HIV-1 Uncoating and Nuclear Import Precede the Completion of Reverse Transcription in Cell Lines and in Primary Macrophages


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cell Lines and Reagents
2.3. Pseudovirus Production and Characterization
2.4. Isolation, Differentiation and Treatment of MDMs
2.5. Single-Cycle Infection Assay
2.6. Fixed-Cell Imaging and Immunofluorescence Assay
2.7. Live-Cell Imaging of HIV-1 Uncoating, Nuclear Entry, and Infection
2.8. Image Acquisition
2.9. Single-Particle Tracking and Image Analysis
2.10. Statistical Analyses
3. Results
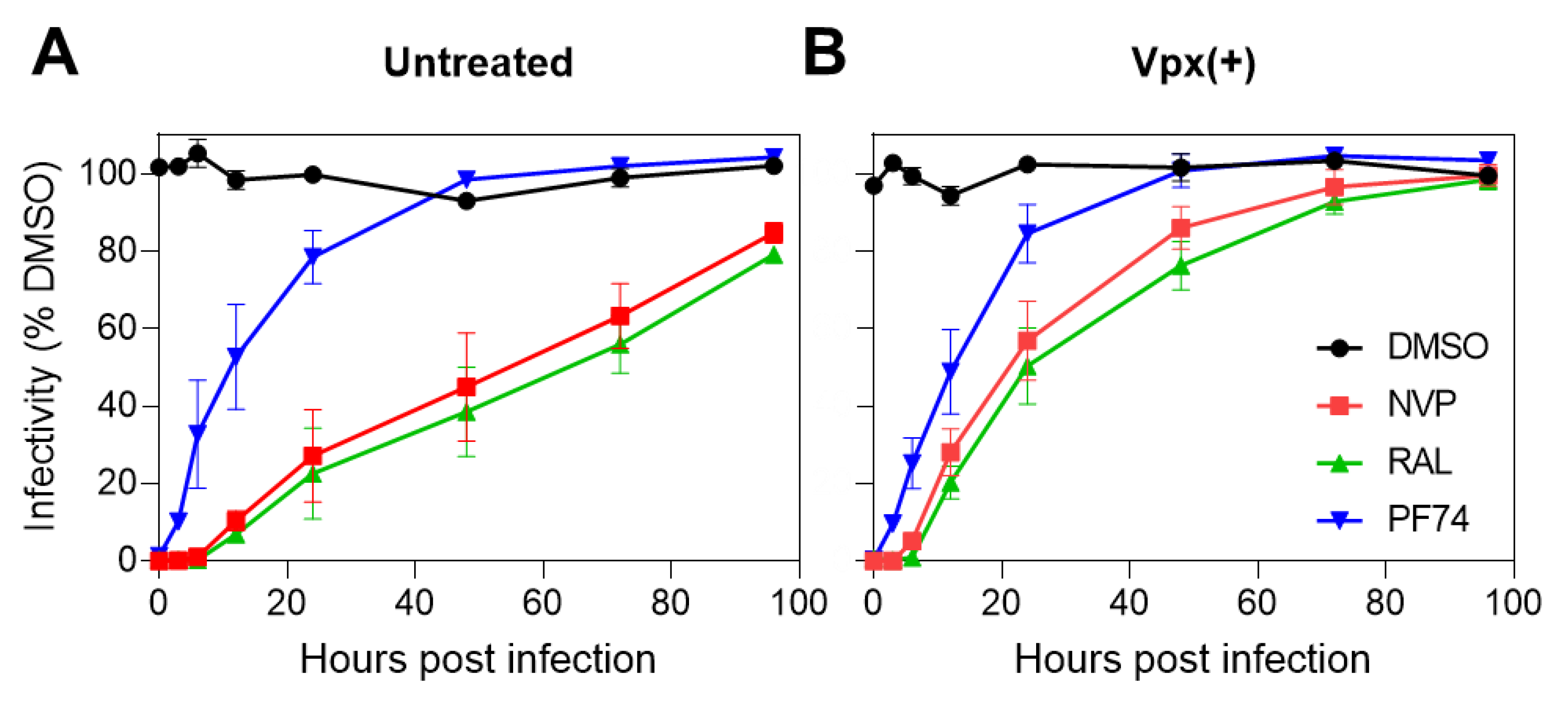
3.1. Productive HIV-1 Uncoating and Nuclear Import Precede the Completion of Reverse Transcription in Cell Lines
3.2. Reverse Transcription Continues Past HIV-1 Escape from CA-Targeting Inhibitor and Is Completed in the Nucleus of MDMs, Independent of SAMHD1 Depletion
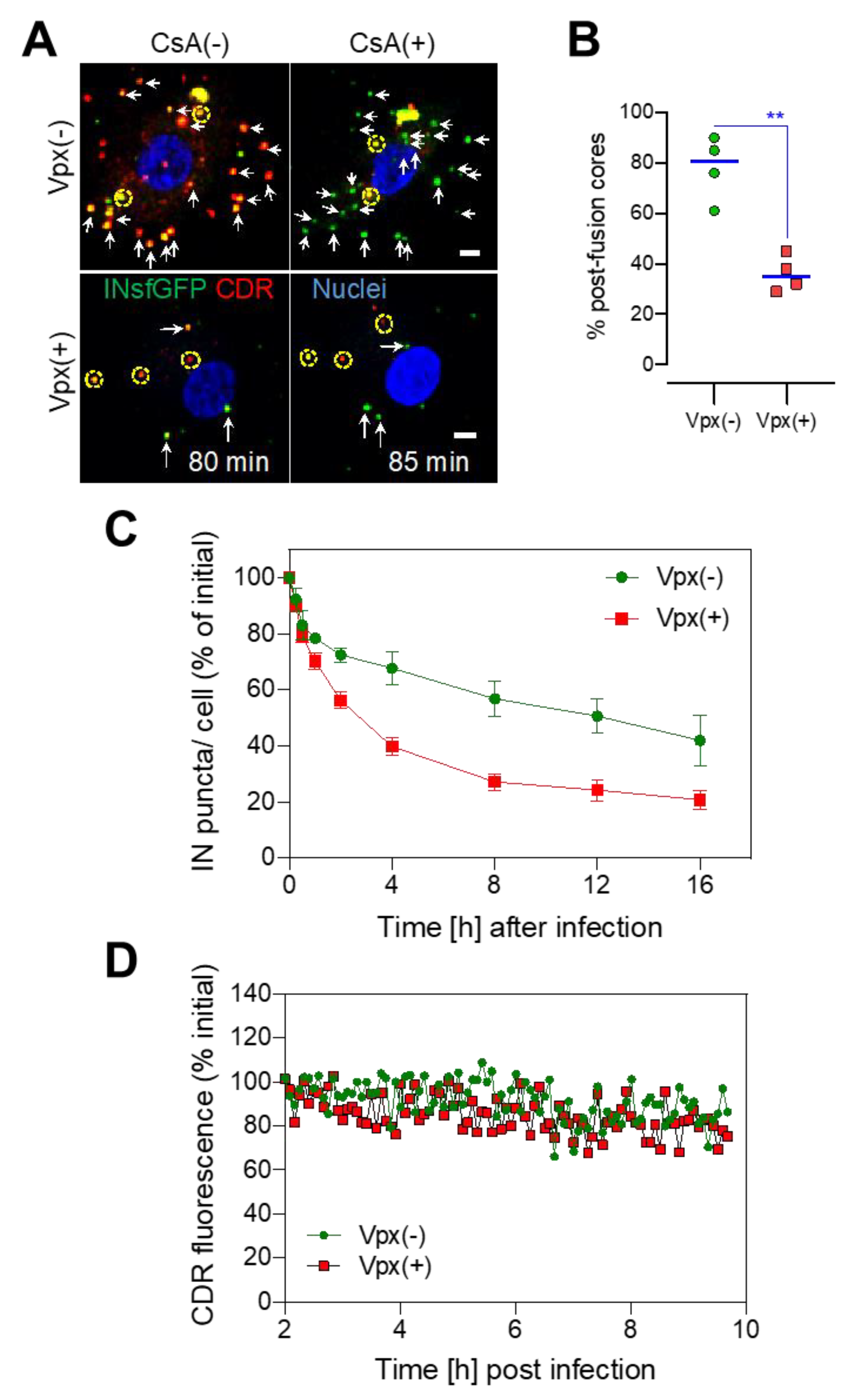
3.3. SAMHD1 Depletion in MDMs Diminishes the Pool of Stable Post-Fusion Cores
3.4. Long-Lived HIV-1 Cores Retain CDR over the Course of Several Hours Irrespective of SAMHD1 Depletion
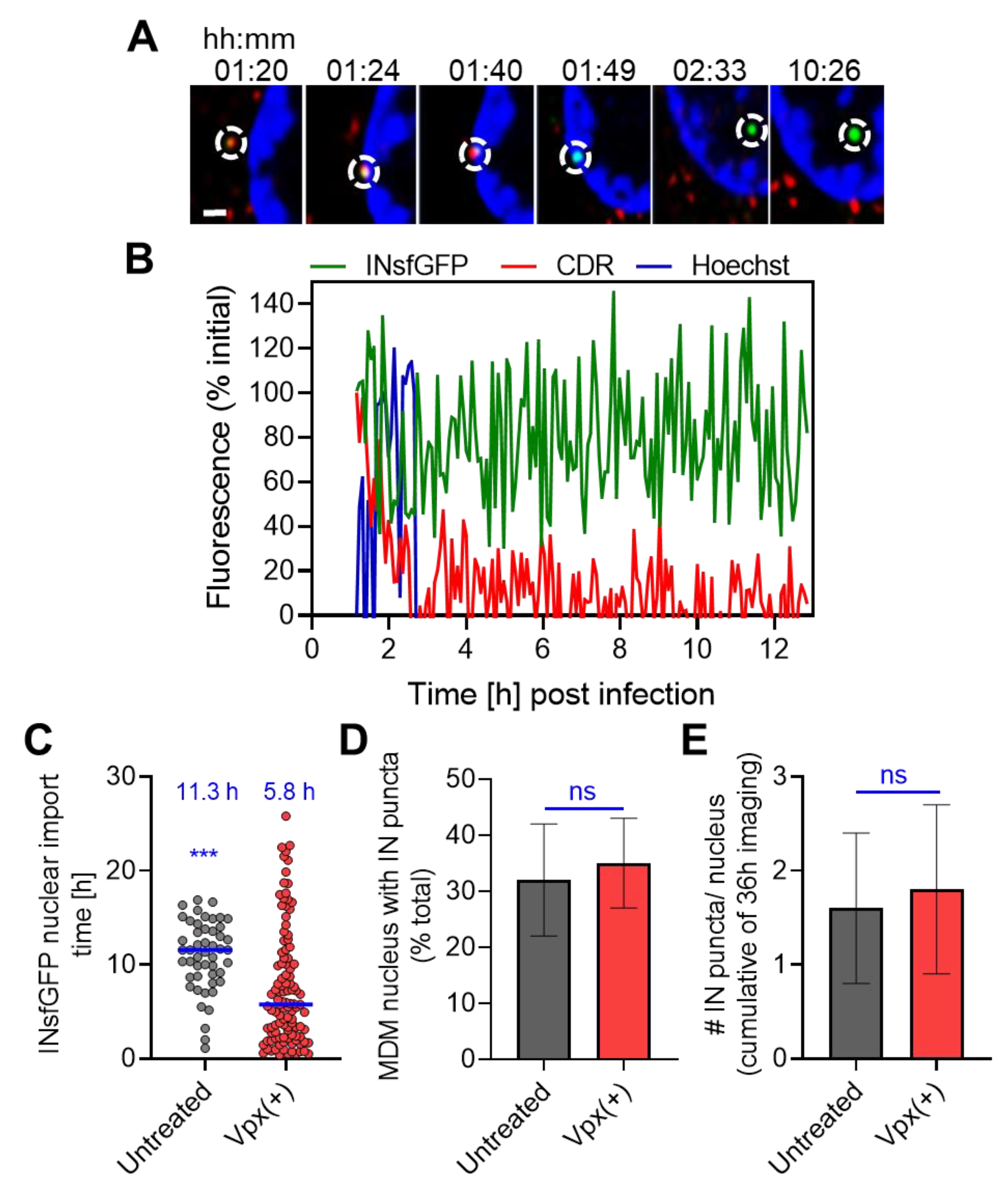
3.5. HIV-1 Nuclear Import in MDMs Progresses through a Loss of CDR at the Nuclear Envelope and Is Accelerated upon SAMHD1 Depletion
3.6. Improved HIV-1 Infection in SAMHD1-Depleted MDMs Correlates with a More Efficient Nuclear Reverse Transcription
3.7. Host Transcriptional Factors Are Enriched at the Sites of vDNA-Containing Nuclear HIV-1 Clusters
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Briggs, J.A.; Krausslich, H.G. The molecular architecture of HIV. J. Mol. Biol. 2011, 410, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.; Wilk, T.; Welker, R.; Krausslich, H.G.; Fuller, S.D. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 2003, 22, 1707–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, E.M.; Hope, T.J. HIV-1 capsid: The multifaceted key player in HIV-1 infection. Nat. Rev. Microbiol. 2015, 13, 471–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassati, A.; Goff, S.P. Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1. J. Virol. 2001, 75, 3626–3635. [Google Scholar] [CrossRef] [Green Version]
- Nermut, M.V.; Fassati, A. Structural analyses of purified human immunodeficiency virus type 1 intracellular reverse transcription complexes. J. Virol. 2003, 77, 8196–8206. [Google Scholar] [CrossRef] [Green Version]
- Farnet, C.M.; Haseltine, W.A. Determination of viral proteins present in the human immunodeficiency virus type 1 preintegration complex. J. Virol. 1991, 65, 1910–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.D.; Farnet, C.M.; Bushman, F.D. Human immunodeficiency virus type 1 preintegration complexes: Studies of organization and composition. J. Virol. 1997, 71, 5382–5390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Francis, A.C.; Marin, M.; Singh, P.K.; Achuthan, V.; Prellberg, M.J.; Palermino-Rowland, K.; Lan, S.; Tedbury, P.R.; Sarafianos, S.G.; Engelman, A.N.; et al. HIV-1 replication complexes accumulate in nuclear speckles and integrate into speckle-associated genomic domains. Nat. Commun. 2020, 11, 3505. [Google Scholar] [CrossRef] [PubMed]
