2.13. Histology
2.14. Statistical Analysis
3. Results
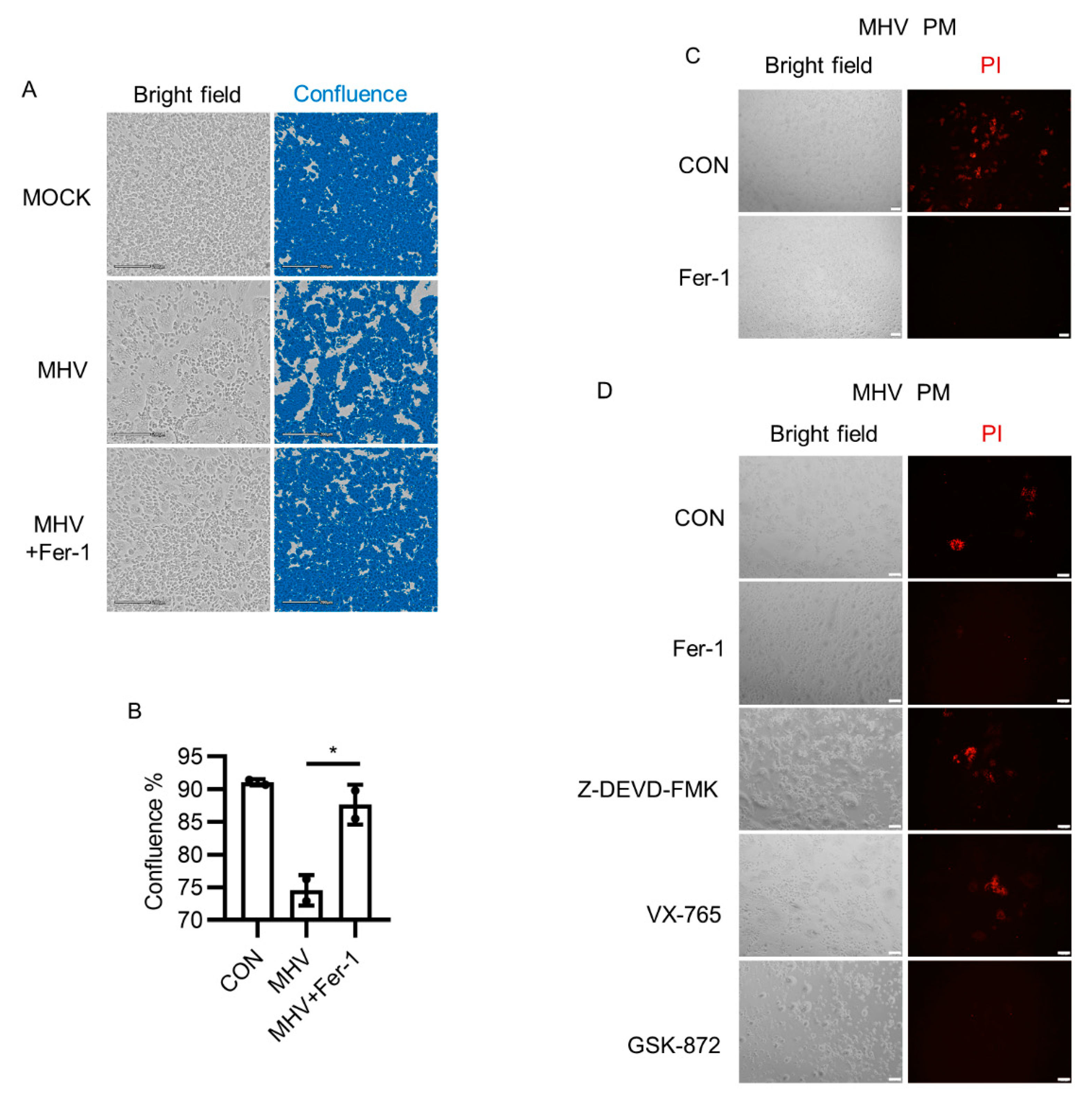
3.1. Ferroptosis Inhibitors Suppressed Syncytia Formation after MHV-A59 Infection
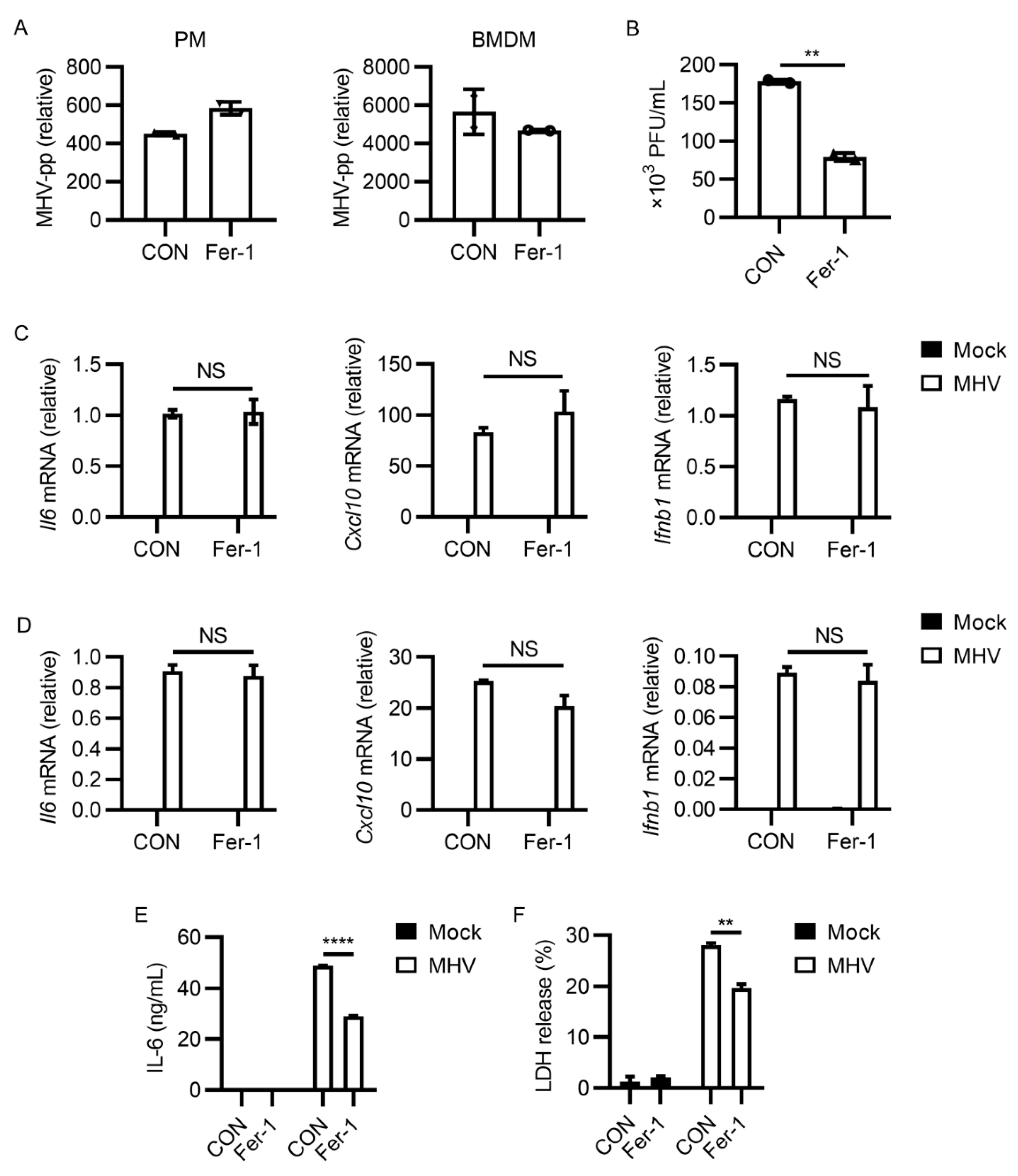
