
Studies on the Efficacy, Potential Cardiotoxicity and Monkey Pharmacokinetics of GLP-26 as a Potent Hepatitis B Virus Capsid Assembly Modulator

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Evaluation of GLP-26 in AD38/Nude Mice
2.3. Measuring the Unbound Fraction of GLP-26 in Cynomolgus Monkey Plasma
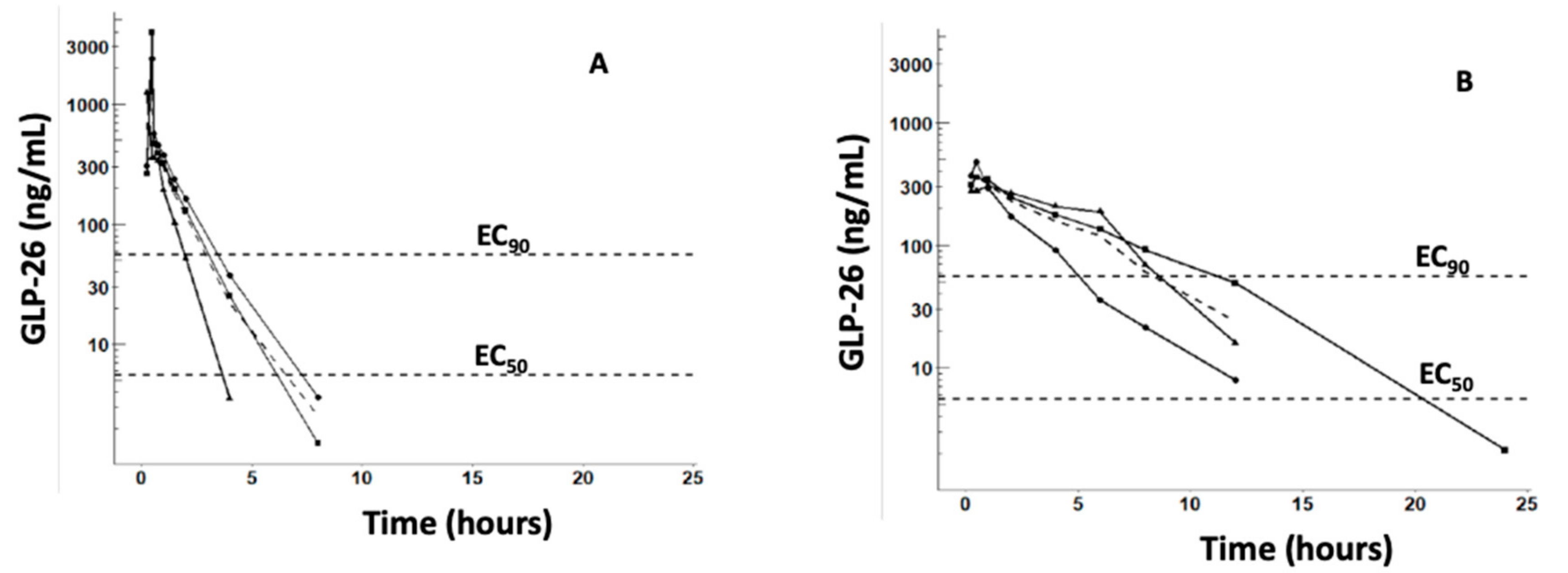
2.4. Pharmacokinetic Studies with Male Cynomolgus Monkeys
PK Analysis
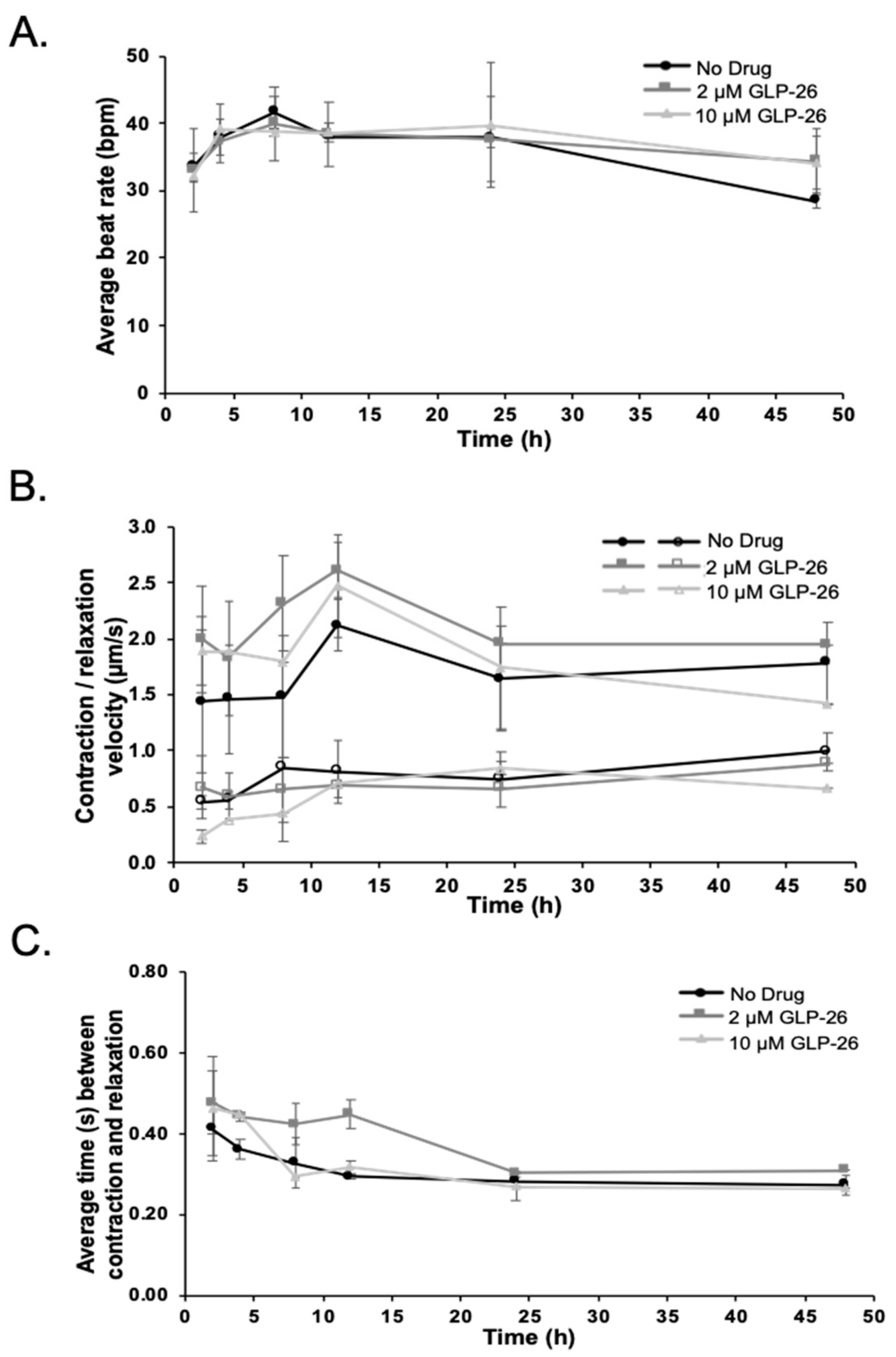
2.5. Effect of GLP-26 on Human Cardiomyocyte Function
Statistical Analysis
3. Results
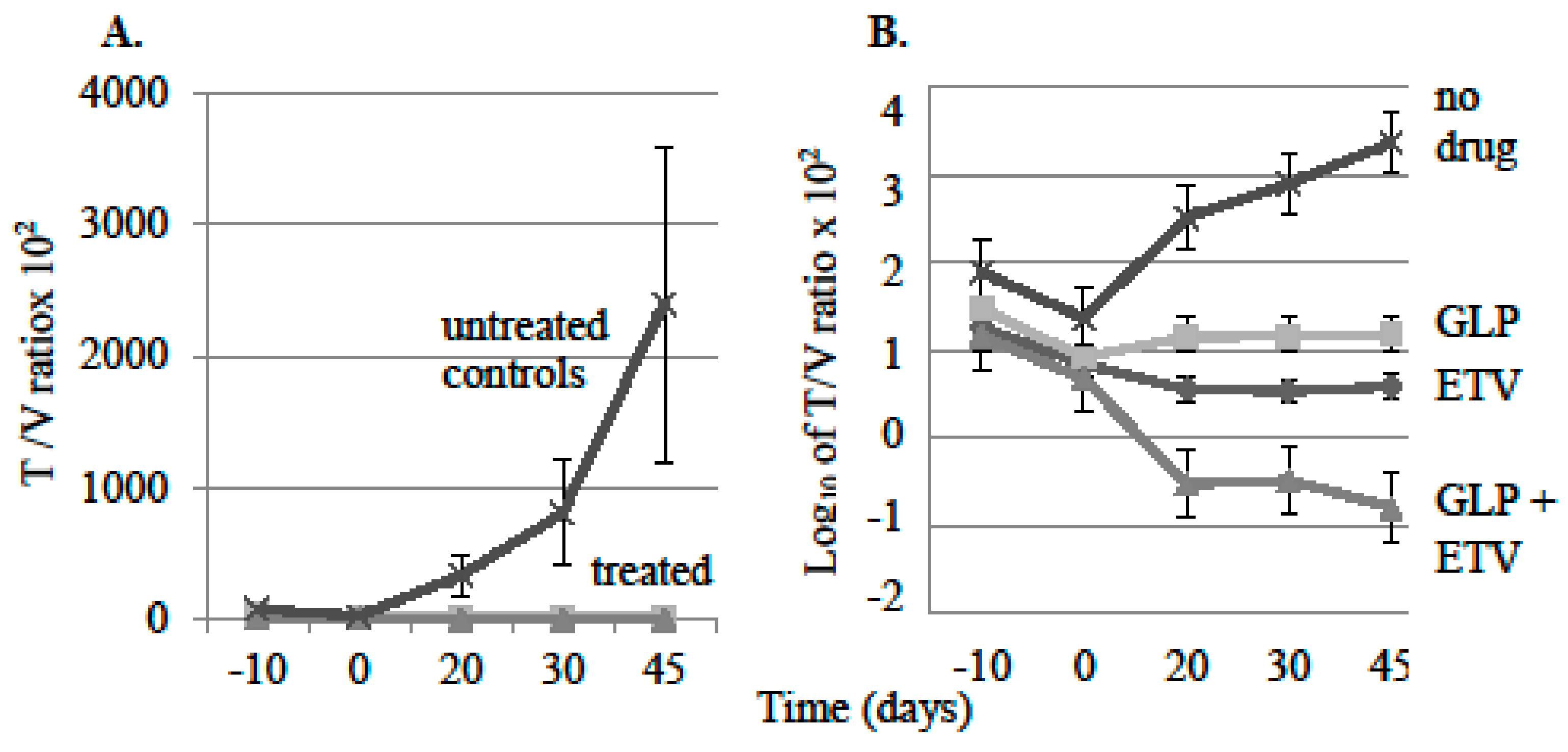
3.1. Effect of Antiviral Monotherapy or Combination Therapy on HBV DNA Copies/mL Serum in Nude Mice Bearing HBV-Infected AD38 Xenografts
3.2. Pharmacokinetic Characterization of GLP-26 in Male Cynomolgus Monkeys
3.3. Effect of GLP-26 on Human Cardiomyocyte Function
4. Discussion
4.1. Effect of Antiviral Monotherapy or Combination Therapy on HBV DNA Copies/mL Serum in Nude Mice Bearing HBV-Infected AD38 Xenografts
4.2. Determining the Mechanism of Action of GLP-26 on Viral Replication Versus Integrated HBV DNA in the AD38 Nude Mouse Xenograft Model
4.3. PK Studies in Cynomolgus Monkeys
4.4. Effect of GLP-26 on Human Cardiomyocyte Function
4.5. Overall Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Nicolini, L.A.; Orsi, A.; Tatarelli, P.; Viscoli, C.; Icardi, G.; Sticchi, L. A global view to HBV chronic infection: Evolving strategies for diagnosis, treatment and prevention in immunocompetent individuals. Int. J. Environ. Res. Public Health 2019, 16, 3307. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Hepatitis B Key Facts. 2019. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 22 July 2020).