- Forshey, B.M.; von Schwedler, U.; Sundquist, W.I.; Aiken, C. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 2002, 76, 5667–5677. [Google Scholar] [CrossRef] [Green Version]
- Hulme, A.E.; Perez, O.; Hope, T.J. Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proc. Natl. Acad. Sci. USA 2011, 108, 9975–9980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosnefroy, O.; Murray, P.J.; Bishop, K.N. HIV-1 capsid uncoating initiates after the first strand transfer of reverse transcription. Retrovirology 2016, 13, 58. [Google Scholar] [CrossRef] [Green Version]
- Francis, A.C.; Marin, M.; Shi, J.; Aiken, C.; Melikyan, G.B. Time-Resolved Imaging of Single HIV-1 Uncoating In Vitro and in Living Cells. PLoS Pathog. 2016, 12, e1005709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamede, J.I.; Cianci, G.C.; Anderson, M.R.; Hope, T.J. Early cytoplasmic uncoating is associated with infectivity of HIV-1. Proc. Natl. Acad. Sci. USA 2017, 114, E7169–E7178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dismuke, D.J.; Aiken, C. Evidence for a functional link between uncoating of the human immunodeficiency virus type 1 core and nuclear import of the viral preintegration complex. J. Virol. 2006, 80, 3712–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Shi, J.; Byeon, I.J.; Ahn, J.; Sheehan, J.H.; Meiler, J.; Gronenborn, A.M.; Aiken, C. Second-site suppressors of HIV-1 capsid mutations: Restoration of intracellular activities without correction of intrinsic capsid stability defects. Retrovirology 2012, 9, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.S.; Hughes, S.H. HIV-1 reverse transcription. Cold Spring Harb. Perspect. Med. 2012, 2, a006882. [Google Scholar] [CrossRef] [Green Version]
- Dharan, A.; Bachmann, N.; Talley, S.; Zwikelmaier, V.; Campbell, E.M. Nuclear pore blockade reveals that HIV-1 completes reverse transcription and uncoating in the nucleus. Nat. Microbiol. 2020, 5, 1088–1095. [Google Scholar] [CrossRef]
- Francis, A.C.; Melikyan, G.B. Single HIV-1 Imaging Reveals Progression of Infection through CA-Dependent Steps of Docking at the Nuclear Pore, Uncoating, and Nuclear Transport. Cell Host Microbe 2018, 23, 536–548.e6. [Google Scholar] [CrossRef] [Green Version]
- Hulme, A.E.; Kelley, Z.; Foley, D.; Hope, T.J. Complementary assays reveal a low level of CA associated with nuclear HIV-1 viral complexes in the Nuclei of HIV-1-infected cells. J. Virol. 2015, 89, 5350–5361. [Google Scholar] [CrossRef] [Green Version]
- Burdick, R.C.; Li, C.; Munshi, M.; Rawson, J.M.O.; Nagashima, K.; Hu, W.S.; Pathak, V.K. HIV-1 uncoats in the nucleus near sites of integration. Proc. Natl. Acad. Sci. USA 2020, 117, 5486–5493. [Google Scholar] [CrossRef] [PubMed]