3.2. Ferroptosis Inhibitors Reduced Viral Load and Inflammatory Cytokine Release after MHV-A59 Infection
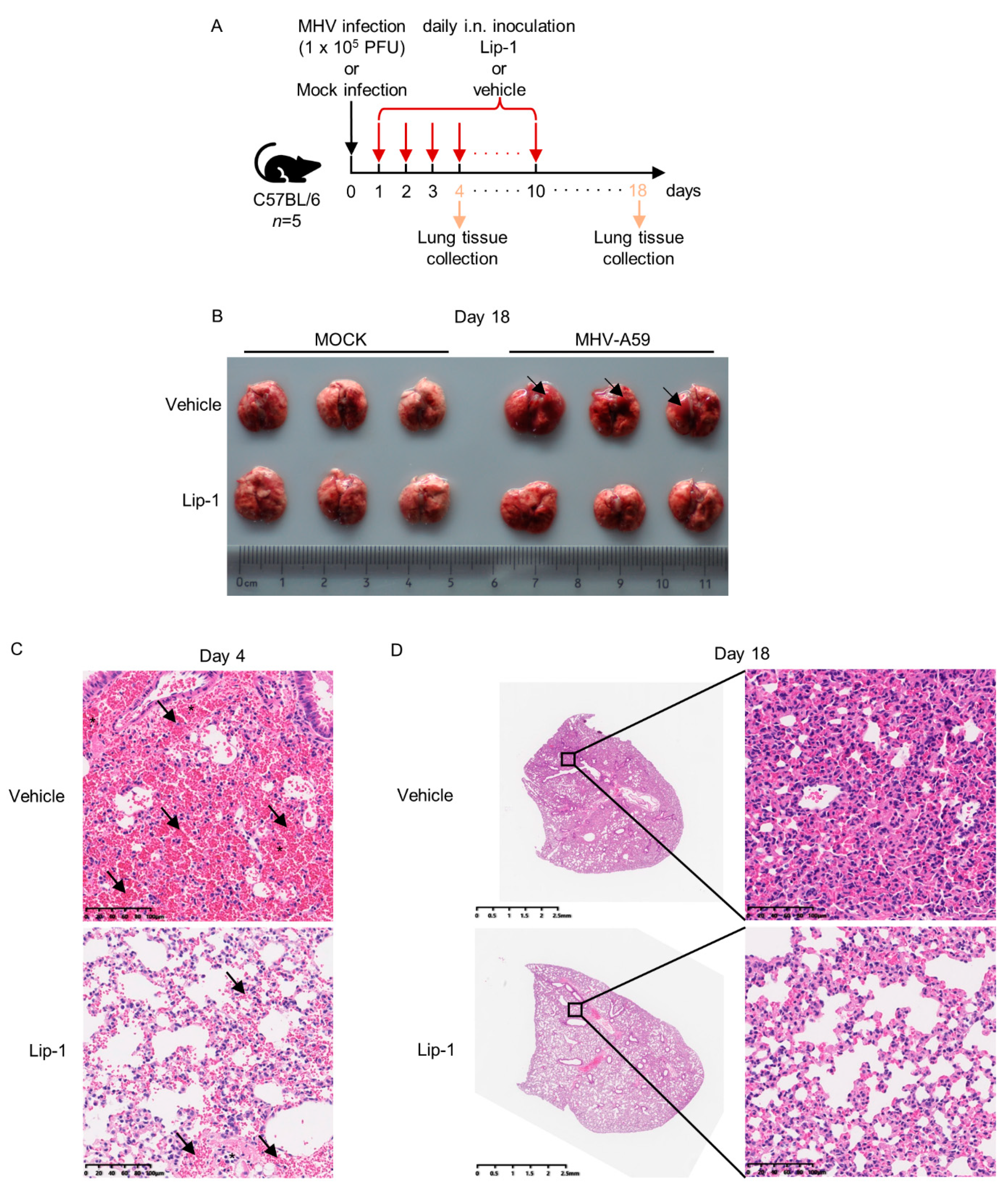
3.3. Liproxstatin-1 Ameliorated MHV-A59 