- Block, T.M.; Guo, H.; Guo, J.T. Molecular virology of hepatitis B virus for clinicians. Clin. Liver. Dis. 2007, 11, 685–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajbhandari, R.; Chung, R.T. Treatment of hepatitis B: A concise review. Clin. Transl. Gastroenterol. 2016, 7, e190. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.R. Emerging therapies toward a functional cure for hepatitis B virus infection. Gastroenterol. Hepatol. 2018, 14, 439–442. [Google Scholar]
- Cole, A.G. Modulators of HBV capsid assembly as an approach to treating hepatitis B virus infection. Curr. Opin. Pharmacol. 2016, 30, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Soriano, V.; Barreiro, P.; Cachay, E.; Kottilil, S.; Fernandez-Montero, J.V.; de Mendoza, C. Advances in hepatitis B therapeutics. Ther. Adv. Infect. Dis. 2020, 7, 1–11. [Google Scholar] [CrossRef]
- Boucle, S.; Bassit, L.; Ehteshami, M.; Schinazi, R.F. Toward elimination of hepatitis B virus using novel drugs, approaches, and combined modalities. Clin. Liver. Dis. 2016, 20, 737–749. [Google Scholar] [CrossRef] [Green Version]
- Amblard, F.; Boucle, S.; Bassit, L.; Cox, B.; Sari, O.; Tao, S.; Chen, Z.; Ozturk, T.; Verma, K.; Russell, O.; et al. Novel hepatitis B virus capsid assembly modulator induces potent antiviral responses in vitro and in humanized mice. Antimicrob. Agents Chemother. 2020, 64, e01701–e01719. [Google Scholar] [CrossRef]
- Ladner, S.K.; Otto, M.J.; Barker, C.S.; Zaifert, K.; Wang, G.H.; Guo, J.T.; Seeger, C.; King, R.W. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: A novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 1997, 41, 1715–1720. [Google Scholar] [CrossRef] [Green Version]
- Feitelson, M.A.; Clayton, M.M.; Sun, B.; Schinazi, R.F. Development of a novel mouse model to evaluate drug candidates against hepatitis B virus. Antivir. Chem. Chemother. 2007, 18, 213–223. [Google Scholar] [CrossRef]
- Schinazi, R.F.; Bassit, L.; Clayton, M.M.; Sun, B.; Kohler, J.J.; Obikhod, A.; Arzumanyan, A.; Feitelson, M.A. Evaluation of single and combination therapies with tenofovir disoproxil fumarate and emtricitabine in vitro and in a robust mouse model supporting high levels of hepatitis B virus replication. Antimicrob. Agents Chemother. 2012, 56, 6186–6191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferdinandy, P.; Baczko, I.; Bencsik, P.; Giricz, Z.; Gorbe, A.; Pacher, P.; Varga, Z.V.; Varro, A.; Schulz, R. Definition of hidden drug cardiotoxicity: Paradigm change in cardiac safety testing and its clinical implications. Eur. Heart J. 2019, 40, 1771–1777. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Siegl, P.; Corsini, A.; Herrmann, J.; Lerman, A.; Benghozi, R. Drug attrition during pre-clinical and clinical development: Understanding and managing drug-induced cardiotoxicity. Pharmacol. Ther. 2013, 138, 470–484. [Google Scholar] [CrossRef] [PubMed]
- Arzumanyan, A.; Sambandam, V.; Clayton, M.M.; Choi, S.S.; Xie, G.; Diehl, A.M.; Yu, D.Y.; Feitelson, M.A. Hedgehog signaling blockade delays hepatocarcinogenesis induced by hepatitis B virus X protein. Cancer Res. 2012, 72, 5912–5920. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, D.; Schmidt, S.; Darendorf, H. Importance of relating efficacy measures to unbound drug concentrations for anti-infective agents. Antimicrob. Agents Chemother. 2013, 26, 274–280. [Google Scholar] [CrossRef] [Green Version]
- Fauchet, F.; Treluyer, J.M.; Illamola, S.M.; Pressiat, C.; Lui, G.; Valade, E.; Mandelbrot, L.; Lechedanec, J.; Delmas, S.; Blanche, S.; et al. Population approach to analyze the pharmacokinetics of free and total lopinavir in HIV-infected pregnant women and consequences for dose adjustment. Antimicrob. Agents Chemother. 2015, 59, 5727–5735. [Google Scholar] [CrossRef] [Green Version]
- Gilead Sciences. Emtriva (Emtricitabine) Capsule and Oral Solution Label, 12-2018. Available online: https://www.gilead.com/-/media/files/pdfs/medicines/hiv/emtriva/emtriva_patient_pi.pdf (accessed on 10 August 2020).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN1 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org (accessed on 10 September 2020)ISBN2 978-3-319-24277-4.