- Zila, V.; Muller, T.G.; Laketa, V.; Muller, B.; Krausslich, H.G. Analysis of CA Content and CPSF6 Dependence of Early HIV-1 Replication Complexes in SupT1-R5 Cells. mBio 2019, 10, e02501-19. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.W.; Engelman, A. The barrier-to-autointegration factor is a component of functional human immunodeficiency virus type 1 preintegration complexes. J. Virol. 2003, 77, 5030–5036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, A.; Oztop, I.; Vandegraaff, N.; Raghavendra, N.K. Quantitative analysis of HIV-1 preintegration complexes. Methods 2009, 47, 283–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farnet, C.M.; Haseltine, W.A. Integration of human immunodeficiency virus type 1 DNA in vitro. Proc. Natl. Acad. Sci. USA 1990, 87, 4164–4168. [Google Scholar] [CrossRef] [Green Version]
- Bushman, F.D.; Craigie, R. Activities of human immunodeficiency virus (HIV) integration protein in vitro: Specific cleavage and integration of HIV DNA. Proc. Natl. Acad. Sci. USA 1991, 88, 1339–1343. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Wei, S.Q.; Engelman, A. Multiple integrase functions are required to form the native structure of the human immunodeficiency virus type I intasome. J. Biol. Chem. 1999, 274, 17358–17364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arhel, N.J.; Souquere-Besse, S.; Munier, S.; Souque, P.; Guadagnini, S.; Rutherford, S.; Prevost, M.C.; Allen, T.D.; Charneau, P. HIV-1 DNA Flap formation promotes uncoating of the pre-integration complex at the nuclear pore. EMBO J. 2007, 26, 3025–3037. [Google Scholar] [CrossRef]
- Zennou, V.; Serguera, C.; Sarkis, C.; Colin, P.; Perret, E.; Mallet, J.; Charneau, P. The HIV-1 DNA flap stimulates HIV vector-mediated cell transduction in the brain. Nat. Biotechnol. 2001, 19, 446–450. [Google Scholar] [CrossRef]
- Zennou, V.; Petit, C.; Guetard, D.; Nerhbass, U.; Montagnier, L.; Charneau, P. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell 2000, 101, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Burdick, R.C.; Hu, W.S.; Pathak, V.K. Nuclear import of APOBEC3F-labeled HIV-1 preintegration complexes. Proc. Natl. Acad. Sci. USA 2013, 110, E4780–E4789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejarano, D.A.; Peng, K.; Laketa, V.; Borner, K.; Jost, K.L.; Lucic, B.; Glass, B.; Lusic, M.; Muller, B.; Krausslich, H.G. HIV-1 nuclear import in macrophages is regulated by CPSF6-capsid interactions at the Nuclear Pore Complex. eLife 2019, 8, e41800. [Google Scholar] [CrossRef] [PubMed]
- De Iaco, A.; Luban, J. Cyclophilin A promotes HIV-1 reverse transcription but its effect on transduction correlates best with its effect on nuclear entry of viral cDNA. Retrovirology 2014, 11, 11. [Google Scholar] [CrossRef] [Green Version]
