
Full-Length Transcriptome Sequencing and Identification of Hsf Genes in Cunninghamia lanceolata (Lamb.) Hook
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction, Library Construction, and SMRT Sequencing
2.3. Data Processing
2.4. Gene Structure Analysis
2.5. CDS, TF, and lncRNA Analyses
2.6. Functional Annotation
2.7. Identification and Multi-Segment Alignments of Hsf Genes
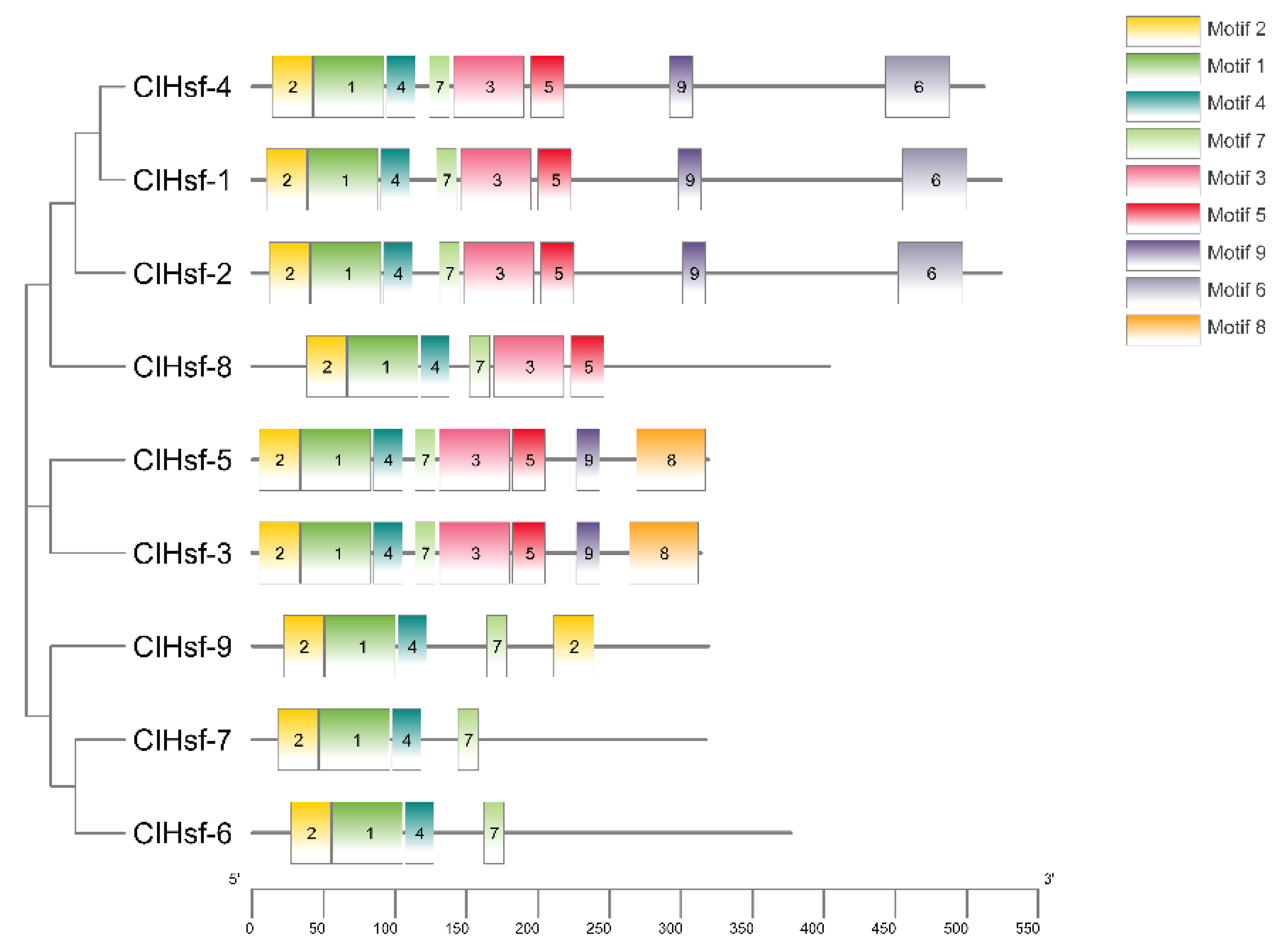
2.8. Motif and Phylogenetic Analyses
2.9. Hsf Expression Analysisusingtranscriptome Data
2.10. Heat Stress and qRT-PCR Analyses
3. Results
3.1. Sequencing Data Statistics and De-Redundancy
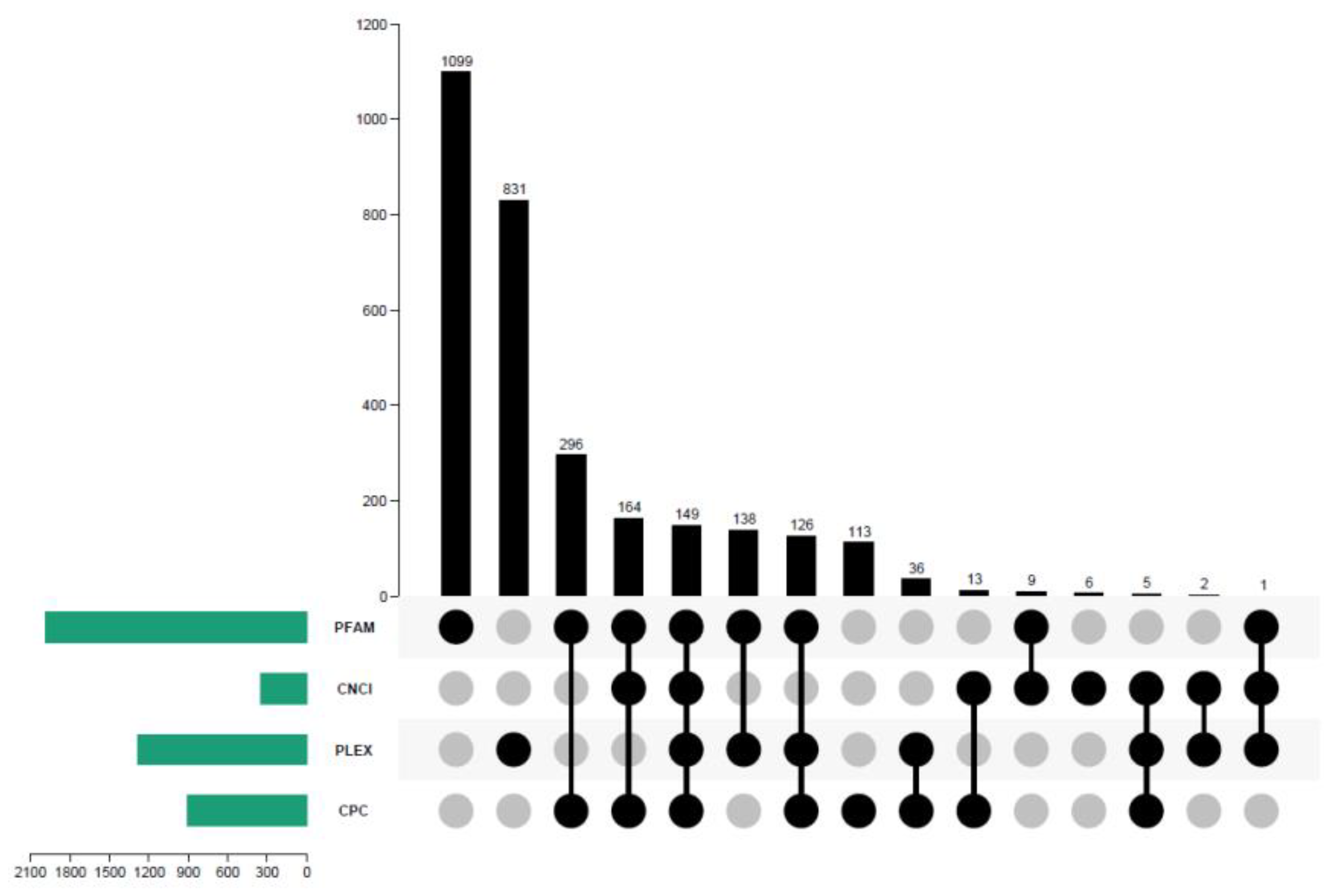
3.2. Transcript Redundancy Analysis
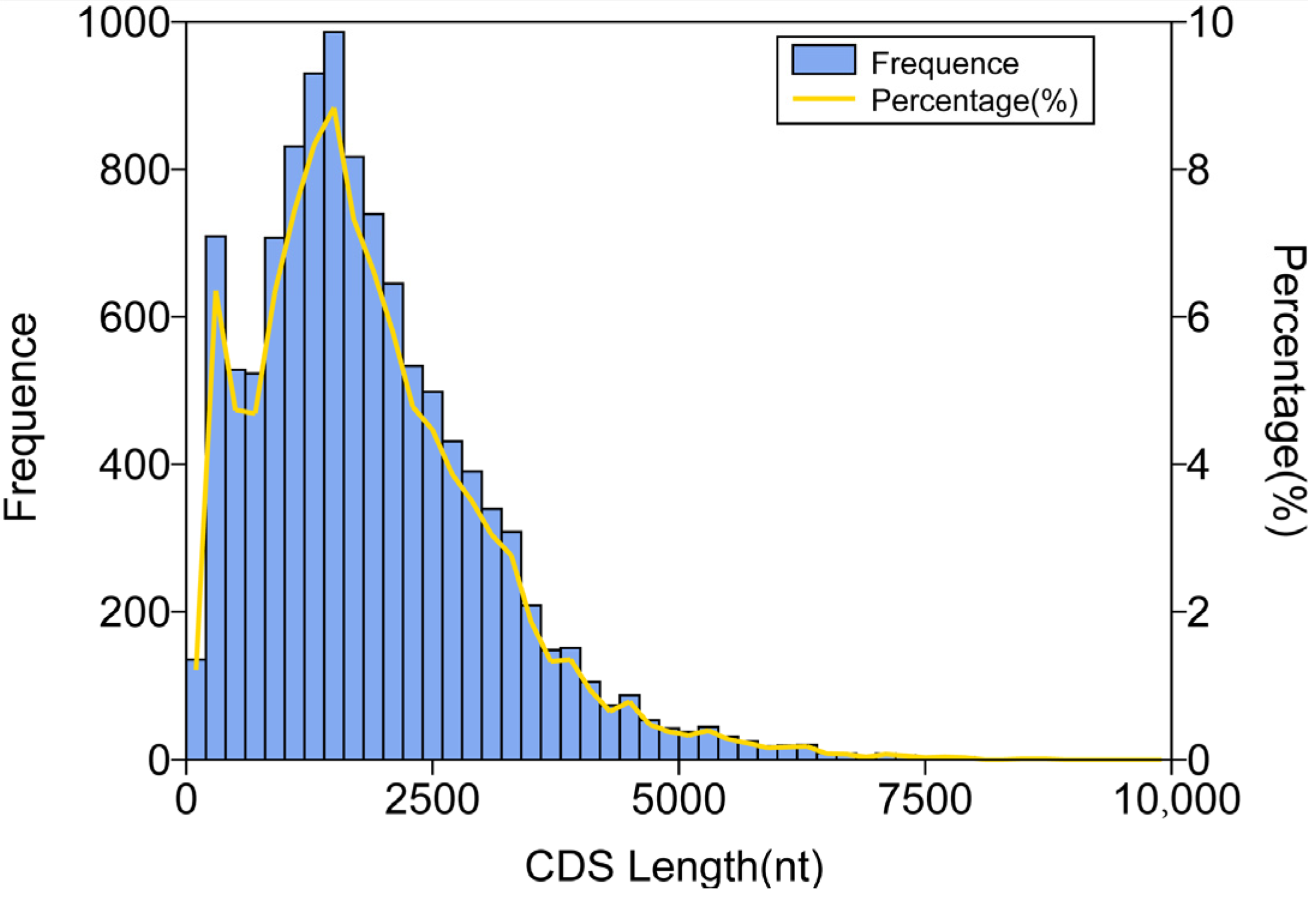
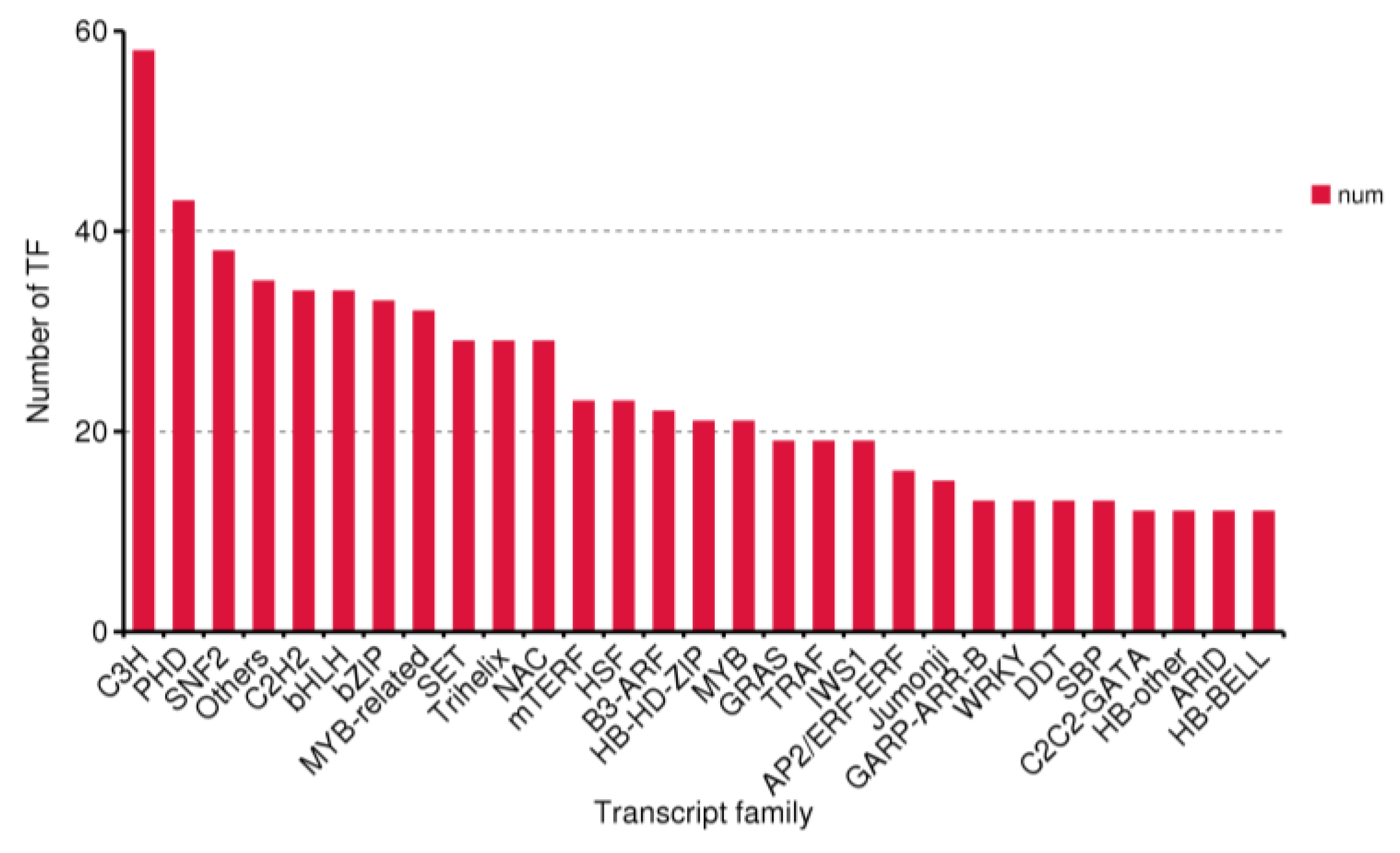
3.3. CDS, TF, and lncRNA Analyses