Infection-Induced Inflammation and Lung Injury
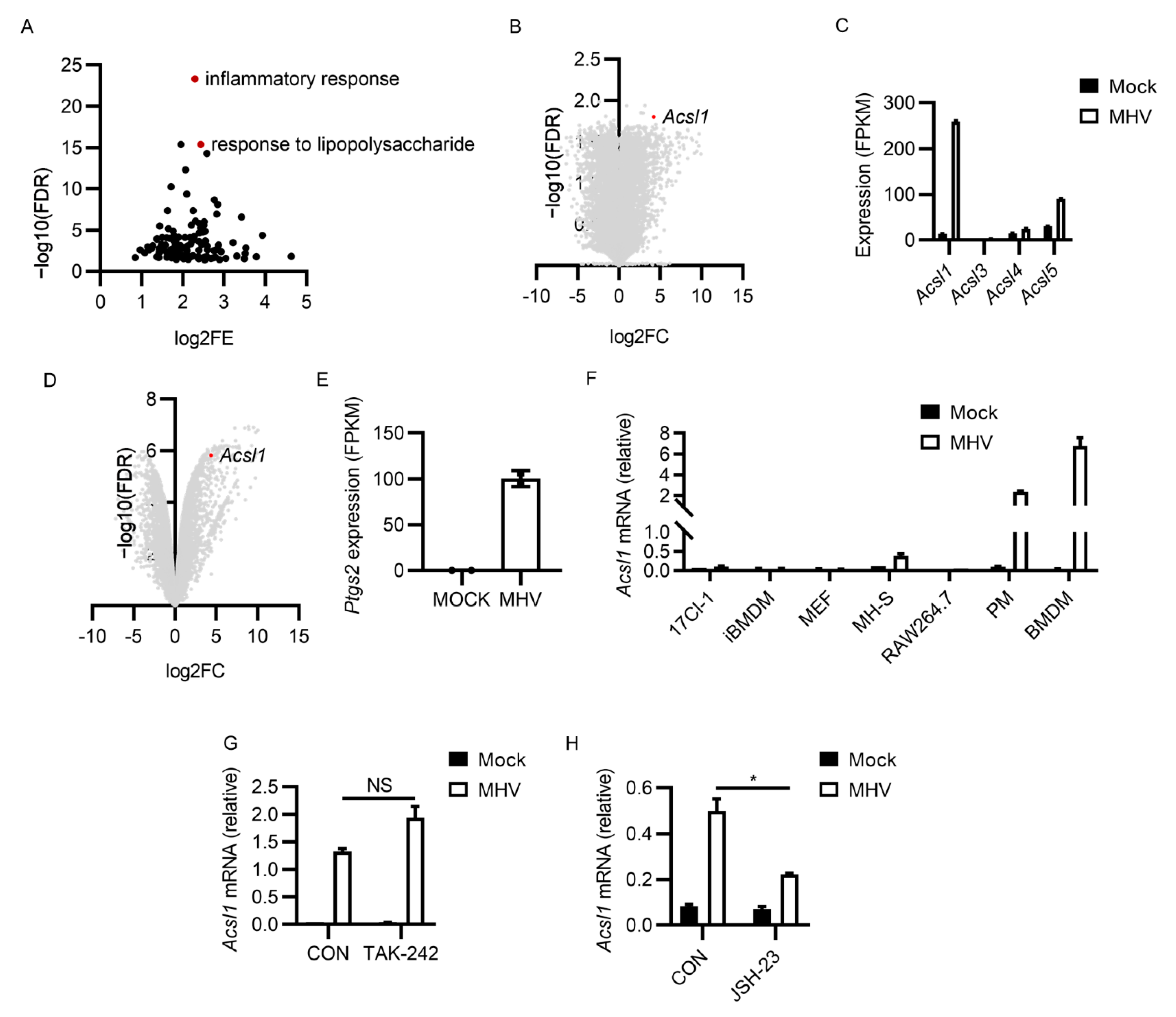
3.4. MHV-A59 Infection Induced Potent ACSL1 Expression
3.5. Inhibiting ACSL1 Suppressed MHV-A59 Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. WHO Coronavirus (COVID-19) Dashboard; WHO Media Centre: Geneva, Switzerland, 2021; Available online: https://covid19.who.int/ (accessed on 23 November 2021).