- Butler, S.L.; Hansen, M.S.; Bushman, F.D. A quantitative assay for HIV DNA integration in vivo. Nat. Med. 2001, 7, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.; Prasad, V.R. Analysis of 2-LTR circle junctions of viral DNA in infected cells. Methods Mol. Biol. 2009, 485, 73–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Price, A.J.; Halambage, U.D.; James, L.C.; Aiken, C. HIV-1 Resistance to the Capsid-Targeting Inhibitor PF74 Results in Altered Dependence on Host Factors Required for Virus Nuclear Entry. J. Virol. 2015, 89, 9068–9079. [Google Scholar] [CrossRef] [Green Version]
- Fricke, T.; Buffone, C.; Opp, S.; Valle-Casuso, J.; Diaz-Griffero, F. BI-2 destabilizes HIV-1 cores during infection and Prevents Binding of CPSF6 to the HIV-1 Capsid. Retrovirology 2014, 11, 120. [Google Scholar] [CrossRef]
- Singh, K.; Gallazzi, F.; Hill, K.J.; Burke, D.H.; Lange, M.J.; Quinn, T.P.; Neogi, U.; Sonnerborg, A. GS-CA Compounds: First-In-Class HIV-1 Capsid Inhibitors Covering Multiple Grounds. Front. Microbiol. 2019, 10, 1227. [Google Scholar] [CrossRef]
- Yamashita, M.; Perez, O.; Hope, T.J.; Emerman, M. Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS Pathog. 2007, 3, 1502–1510. [Google Scholar] [CrossRef] [Green Version]
- Schaller, T.; Ocwieja, K.E.; Rasaiyaah, J.; Price, A.J.; Brady, T.L.; Roth, S.L.; Hue, S.; Fletcher, A.J.; Lee, K.; KewalRamani, V.N.; et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011, 7, e1002439. [Google Scholar] [CrossRef]
- Lee, K.; Ambrose, Z.; Martin, T.D.; Oztop, I.; Mulky, A.; Julias, J.G.; Vandegraaff, N.; Baumann, J.G.; Wang, R.; Yuen, W.; et al. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 2010, 7, 221–233. [Google Scholar] [CrossRef] [Green Version]
- Delaney, M.K.; Malikov, V.; Chai, Q.; Zhao, G.; Naghavi, M.H. Distinct functions of diaphanous-related formins regulate HIV-1 uncoating and transport. Proc. Natl. Acad. Sci. USA 2017, 114, E6932–E6941. [Google Scholar] [CrossRef] [Green Version]
- Bonisch, I.Z.; Dirix, L.; Lemmens, V.; Borrenberghs, D.; De Wit, F.; Vernaillen, F.; Rocha, S.; Christ, F.; Hendrix, J.; Hofkens, J.; et al. Capsid-Labelled HIV to Investigate the Role of Capsid during Nuclear Import and Integration. J. Virol. 2020, 94, e01024–19. [Google Scholar] [CrossRef]
- Kumar, A.; Herbein, G. The macrophage: A therapeutic target in HIV-1 infection. Mol. Cell. Ther. 2014, 2, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasaiyaah, J.; Tan, C.P.; Fletcher, A.J.; Price, A.J.; Blondeau, C.; Hilditch, L.; Jacques, D.A.; Selwood, D.L.; James, L.C.; Noursadeghi, M.; et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 2013, 503, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Dauphin, A.; Komurlu, S.; McCauley, S.M.; Yurkovetskiy, L.; Carbone, C.; Diehl, W.E.; Strambio-De-Castillia, C.; Campbell, E.M.; Luban, J. Cyclophilin A protects HIV-1 from restriction by human TRIM5alpha. Nat. Microbiol. 2019, 4, 2044–2051. [Google Scholar] [CrossRef]