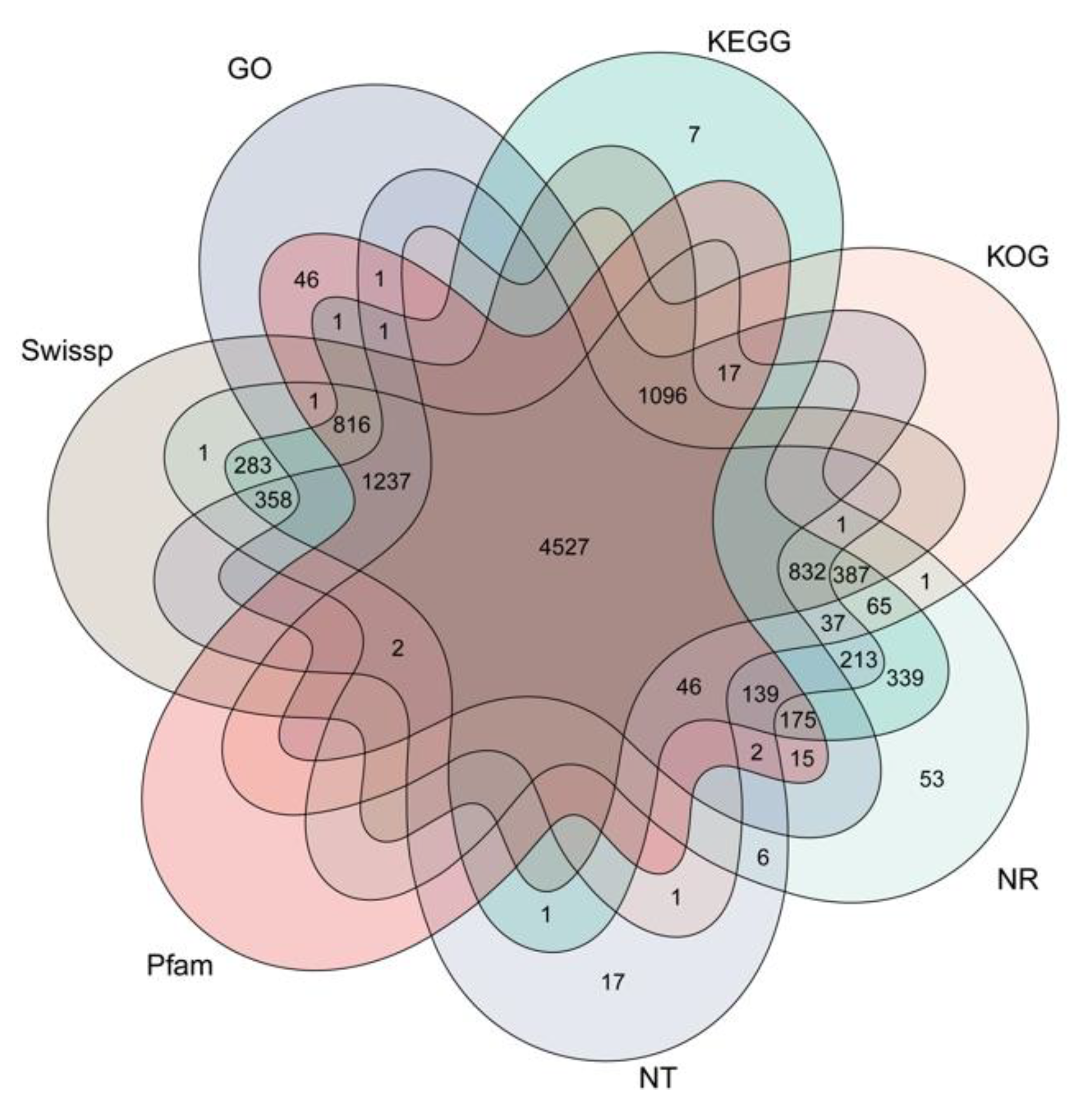
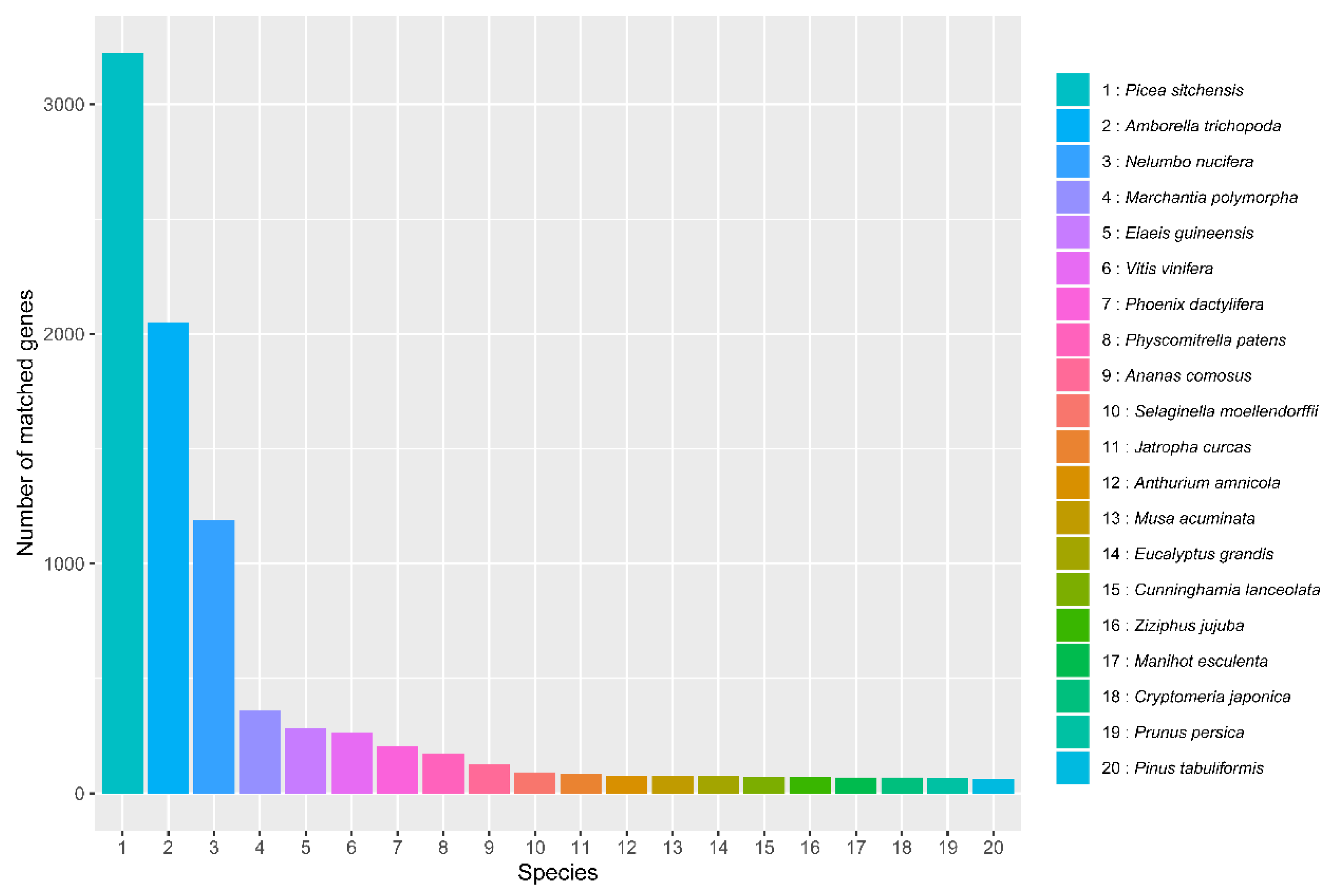
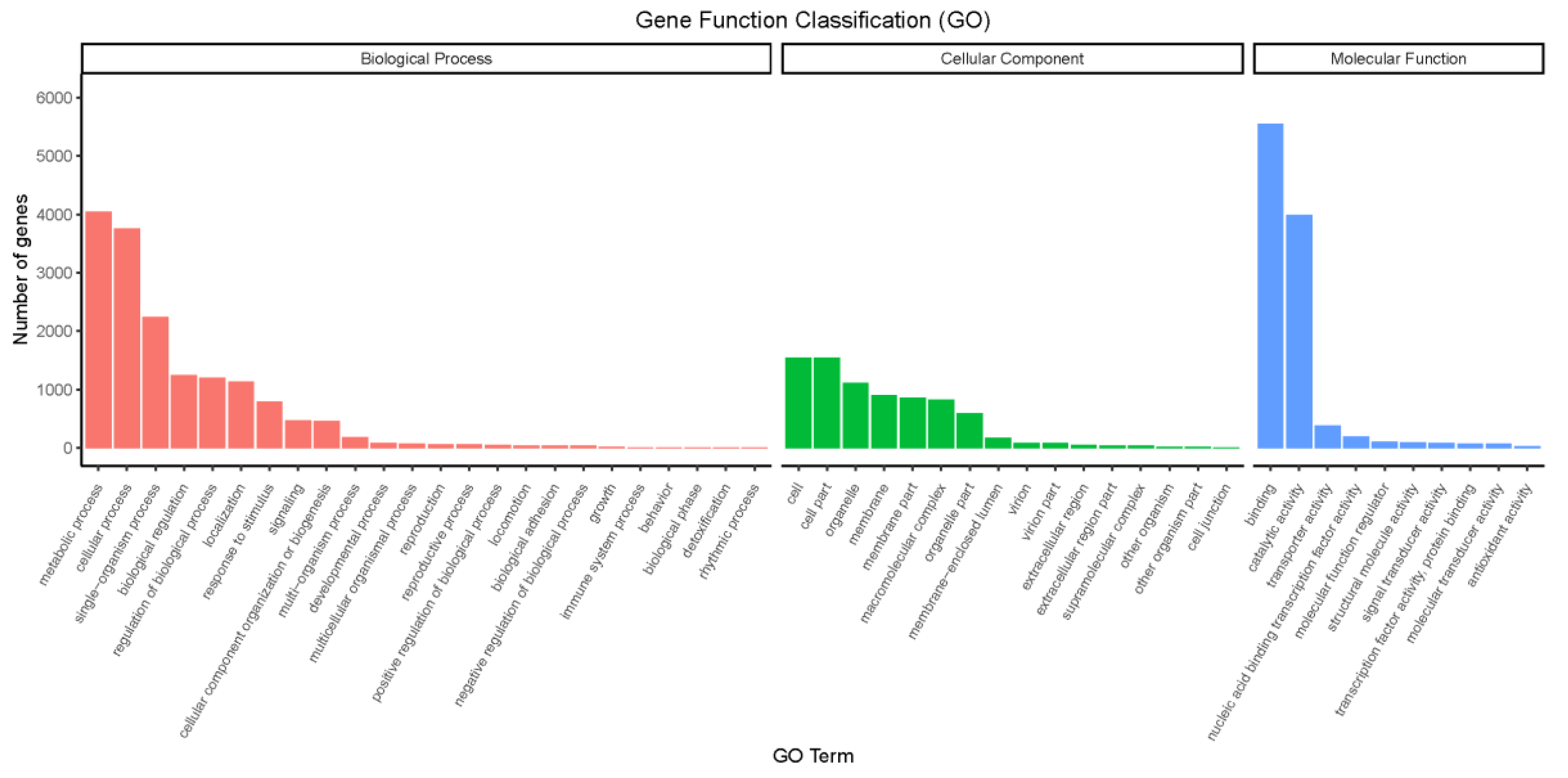
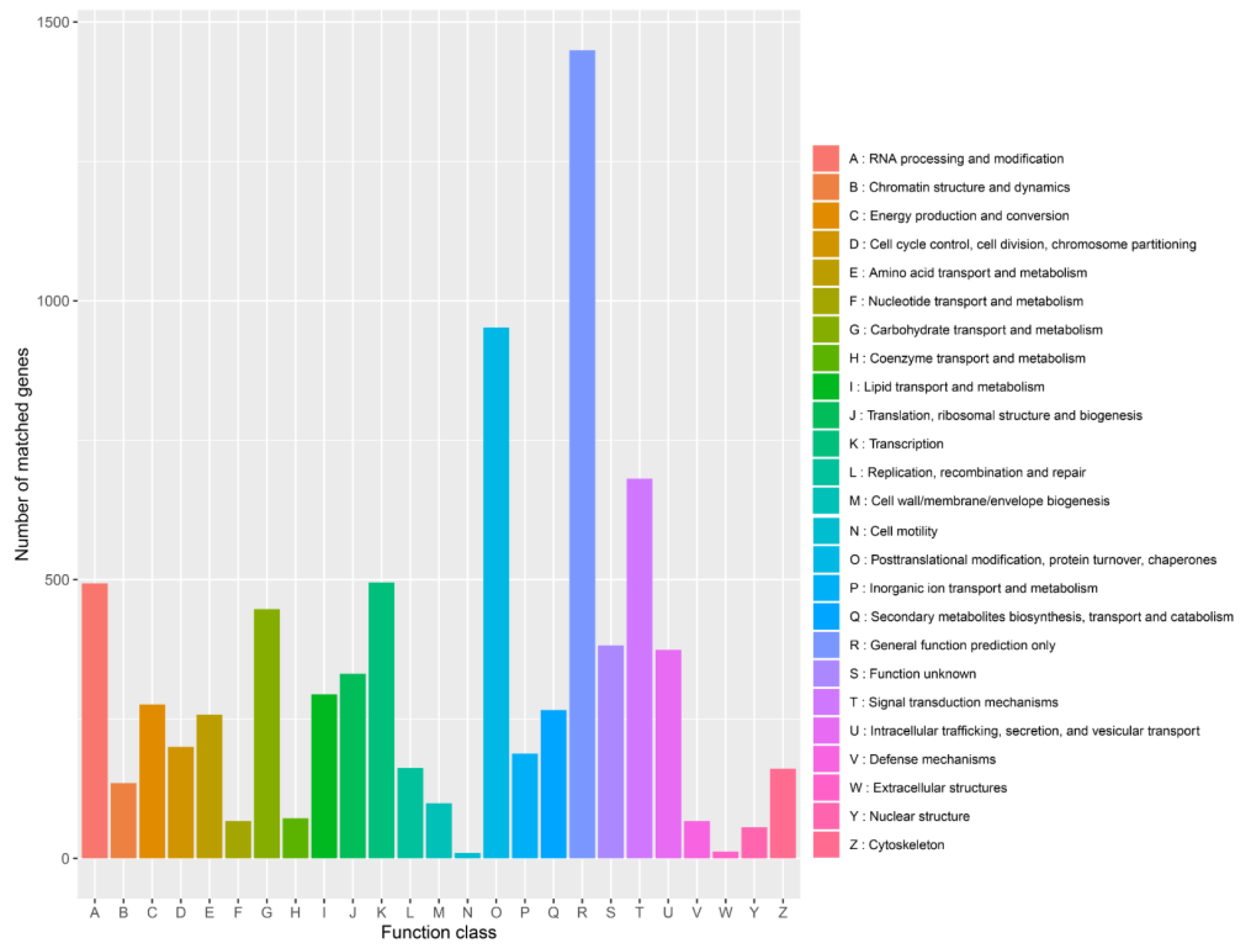
3.4. Functional Annotation of Genes
3.5. Identification of Hsf Genes Using the Full-Length Transcriptome Data
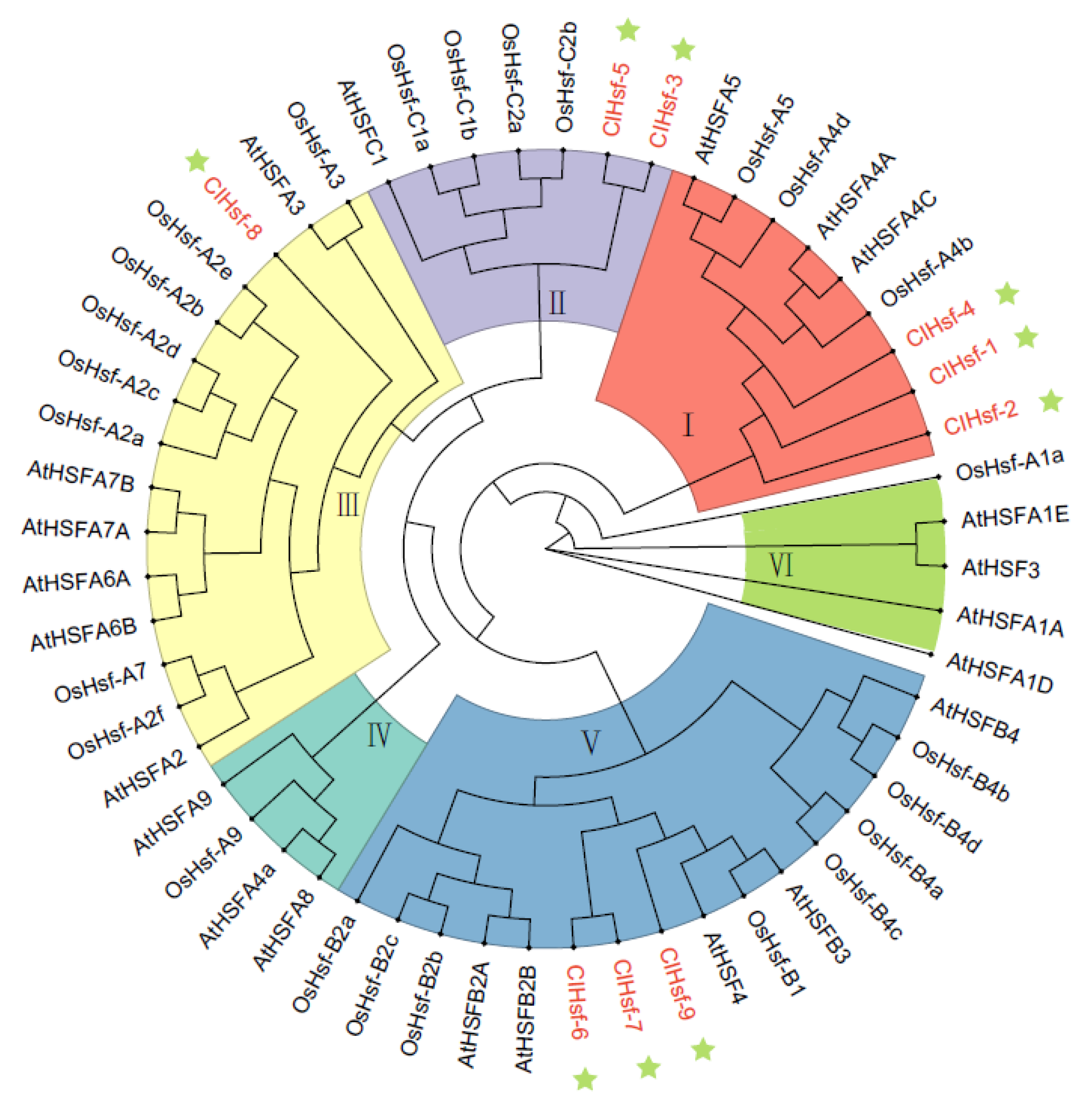
3.6. Conserved Domains and Phylogenetic Analysis
3.7. Expression of Hsfs in Transcriptomes under Heat Stress
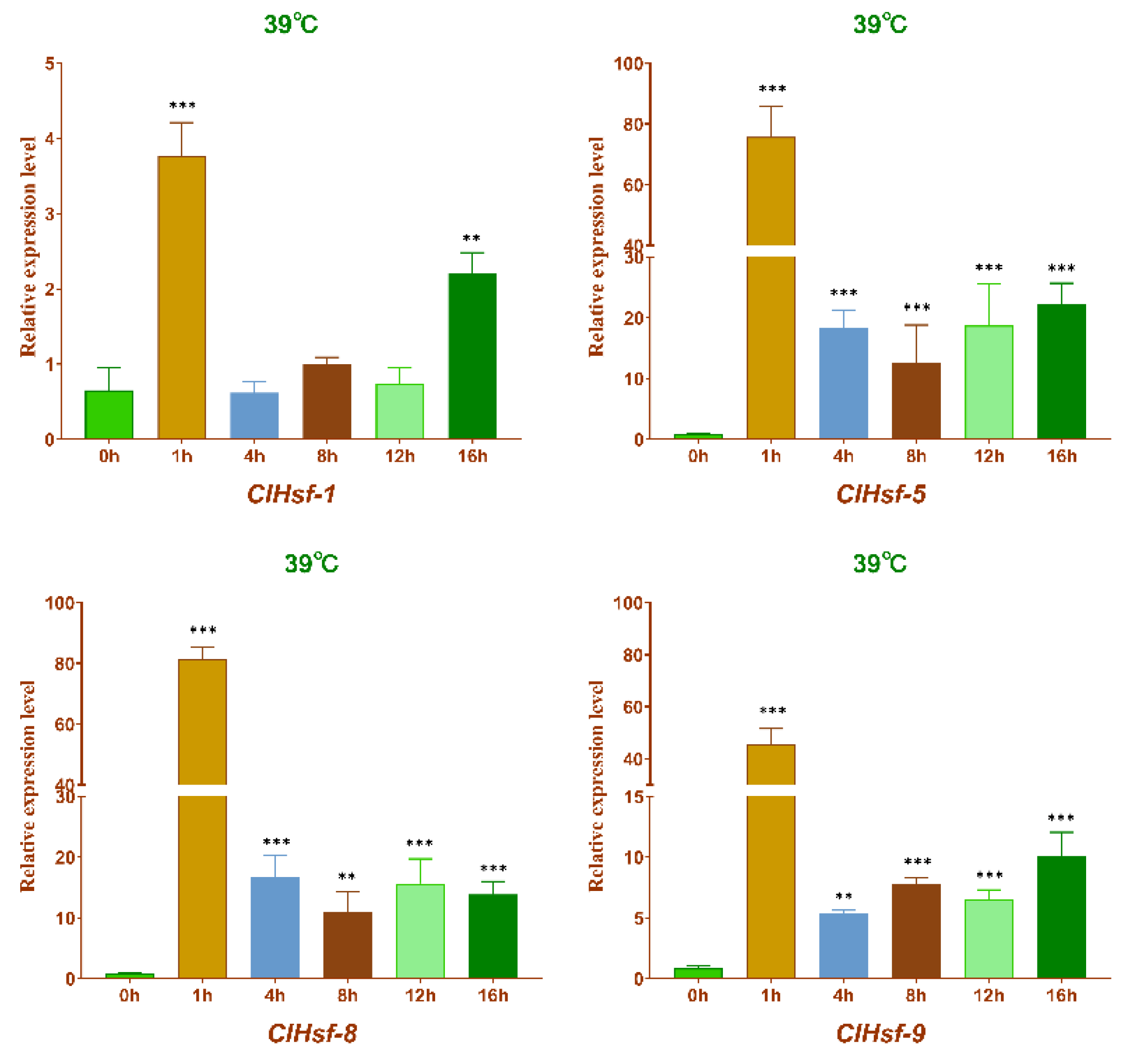
3.8. Expression Patterns of Hsf Genes in Heat Stress and Different Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gilman, E.F.; Watson, D.G. Cunninghamia lanceolata: China Fir; Environmental Horticulture Department, University of Florida: Gainesville, FL, USA, 2014; pp. 1–3. [Google Scholar]