- Salton, F.; Confalonieri, P.; Meduri, G.U.; Santus, P.; Harari, S.; Scala, R.; Lanini, S.; Vertui, V.; Oggionni, T.; Caminati, A.; et al. Prolonged low-dose methylprednisolone in patients with severe COVID-19 pneumonia. Open Forum Infect. Dis. 2020, 7, ofaa421. [Google Scholar] [CrossRef] [PubMed]
- Baratella, E.; Ruaro, B.; Marrocchio, C.; Starvaggi, N.; Salton, F.; Giudici, F.; Quaia, E.; Confalonieri, M.; Cova, M.A. Interstitial lung disease at high resolution CT after SARS-CoV-2-related acute respiratory distress syndrome according to pulmonary segmental anatomy. J. Clin. Med. 2021, 10, 3985. [Google Scholar] [CrossRef] [PubMed]
- Lavi, E.; Gilden, D.H.; Wroblewska, Z.; Rorke, L.B.; Weiss, S.R. Experimental demyelination produced by the A59 strain of mouse hepatitis virus. Neurology 1984, 34, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Du, J.; Chen, G.; Zhao, J.; Yang, X.; Su, L.; Cheng, G.; Tang, H. Coronavirus MHV-A59 infects the lung and causes severe pneumonia in C57BL/6 mice. Virol. Sin. 2014, 29, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Ryu, S.; Shchukina, I.; Youm, Y.H.; Qing, H.; Hilliard, B.; Dlugos, T.; Zhang, X.; Yasumoto, Y.; Booth, C.J.; Fernández-Hernando, C.; et al. Ketogenic diet restrains aging-induced exacerbation of coronavirus infection in mice. eLife 2021, 10, e66522. [Google Scholar] [CrossRef]
- Guo, Q.; Zhao, Y.; Li, J.; Liu, J.; Yang, X.; Guo, X.; Kuang, M.; Xia, H.; Zhang, Z.; Cao, L.; et al. Induction of alarmin S100A8/A9 mediates activation of aberrant neutrophils in the pathogenesis of COVID-19. Cell Host Microbe 2021, 29, 222–235. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 2017, 171, 628–641. [Google Scholar] [CrossRef] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Beatty, A.; Singh, T.; Tyurina, Y.Y.; Tyurin, V.A.; Samovich, S.; Nicolas, E.; Maslar, K.; Zhou, Y.; Cai, K.Q.; Tan, Y.; et al. Ferroptotic cell death triggered by conjugated linolenic acids is mediated by ACSL1. Nat. Commun. 2021, 12, 2244. [Google Scholar] [CrossRef] [PubMed]
- Al-Rashed, F.; Thomas, R.; Al-Roub, A.; Al-Mulla, F.; Ahmad, R. LPS induces GM-CSF production by breast cancer MDA-MB-231 cells via long-chain Acyl-CoA synthetase 1. Molecules 2020, 25, 4709. [Google Scholar] [CrossRef]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 2021, 184, 149–168. [Google Scholar] [CrossRef]
- Zheng, M.; Williams, E.P.; Malireddi, R.K.S.; Karki, R.; Banoth, B.; Burton, A.; Webby, R.; Channappanavar, R.; Jonsson, C.B.; Kanneganti, T.D. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J. Biol. Chem. 2020, 295, 14040–14052. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target. Ther. 2020, 5, 235. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Soares, V.C.; de Azevedo-Quintanilha, I.G.; Dias, S.; Fintelman-Rodrigues, N.; Sacramento, C.Q.; Mattos, M.; de Freitas, C.S.; Temerozo, J.R.; Teixeira, L.; et al. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov. 2021, 7, 43. [Google Scholar] [CrossRef]
- Ma, H.; Zhu, Z.; Lin, H.; Wang, S.; Zhang, P.; Li, Y.; Li, L.; Wang, J.; Zhao, Y.; Han, J. Pyroptosis of syncytia formed by fusion of SARS-CoV-2 spike and ACE2-expressing cells. Cell Discov. 2021, 7, 73. [Google Scholar] [CrossRef]
- Kan, X.; Yin, Y.; Song, C.; Tan, L.; Qiu, X.; Liao, Y.; Liu, W.; Meng, S.; Sun, Y.; Ding, C. Newcastle-disease-virus-induced ferroptosis through nutrient deprivation and ferritinophagy in tumor cells. iScience 2021, 24, 102837. [Google Scholar] [CrossRef] [PubMed]