- Jimenez-Guardeno, J.M.; Apolonia, L.; Betancor, G.; Malim, M.H. Immunoproteasome activation enables human TRIM5alpha restriction of HIV-1. Nat. Microbiol. 2019, 4, 933–940. [Google Scholar] [CrossRef]
- Gavegnano, C.; Kennedy, E.M.; Kim, B.; Schinazi, R.F. The Impact of Macrophage Nucleotide Pools on HIV-1 Reverse Transcription, Viral Replication, and the Development of Novel Antiviral Agents. Mol. Biol. Int. 2012, 2012, 625983. [Google Scholar] [CrossRef]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Peng, K.; Muranyi, W.; Glass, B.; Laketa, V.; Yant, S.R.; Tsai, L.; Cihlar, T.; Muller, B.; Krausslich, H.G. Quantitative microscopy of functional HIV post-entry complexes reveals association of replication with the viral capsid. eLife 2014, 3, e04114. [Google Scholar] [CrossRef]
- Francis, A.C.; Di Primio, C.; Quercioli, V.; Valentini, P.; Boll, A.; Girelli, G.; Demichelis, F.; Arosio, D.; Cereseto, A. Second generation imaging of nuclear/cytoplasmic HIV-1 complexes. AIDS Res. Hum. Retrovir. 2014, 30, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, H.; Logue, E.C.; Bloch, N.; Daddacha, W.; Polsky, S.B.; Schultz, M.L.; Kim, B.; Landau, N.R. The Vpx lentiviral accessory protein targets SAMHD1 for degradation in the nucleus. J. Virol. 2012, 86, 12552–12560. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Choe, S.; Walker, R.; Di Marzio, P.; Morgan, D.O.; Landau, N.R. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 1995, 69, 6705–6711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, R.I.; Chen, B.K.; Choe, S.; Landau, N.R. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 1995, 206, 935–944. [Google Scholar] [CrossRef] [Green Version]
- Platt, E.J.; Wehrly, K.; Kuhmann, S.E.; Chesebro, B.; Kabat, D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 1998, 72, 2855–2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simm, M.; Shahabuddin, M.; Chao, W.; Allan, J.S.; Volsky, D.J. Aberrant Gag protein composition of a human immunodeficiency virus type 1 vif mutant produced in primary lymphocytes. J. Virol. 1995, 69, 4582–4586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzato, M.; Erlwein, O.; Bonsall, D.; Kaye, S.; Muir, D.; McClure, M.O. A one-step SYBR Green I-based product-enhanced reverse transcriptase assay for the quantitation of retroviruses in cell culture supernatants. J. Virol. Methods 2009, 156, 1–7. [Google Scholar] [CrossRef]
- de Chaumont, F.; Dallongeville, S.; Chenouard, N.; Herve, N.; Pop, S.; Provoost, T.; Meas-Yedid, V.; Pankajakshan, P.; Lecomte, T.; Le Montagner, Y.; et al. Icy: An open bioimage informatics platform for extended reproducible research. Nat. Methods 2012, 9, 690–696. [Google Scholar] [CrossRef]
- Francis, A.C.; Melikyan, G.B. Live-Cell Imaging of Early Steps of Single HIV-1 Infection. Viruses 2018, 10, 275. [Google Scholar] [CrossRef] [Green Version]