- Lu, Y.; Coops, N.C.; Wang, T.; Wang, G. A process-based approach to estimate Chinese fir (Cunninghamia lanceolata) distribution and productivity in southern China under climate change. Forests 2015, 6, 360–379. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yu, X.; Liu, Y.; Shi, Z.; Li, L.; Xie, S.; Zhu, G.; Zhao, P. Comparative metabolomics analysis reveals the color variation between heartwood and sapwood of Chinese fir (Cunninghamia lanceolata (Lamb.) Hook. Ind. Crop. Prod. 2021, 169, 113656. [Google Scholar] [CrossRef]
- Wu, H.; Xiang, W.; Chen, L.; Ouyang, S.; Xiao, W.; Li, S.; Forrester, D.I.; Lei, P.; Zeng, Y.; Deng, X.; et al. Soil phosphorus bioavailability and recycling increased with stand age in Chinese fir plantations. Ecosystems 2020, 23, 973–988. [Google Scholar] [CrossRef]
- Yi, C.; Hendrey, G.; Niu, S.; McDowell, N.; Allen, C.D. Tree mortality in a warming world: Causes, patterns, and implications. Environ. Res. Lett. 2022, 17, 030201. [Google Scholar] [CrossRef]
- Allen, C.D.; Macalady, A.K.; Chenchouni, H.; Bachelet, D.; McDowell, N.; Vennetier, M.; Cobb, N. A global overview of drought and heat-induced tree mortality reveals emerging climate change risks for forests. For. Ecol. Manag. 2010, 259, 660–684. [Google Scholar] [CrossRef] [Green Version]
- Camarero, J.J. The drought-dieback-death conundrum in trees and forests. Plant Ecol. Divers. 2021, 14, 1–12. [Google Scholar] [CrossRef]
- Wu, H.; Zheng, R.; Hao, Z.; Meng, Y.; Weng, Y.; Zhou, X.; Chen, J. Cunninghamia lanceolata PSK peptide hormone genes promote primary root growth and adventitious root formation. Plants 2019, 8, 520. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.; Zhuang, H.; Yu, J.; Liu, X.; Huang, H.; Zhu, M.; Tong, Z. Genome survey of Chinese fir (Cunninghamia lanceolata): Identification of genomic SSRs and demonstration of their utility in genetic diversity analysis. Sci. Rep. 2020, 10, 4698. [Google Scholar] [CrossRef]
- Ji, Y.; Zhu, L.; Hao, Z.; Su, S.; Zheng, X.; Shi, J.; Zheng, R.; Chen, J. Exploring the Cunninghamia lanceolata (Lamb.) Hook Genome by BAC Sequencing. Front. Bioeng. Biotechnol. 2022, 10, 854130. [Google Scholar] [CrossRef]
- Zheng, W.; Chen, J.; Hao, Z.; Shi, J. Comparative analysis of the chloroplast genomic information of Cunninghamia lanceolata (Lamb.) Hook with sibling species from the Genera Cryptomeria D. Don, Taiwania Hayata, and Calocedrus Kurz. Int. J. Mol. Sci. 2016, 17, 1084. [Google Scholar] [CrossRef] [Green Version]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H. The review of transcriptome sequencing: Principles, history and advances. IOP Conf. Ser. Earth Environ. Sci. 2019, 332, 042003. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child.-Educ. Pract. 2013, 98, 236–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.C.; Ahn, D.H.; Kim, S.J.; Lee, H.; Oh, T.; Lee, J.E.; Park, H. Advantages of single-molecule real-time sequencing in high-GC content genomes. PLoS ONE 2013, 8, e68824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; Su, Y.; Wang, T. Full-length transcriptome analysis of four different tissues of Cephalotaxus oliveri. Int. J. Mol. Sci. 2021, 22, 787. [Google Scholar] [CrossRef]
- Zhu, L.; Lu, L.; Yang, L.; Hao, Z.; Chen, J.; Cheng, T. The full-length transcriptome sequencing and identification of Na+/H+ antiporter genes in halophyte Nitraria tangutorum Bobrov. Genes 2021, 12, 836. [Google Scholar] [CrossRef]
- Ye, J.; Cheng, S.; Zhou, X.; Chen, Z.; Kim, S.U.; Tan, J.; Zheng, J.; Xu, F.; Zhang, W.; Liao, Y. A global survey of full-length transcriptome of Ginkgo biloba reveals transcript variants involved in flavonoid biosynthesis. Ind. Crop. Prod. 2019, 139, 111547. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Kang, Y.; Yang, D.; Kong, L.; Hou, M.; Meng, Y.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. The clustal omega multiple alignment package. In Multiple Sequence Alignment. Methods in Molecular Biology; Humana: New York, NY, USA, 2021; pp. 3–16. [Google Scholar]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree v1. 3.1. 2009. Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 5 July 2022).