- Armitage, A.E.; Stacey, A.R.; Giannoulatou, E.; Marshall, E.; Sturges, P.; Chatha, K.; Smith, N.M.; Huang, X.; Xu, X.; Pasricha, S.R.; et al. Distinct patterns of hepcidin and iron regulation during HIV-1, HBV, and HCV infections. Proc. Natl. Acad. Sci. USA 2014, 111, 12187–12192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, J.K.; Werner, B.G.; Ruthazer, R.; Snydman, D.R. Increased serum iron levels and infectious complications after liver transplantation. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2010, 51, e16–e23. [Google Scholar] [CrossRef]
- Jia, M.; Qin, D.; Zhao, C.; Chai, L.; Yu, Z.; Wang, W.; Tong, L.; Lv, L.; Wang, Y.; Rehwinkel, J.; et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat. Immunol. 2020, 21, 727–735. [Google Scholar] [CrossRef]
- Li, P.; Jiang, M.; Li, K.; Li, H.; Zhou, Y.; Xiao, X.; Xu, Y.; Krishfield, S.; Lipsky, P.E.; Tsokos, G.C.; et al. Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity. Nat. Immunol. 2021, 22, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Chen, Z.; Zhang, H.; Chen, C.; Zeng, M.; Yunis, J.; Wei, Y.; Wan, Y.; Wang, N.; Zhou, M.; et al. Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat. Immunol. 2021, 22, 1127–1139. [Google Scholar] [CrossRef]
- Wen, Q.; Liu, J.; Kang, R.; Zhou, B.; Tang, D. The release and activity of HMGB1 in ferroptosis. Biochem. Biophys. Res. Commun. 2019, 510, 278–283. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Liu, J.; Kang, R.; Tang, D. The circadian clock protects against ferroptosis-induced sterile inflammation. Biochem. Biophys. Res. Commun. 2020, 525, 620–625. [Google Scholar] [CrossRef]
- Huang, L.; Shi, Y.; Gong, B.; Jiang, L.; Zhang, Z.; Liu, X.; Yang, J.; He, Y.; Jiang, Z.; Zhong, L.; et al. Dynamic blood single-cell immune responses in patients with COVID-19. Signal Transduct. Target. Ther. 2021, 6, 110. [Google Scholar] [CrossRef]
- Girelli, D.; Marchi, G.; Busti, F.; Vianello, A. Iron metabolism in infections: Focus on COVID-19. Semin. Hematol. 2021, 58, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Lai, C.L. SARS-CoV-2 infection: Can ferroptosis be a potential treatment target for multiple organ involvement? Cell Death Discov. 2020, 6, 130. [Google Scholar] [CrossRef]
- Zhao, K.; Huang, J.; Dai, D.; Feng, Y.; Liu, L.; Nie, S. Serum iron level as a potential predictor of Coronavirus disease 2019 severity and mortality: A retrospective study. Open Forum Infect. Dis. 2020, 7, ofaa250. [Google Scholar] [CrossRef] [PubMed]
- Chakurkar, V.; Rajapurkar, M.; Lele, S.; Mukhopadhyay, B.; Lobo, V.; Injarapu, R.; Sheikh, M.; Dholu, B.; Ghosh, A.; Jha, V. Increased serum catalytic iron may mediate tissue injury and death in patients with COVID-19. Sci. Rep. 2021, 11, 19618. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, J.; Sun, Y.; Stubbs, D.; He, J.; Li, W.; Wang, F.; Liu, Z.; Ruzicka, J.A.; Taylor, E.W.; et al. SARS-CoV-2 suppresses mRNA expression of selenoproteins associated with ferroptosis, endoplasmic reticulum stress and DNA synthesis. Food Chem. Toxicol. 2021, 153, 112286. [Google Scholar] [CrossRef]
- Buchrieser, J.; Dufloo, J.; Hubert, M.; Monel, B.; Planas, D.; Rajah, M.M.; Planchais, C.; Porrot, F.; Guivel-Benhassine, F.; Van der Werf, S.; et al. Syncytia formation by SARS-CoV-2-infected cells. EMBO J. 2020, 39, e106267. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.W.; Jumper, C.C.; Ackerman, P.J.; Bracha, D.; Donlic, A.; Kim, H.; Kenney, D.; Castello-Serrano, I.; Suzuki, S.; Tamura, T.; et al. SARS-CoV-2 requires cholesterol for viral entry and pathological syncytia formation. eLife 2021, 10, e65962. [Google Scholar] [CrossRef]
- Daniloski, Z.; Jordan, T.X.; Wessels, H.H.; Hoagland, D.A.; Kasela, S.; Legut, M.; Maniatis, S.; Mimitou, E.P.; Lu, L.; Geller, E.; et al. Identification of required host factors for SARS-CoV-2 infection in human cells. Cell 2021, 184, 92–105. [Google Scholar] [CrossRef]