- Watari, N.; Benet, L.Z. Determination of mean input time, mean residence time, and steady-state volume of distribution with multiple drug inputs. J. Pharmacokinet. Biopharm. 1989, 17, 593–599. [Google Scholar] [CrossRef]
- Tao, S.; Zhou, L.; Zhang, H.; Zhou, S.; Amiralaei, S.; Shelton, J.; Ehteshami, M.; Jiang, Y.; Amblard, F.; Coats, S.J.; et al. Intracellular metabolism and potential cardiotoxicity of a beta-D-2’-C-methyl-2,6-diaminopurine ribonucleoside phosphoramidate that inhibits hepatitis C virus replication. Nucleosides Nucleotides Nucleic Acids 2020, 39, 204–224. [Google Scholar] [CrossRef]
- Huebsch, N.; Loskill, P.; Mandegar, M.A.; Marks, N.C.; Sheehan, A.S.; Ma, Z.; Mathur, A.; Nguyen, T.N.; Yoo, J.C.; Judge, L.M.; et al. Automated video-based analysis of contractility and calcium flux in human-induced pluripotent stem cell-derived cardiomyocytes cultured over different spatial scales. Tissue Eng. Part C Methods 2015, 21, 467–479. [Google Scholar] [CrossRef]
- Ko, C.; Chakraborty, A.; Chou, W.M.; Hasreiter, J.; Wettengel, J.M.; Stadler, D.; Bester, R.; Asen, T.; Zhang, K.; Wisskirchen, K.; et al. Hepatitis B virus genome recycling and de novo secondary infection events maintain stable cccDNA levels. J. Hepatol. 2018, 69, 1231–1241. [Google Scholar] [CrossRef]
- Ahmad, T.; Yin, P.; Saffitz, J.; Pockros, P.J.; Lalezari, J.; Shiffman, M.; Freilich, B.; Zamparo, J.; Brown, K.; Dimitrova, D.; et al. Cardiac dysfunction associated with a nucleotide polymerase inhibitor for treatment of hepatitis, C. Hepatology 2015, 62, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Ucciferri, C.; Occhionero, A.; Vecchiet, J.; Falasca, K. Cardiac toxicity associated with HCV direct antiviral agents. Mediterr. J. Hematol. Infect. Dis. 2018, 10, e2018069. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, R.; Auerbach, W. Regional differences in the growth of normal and neoplastic cells. Science 1982, 215, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Kyriazis, A.A.; Kyriazis, A.P. Preferential sites of growth of human tumors in nude mice following subcutaneous transplantation. Cancer Res. 1980, 40, 4509–4511. [Google Scholar] [PubMed]
- Anderson, B.F.; Holford, N.H.G. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug. Metab. Pharmacokinet. 2009, 24, 25–36. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 mg/kg IV 1 h Infusion | 5 mg/kg, PO | |||||
|---|---|---|---|---|---|---|
| Mean (n = 3) | SD | % CV | Mean (n = 3) | SD | % CV | |
| Cmax, ng·mL−1 | 2513 | 1339 | 53.3 | 380.7 | 88.21 | 23.2 |
| Tmax, h | 0.40 | 0.13 | 32.9 | 0.67 | 0.29 | 43.30 |
| AUCtotal, ng·h·mL−1 | 986.6 | 316.42 | 32.1 | 1660 | 530.05 | 31.9 |
| AUMC, ng·mL−1·h2 | 1058 | 525.5 | 49.7 | 7435 | 4123 | 56.5 |
| t1/2, h | 0.77 | 0.22 | 29.1 | 2.44 | 0.57 | 23.2 |
| MRT, h | 1.2 | 0.27 | 26.6 | 4.19 | 1.38 | 33.0 |
| CL, L·h·kg−1 | 1.106 | 0.43 | 39.1 | 3,285 | 1.28 | 39.1 |
| Vss, Lkg−1 | 1.065 | 0.18 | 16.6 | NA | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hurwitz, S.J.; McBrearty, N.; Arzumanyan, A.; Bichenkov, E.; Tao, S.; Bassit, L.; Chen, Z.; Kohler, J.J.; Amblard, F.; Feitelson, M.A.; et al. Studies on the Efficacy, Potential Cardiotoxicity and Monkey Pharmacokinetics of GLP-26 as a Potent Hepatitis B Virus Capsid Assembly Modulator. Viruses 2021, 13, 114. https://doi.org/10.3390/v13010114
Hurwitz SJ, McBrearty N, Arzumanyan A, Bichenkov E, Tao S, Bassit L, Chen Z, Kohler JJ, Amblard F, Feitelson MA, et al. Studies on the Efficacy, Potential Cardiotoxicity and Monkey Pharmacokinetics of GLP-26 as a Potent Hepatitis B Virus Capsid Assembly Modulator. Viruses. 2021; 13(1):114. https://doi.org/10.3390/v13010114
Chicago/Turabian StyleHurwitz, Selwyn J., Noreen McBrearty, Alla Arzumanyan, Eugene Bichenkov, Sijia Tao, Leda Bassit, Zhe Chen, James J. Kohler, Franck Amblard, Mark A. Feitelson, and et al. 2021. "Studies on the Efficacy, Potential Cardiotoxicity and Monkey Pharmacokinetics of GLP-26 as a Potent Hepatitis B Virus Capsid Assembly Modulator" Viruses 13, no. 1: 114. https://doi.org/10.3390/v13010114