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Shimizu, K.; Adachi, J.; Muraoka, Y. ANGLE: A sequencing errors resistant program for predicting protein coding regions in unfinished cDNA. J. Bioinform. Comput. Biol. 2006, 4, 649–664. [Google Scholar] [CrossRef]
- Yao, S.; Wu, F.; Hao, Q.; Ji, K. Transcriptome-wide identification of WRKY transcription factors and their expression profiles under different types of biological and abiotic stress in Pinus massoniana lamb. Genes 2020, 11, 1386. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tang, X.; Ren, C.; Wei, B.; Wu, Y.; Wu, Q.; Pei, J. Full-length transcriptome sequences and the identification of putative genes for flavonoid biosynthesis in safflower. BMC Genom. 2018, 19, 548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, J.; Wang, R.; Ma, X.; Liu, J.; Lu, X.; Thakar, S.B.; An, N.; Liu, J.; Xia, E.; Liu, Y. Full-length transcriptome sequencing provides insights into the evolution of apocarotenoid biosynthesis in Crocus sativus. Comput. Struct. Biotechnol. J. 2020, 18, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, J.; He, N.; Guo, F. Metabolic reprogramming in chloroplasts under heat stress in plants. Int. J. Mol. Sci. 2018, 19, 849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Ding, Y.; Zhu, C. Sensitivity and responses of chloroplasts to heat stress in plants. Front. Plant Sci. 2020, 11, 375. [Google Scholar]
- Jagadish, S.K.; Way, D.A.; Sharkey, T.D. Plant heat stress: Concepts directing future research. Plant Cell Environ. 2021, 44, 1992–2005. [Google Scholar] [CrossRef]
- Xue, G.; Drenth, J.; McIntyre, C.L. TaHsfA6f is a transcriptional activator that regulates a suite of heat stress protection genes in wheat (Triticum aestivum L.) including previously unknown Hsf targets. J. Exp. Bot. 2015, 66, 1025–1039. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Niu, C.; Yang, C.; Jinn, T. The heat stress factor HSFA6b connects ABA signaling and ABA-mediated heat responses. Plant Physiol. 2016, 172, 1182–1199. [Google Scholar] [CrossRef]
- Liu, M.; Huang, Q.; Sun, W.; Ma, Z.; Huang, L.; Wu, Q.; Tang, Z.; Bu, T.; Li, C.; Chen, H. Genome-wide investigation of the heat shock transcription factor (Hsf) gene family in Tartary buckwheat (Fagopyrum tataricum). BMC Genom. 2019, 20, 871. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, H.; Chu, Z.; Tang, X.; Zhu, S.; Cheng, B. Genome-wide identification, classification and analysis of heat shock transcription factor family in maize. BMC Genom. 2011, 12, 76. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Song, H.; Li, C.; Li, P.; Li, A.; Guan, H.; Hou, L.; Wang, X. Genome-wide dissection of the heat shock transcription factor family genes in Arachis. Front. Plant Sci. 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Subreads Base.G. | Subreads Number | Average Length | N50 |
|---|---|---|---|---|
| Chinese fir | 20.62 | 6,747,129 | 3057 | 3420 |
| Stage | <500 bp | 500 bp–1 kb | 1–2 kb | 2–3 kb | >3 kb | Total |
|---|---|---|---|---|---|---|
| Before | 6 | 107 | 3659 | 6718 | 10,841 | 21,331 |
| After | 3 | 78 | 2103 | 3461 | 5449 | 11,094 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, Y.; Wu, H.; Zheng, X.; Zhu, L.; Zhu, Z.; Chen, Y.; Shi, J.; Zheng, R.; Chen, J. Full-Length Transcriptome Sequencing and Identification of Hsf Genes in Cunninghamia lanceolata (Lamb.) Hook. Forests 2023, 14, 684. https://doi.org/10.3390/f14040684
Ji Y, Wu H, Zheng X, Zhu L, Zhu Z, Chen Y, Shi J, Zheng R, Chen J. Full-Length Transcriptome Sequencing and Identification of Hsf Genes in Cunninghamia lanceolata (Lamb.) Hook. Forests. 2023; 14(4):684. https://doi.org/10.3390/f14040684
Chicago/Turabian StyleJi, Yuan, Hua Wu, Xueyan Zheng, Liming Zhu, Zeli Zhu, Ya Chen, Jisen Shi, Renhua Zheng, and Jinhui Chen. 2023. "Full-Length Transcriptome Sequencing and Identification of Hsf Genes in Cunninghamia lanceolata (Lamb.) Hook" Forests 14, no. 4: 684. https://doi.org/10.3390/f14040684