- Borrenberghs, D.; Dirix, L.; De Wit, F.; Rocha, S.; Blokken, J.; De Houwer, S.; Gijsbers, R.; Christ, F.; Hofkens, J.; Hendrix, J.; et al. Dynamic Oligomerization of Integrase Orchestrates HIV Nuclear Entry. Sci. Rep. 2016, 6, 36485. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Alam, S.L.; Fricke, T.; Zadrozny, K.; Sedzicki, J.; Taylor, A.B.; Demeler, B.; Pornillos, O.; Ganser-Pornillos, B.K.; Diaz-Griffero, F.; et al. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc. Natl. Acad. Sci. USA 2014, 111, 18625–18630. [Google Scholar] [CrossRef] [Green Version]
- Price, A.J.; Fletcher, A.J.; Schaller, T.; Elliott, T.; Lee, K.; KewalRamani, V.N.; Chin, J.W.; Towers, G.J.; James, L.C. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog. 2012, 8, e1002896. [Google Scholar] [CrossRef] [Green Version]
- Price, A.J.; Jacques, D.A.; McEwan, W.A.; Fletcher, A.J.; Essig, S.; Chin, J.W.; Halambage, U.D.; Aiken, C.; James, L.C. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 2014, 10, e1004459. [Google Scholar] [CrossRef] [PubMed]
- Marquez, C.L.; Lau, D.; Walsh, J.; Shah, V.; McGuinness, C.; Wong, A.; Aggarwal, A.; Parker, M.W.; Jacques, D.A.; Turville, S.; et al. Kinetics of HIV-1 capsid uncoating revealed by single-molecule analysis. eLife 2018, 7, e34772. [Google Scholar] [CrossRef]
- Kim, B.; Nguyen, L.A.; Daddacha, W.; Hollenbaugh, J.A. Tight interplay among SAMHD1 protein level, cellular dNTP levels, and HIV-1 proviral DNA synthesis kinetics in human primary monocyte-derived macrophages. J. Biol. Chem. 2012, 287, 21570–21574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, T.L.; Roshal, M.; Jamburuthugoda, V.K.; Reynolds, H.M.; Merriam, A.R.; Lee, K.Y.; Balakrishnan, M.; Bambara, R.A.; Planelles, V.; Dewhurst, S.; et al. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J. Biol. Chem. 2004, 279, 51545–51553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traut, T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994, 140, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.; Saito, A.; Halambage, U.D.; Ferhadian, D.; Fischer, D.K.; Francis, A.C.; Melikyan, G.B.; Ambrose, Z.; Aiken, C.; Yamashita, M. A Novel Phenotype Links HIV-1 Capsid Stability to cGAS-Mediated DNA Sensing. J. Virol. 2019, 93, e00706-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, P.; Wolf, K.; Lammerding, J. Nuclear mechanics during cell migration. Curr. Opin. Cell Biol. 2011, 23, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Krause, M.; Yang, F.W.; Te Lindert, M.; Isermann, P.; Schepens, J.; Maas, R.J.A.; Venkataraman, C.; Lammerding, J.; Madzvamuse, A.; Hendriks, W.; et al. Cell migration through three-dimensional confining pores: Speed accelerations by deformation and recoil of the nucleus. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180225. [Google Scholar] [CrossRef]