- Lamers, M.M.; Beumer, J.; van der Vaart, J.; Knoops, K.; Puschhof, J.; Breugem, T.I.; Ravelli, R.B.G.; Paul van Schayck, J.; Mykytyn, A.Z.; Duimel, H.Q.; et al. SARS-CoV-2 productively infects human gut enterocytes. Science 2020, 369, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.A.; Morales-Nebreda, L.; Markov, N.S.; Swaminathan, S.; Querrey, M.; Guzman, E.R.; Abbott, D.A.; Donnelly, H.K.; Donayre, A.; Goldberg, I.A.; et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature 2021, 590, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.K.; Jiang, G.S.; Holmes, K.V. Receptor for mouse hepatitis virus is a member of the carcinoembryonic antigen family of glycoproteins. Proc. Natl. Acad. Sci. USA 1991, 88, 5533–5536. [Google Scholar] [CrossRef] [Green Version]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Gao, J.; Mei, H.; Sun, J.; Li, H.; Huang, Y.; Tang, Y.; Duan, L.; Liu, D.; Wang, Q.; Gao, Y.; et al. Neuropilin-1 mediates SARS-CoV-2 infection in bone marrow-derived macrophages. bioRxiv 2021, 439793. [Google Scholar] [CrossRef]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Central Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Ferroptosis in infection, inflammation, and immunity. J. Exp. Med. 2021, 218, e20210518. [Google Scholar] [CrossRef]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef]
- Hue, S.; Beldi-Ferchiou, A.; Bendib, I.; Surenaud, M.; Fourati, S.; Frapard, T.; Rivoal, S.; Razazi, K.; Carteaux, G.; Delfau-Larue, M.H.; et al. Uncontrolled innate and impaired adaptive immune responses in patients with COVID-19 acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2020, 202, 1509–1519. [Google Scholar] [CrossRef]
- Li, Q.; Han, X.; Lan, X.; Gao, Y.; Wan, J.; Durham, F.; Cheng, T.; Yang, J.; Wang, Z.; Jiang, C.; et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2017, 2, e90777. [Google Scholar] [CrossRef] [Green Version]
- Roelands, J.; Garand, M.; Hinchcliff, E.; Ma, Y.; Shah, P.; Toufiq, M.; Alfaki, M.; Hendrickx, W.; Boughorbel, S.; Rinchai, D.; et al. Long-chain acyl-CoA synthetase 1 role in sepsis and immunity: Perspectives from a parallel review of public transcriptome datasets and of the literature. Front. Immunol. 2019, 10, 2410. [Google Scholar] [CrossRef] [Green Version]
- Rubinow, K.B.; Wall, V.Z.; Nelson, J.; Mar, D.; Bomsztyk, K.; Askari, B.; Lai, M.A.; Smith, K.D.; Han, M.S.; Vivekanandan-Giri, A.; et al. Acyl-CoA synthetase 1 is induced by Gram-negative bacteria and lipopolysaccharide and is required for phospholipid turnover in stimulated macrophages. J. Biol. Chem. 2013, 288, 9957–9970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.L.; Morales-Rosado, J.; Ray, J.; Myers, T.G.; Kho, T.; Lu, M.; Munford, R.S. Toll-like receptor agonists promote prolonged triglyceride storage in macrophages. J. Biol. Chem. 2014, 289, 3001–3012. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Wang, Y.; Feng, J.; Liu, Y.; Wang, T.; Zhao, M.; Ye, L.; Zhang, X. Aspirin suppresses the abnormal lipid metabolism in liver cancer cells via disrupting an NFκB-ACSL1 signaling. Biochem. Biophys. Res. Commun. 2017, 486, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Santos-Beneit, F.; Raškevičius, V.; Skeberdis, V.A.; Bordel, S. A metabolic modeling approach reveals promising therapeutic targets and antiviral drugs to combat COVID-19. Sci. Rep. 2021, 11, 11982. [Google Scholar] [CrossRef]
- Williams, C.G.; Jureka, A.S.; Silvas, J.A.; Nicolini, A.M.; Chvatal, S.A.; Carlson-Stevermer, J.; Oki, J.; Holden, K.; Basler, C.F. Inhibitors of VPS34 and fatty-acid metabolism suppress SARS-CoV-2 replication. Cell Rep. 2021, 36, 109479. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, T.X.; Huang, X.; Li, Y.; Sun, T.; Zang, S.; Guan, K.L.; Xiong, Y.; Liu, J.; Yuan, H.X. Targeting ferroptosis alleviates methionine-choline deficient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int. 2020, 40, 1378–1394. [Google Scholar] [CrossRef]