- Stultz, R.D.; Cenker, J.J.; McDonald, D. Imaging HIV-1 Genomic DNA from Entry through Productive Infection. J. Virol. 2017, 91, e00034-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rensen, E.; Mueller, F.; Scoca, V.; Parmar, J.; Souque, P.; Zimmer, C.; Di Nunzio, F. Clustering and reverse transcription of HIV-1 genomes in nuclear niches of macrophages. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Peterlin, B.M.; Price, D.H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Sloan, R.D.; Wainberg, M.A. The role of unintegrated DNA in HIV infection. Retrovirology 2011, 8, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillim-Ross, L.; Cara, A.; Klotman, M.E. HIV-1 extrachromosomal 2-LTR circular DNA is long-lived in human macrophages. Viral Immunol. 2005, 18, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Kootstra, N.A.; Zwart, B.M.; Schuitemaker, H. Diminished human immunodeficiency virus type 1 reverse transcription and nuclear transport in primary macrophages arrested in early G(1) phase of the cell cycle. J. Virol. 2000, 74, 1712–1717. [Google Scholar] [CrossRef] [Green Version]
- Selyutina, A.; Persaud, M.; Lee, K.; KewalRamani, V.; Diaz-Griffero, F. Nuclear Import of the HIV-1 Core Precedes Reverse Transcription and Uncoating. Cell Rep. 2020, 32, 108201. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.M.; Marin, M.; Sood, C.; Shi, J.; Nawaz, F.; Aiken, C.; Melikyan, G.B. Fluorescent protein-tagged Vpr dissociates from HIV-1 core after viral fusion and rapidly enters the cell nucleus. Retrovirology 2015, 12, 88. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.M.; Hope, T.J. Live cell imaging of the HIV-1 life cycle. Trends Microbiol. 2008, 16, 580–587. [Google Scholar] [CrossRef] [Green Version]
- De Rijck, J.; Vandekerckhove, L.; Gijsbers, R.; Hombrouck, A.; Hendrix, J.; Vercammen, J.; Engelborghs, Y.; Christ, F.; Debyser, Z. Overexpression of the lens epithelium-derived growth factor/p75 integrase binding domain inhibits human immunodeficiency virus replication. J. Virol. 2006, 80, 11498–11509. [Google Scholar] [CrossRef] [Green Version]
- Meehan, A.M.; Saenz, D.T.; Morrison, J.; Hu, C.; Peretz, M.; Poeschla, E.M. LEDGF dominant interference proteins demonstrate prenuclear exposure of HIV-1 integrase and synergize with LEDGF depletion to destroy viral infectivity. J. Virol. 2011, 85, 3570–3583. [Google Scholar] [CrossRef] [Green Version]
- da Silva, E.S.; Shanmugapriya, S.; Malikov, V.; Gu, F.; Delaney, M.K.; Naghavi, M.H. HIV-1 capsids mimic a microtubule regulator to coordinate early stages of infection. EMBO J. 2020, 39, e104870. [Google Scholar] [CrossRef]
- Matreyek, K.A.; Yucel, S.S.; Li, X.; Engelman, A. Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog. 2013, 9, e1003693. [Google Scholar] [CrossRef] [Green Version]
- Mallery, D.L.; Marquez, C.L.; McEwan, W.A.; Dickson, C.F.; Jacques, D.A.; Anandapadamanaban, M.; Bichel, K.; Towers, G.J.; Saiardi, A.; Bocking, T.; et al. IP6 is an HIV pocket factor that prevents capsid collapse and promotes DNA synthesis. eLife 2018, 7, e35335. [Google Scholar] [CrossRef] [PubMed]
- Dick, R.A.; Zadrozny, K.K.; Xu, C.; Schur, F.K.M.; Lyddon, T.D.; Ricana, C.L.; Wagner, J.M.; Perilla, J.R.; Ganser-Pornillos, B.K.; Johnson, M.C.; et al. Inositol phosphates are assembly co-factors for HIV-1. Nature 2018, 560, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Mallery, D.L.; Faysal, K.M.R.; Kleinpeter, A.; Wilson, M.S.C.; Vaysburd, M.; Fletcher, A.J.; Novikova, M.; Bocking, T.; Freed, E.O.; Saiardi, A.; et al. Cellular IP6 Levels Limit HIV Production while Viruses that Cannot Efficiently Package IP6 Are Attenuated for Infection and Replication. Cell Rep. 2019, 29, 3983–3996.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achuthan, V.; Perreira, J.M.; Sowd, G.A.; Puray-Chavez, M.; McDougall, W.M.; Paulucci-Holthauzen, A.; Wu, X.; Fadel, H.J.; Poeschla, E.M.; Multani, A.S.; et al. Capsid-CPSF6 Interaction Licenses Nuclear HIV-1 Trafficking to Sites of Viral DNA Integration. Cell Host Microbe 2018, 24, 392–404.e8. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Rodriguez, G.; Gazi, A.; Monel, B.; Frabetti, S.; Scoca, V.; Mueller, F.; Schwartz, O.; Krijnse-Locker, J.; Charneau, P.; Di Nunzio, F. Remodeling of the Core Leads HIV-1 Preintegration Complex into the Nucleus of Human Lymphocytes. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Chin, C.R.; Perreira, J.M.; Savidis, G.; Portmann, J.M.; Aker, A.M.; Feeley, E.M.; Smith, M.C.; Brass, A.L. Direct Visualization of HIV-1 Replication Intermediates Shows that Capsid and CPSF6 Modulate HIV-1 Intra-nuclear Invasion and Integration. Cell Rep. 2015, 13, 1717–1731. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Sokolskaja, E.; Jolly, C.; James, W.; Cowley, S.A.; Fassati, A. Transportin 3 promotes a nuclear maturation step required for efficient HIV-1 integration. PLoS Pathog. 2011, 7, e1002194. [Google Scholar] [CrossRef]
- Zila, V.; Margiotta, E.; Turonova, B.; Müller, T.G.; Zimmerli, C.E.; Mattei, S.; Allegretti, M.; Börner, K.; Rada, J.; Müller, B.; et al. Cone-shaped HIV-1 capsids are transported through intact nuclear pores. bioRxiv 2020. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francis, A.C.; Marin, M.; Prellberg, M.J.; Palermino-Rowland, K.; Melikyan, G.B. HIV-1 Uncoating and Nuclear Import Precede the Completion of Reverse Transcription in Cell Lines and in Primary Macrophages. Viruses 2020, 12, 1234. https://doi.org/10.3390/v12111234
Francis AC, Marin M, Prellberg MJ, Palermino-Rowland K, Melikyan GB. HIV-1 Uncoating and Nuclear Import Precede the Completion of Reverse Transcription in Cell Lines and in Primary Macrophages. Viruses. 2020; 12(11):1234. https://doi.org/10.3390/v12111234
Chicago/Turabian StyleFrancis, Ashwanth C., Mariana Marin, Mathew J. Prellberg, Kristina Palermino-Rowland, and Gregory B. Melikyan. 2020. "HIV-1 Uncoating and Nuclear Import Precede the Completion of Reverse Transcription in Cell Lines and in Primary Macrophages" Viruses 12, no. 11: 1234. https://doi.org/10.3390/v12111234