- Li, N.; Wang, W.; Zhou, H.; Wu, Q.; Duan, M.; Liu, C.; Wu, H.; Deng, W.; Shen, D.; Tang, Q. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic. Biol. Med. 2020, 160, 303–318. [Google Scholar] [CrossRef]
- Matsushita, M.; Freigang, S.; Schneider, C.; Conrad, M.; Bornkamm, G.W.; Kopf, M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 2015, 212, 555–568. [Google Scholar] [CrossRef] [Green Version]
- Amaral, E.P.; Costa, D.L.; Namasivayam, S.; Riteau, N.; Kamenyeva, O.; Mittereder, L.; Mayer-Barber, K.D.; Andrade, B.B.; Sher, A. A major role for ferroptosis in Mycobacterium tuberculosis-induced cell death and tissue necrosis. J. Exp. Med. 2019, 216, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, D.; Stabile, A.M.; Bastianelli, S.; Giustarini, D.; Pierucci, S.; Busti, C.; Vacca, C.; Gidari, A.; Francisci, D.; Castronari, R.; et al. SARS-CoV2 infection impairs the metabolism and redox function of cellular glutathione. Redox Biol. 2021, 45, 102041. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, H.; Perl, A.; Smith, D.; Lewis, T.; Kon, Z.; Goldenberg, R.; Yarta, K.; Staniloae, C.; Williams, M. Therapeutic blockade of inflammation in severe COVID-19 infection with intravenous N-acetylcysteine. Clin. Immunol. 2020, 219, 108544. [Google Scholar] [CrossRef] [PubMed]
- de Alencar, J.C.G.; Moreira, C.L.; Müller, A.D.; Chaves, C.E.; Fukuhara, M.A.; da Silva, E.A.; Miyamoto, M.F.S.; Pinto, V.B.; Bueno, C.G.; Lazar Neto, F.; et al. Double-blind, randomized, placebo-controlled trial with N-acetylcysteine for Treatment of severe acute respiratory syndrome caused by Coronavirus disease 2019 (COVID-19). Clin. Infect. Dis. 2021, 72, e736–e741. [Google Scholar] [CrossRef]
- Kanter, J.E.; Bornfeldt, K.E. Inflammation and diabetes-accelerated atherosclerosis: Myeloid cell mediators. Trends Endocrinol. Metab. 2013, 24, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Chu, J.; Xing, C.; Du, Y.; Duan, T.; Liu, S.; Zhang, P.; Cheng, C.; Henley, J.; Liu, X.; Qian, C.; et al. Pharmacological inhibition of fatty acid synthesis blocks SARS-CoV-2 replication. Nat. Metab. 2021, 3, 1466–1475. [Google Scholar] [CrossRef]
- Ren, G.; Bhatnagar, S.; Hahn, D.J.; Kim, J.A. Long-chain acyl-CoA synthetase-1 mediates the palmitic acid-induced inflammatory response in human aortic endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E893–E903. [Google Scholar] [CrossRef]
- Bleau, C.; Burnette, M.; Filliol, A.; Piquet-Pellorce, C.; Samson, M.; Lamontagne, L. Toll-like receptor-2 exacerbates murine acute viral hepatitis. Immunology 2016, 149, 204–224. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Kondo, N.; Omori, N.; Yoshimoto, R.; Hato, M.; Shigaki, S.; Nagasawa, A.; Ito, M.; Okuno, T. Discovery of a benzimidazole series as the first highly potent and selective ACSL1 inhibitors. Bioorgan. Med. Chem. Lett. 2021, 33, 127722. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, H.; Zhang, Z.; You, F. Inhibiting ACSL1-Related Ferroptosis Restrains Murine Coronavirus Infection. Viruses 2021, 13, 2383. https://doi.org/10.3390/v13122383
Xia H, Zhang Z, You F. Inhibiting ACSL1-Related Ferroptosis Restrains Murine Coronavirus Infection. Viruses. 2021; 13(12):2383. https://doi.org/10.3390/v13122383
Chicago/Turabian StyleXia, Huawei, Zeming Zhang, and Fuping You. 2021. "Inhibiting ACSL1-Related Ferroptosis Restrains Murine Coronavirus Infection" Viruses 13, no. 12: 2383. https://doi.org/10.3390/v13122383