
Genetic Diversity of Nanmu (Phoebe zhennan S. Lee. et F. N. Wei) Breeding Population and Extraction of Core Collection Using nSSR, cpSSR and Phenotypic Markers


Abstract
:1. Introduction
2. Materials and Methods
2.1. Germplasm Materials
2.2. DNA Isolation and SSRs Amplification
2.3. Genetic Diversity Analyses
2.4. Growth and Leaf Trait Measurement
2.5. Phenotypic Traits Analysis
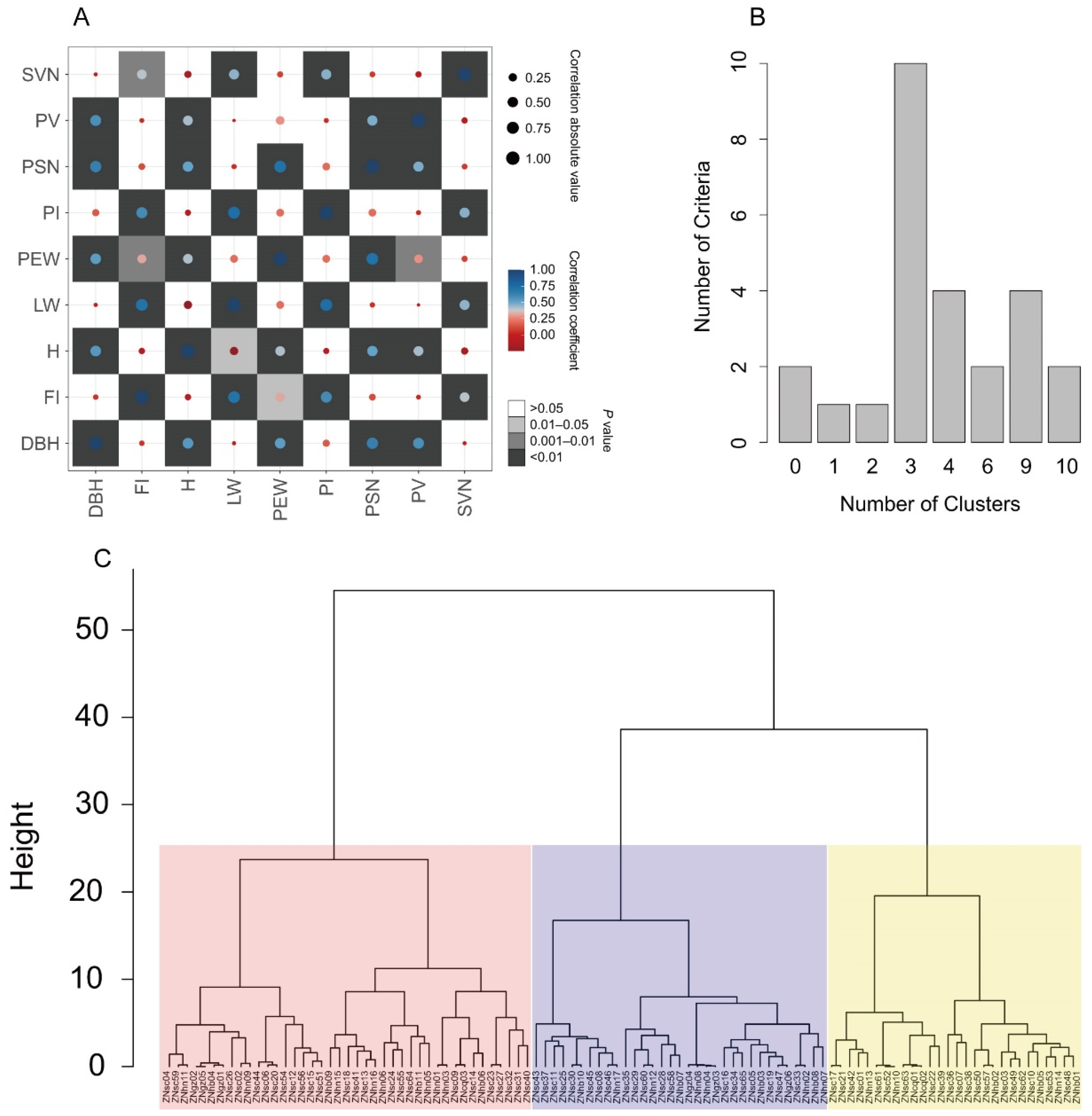
2.6. Core Collection Extraction
3. Results
3.1. Genetic Diversity of the Breeding Population
3.1.1. Nuclear Genetic Diversity
3.1.2. cpSSR Polymorphism
3.2. Genetic Structure of Germplasms
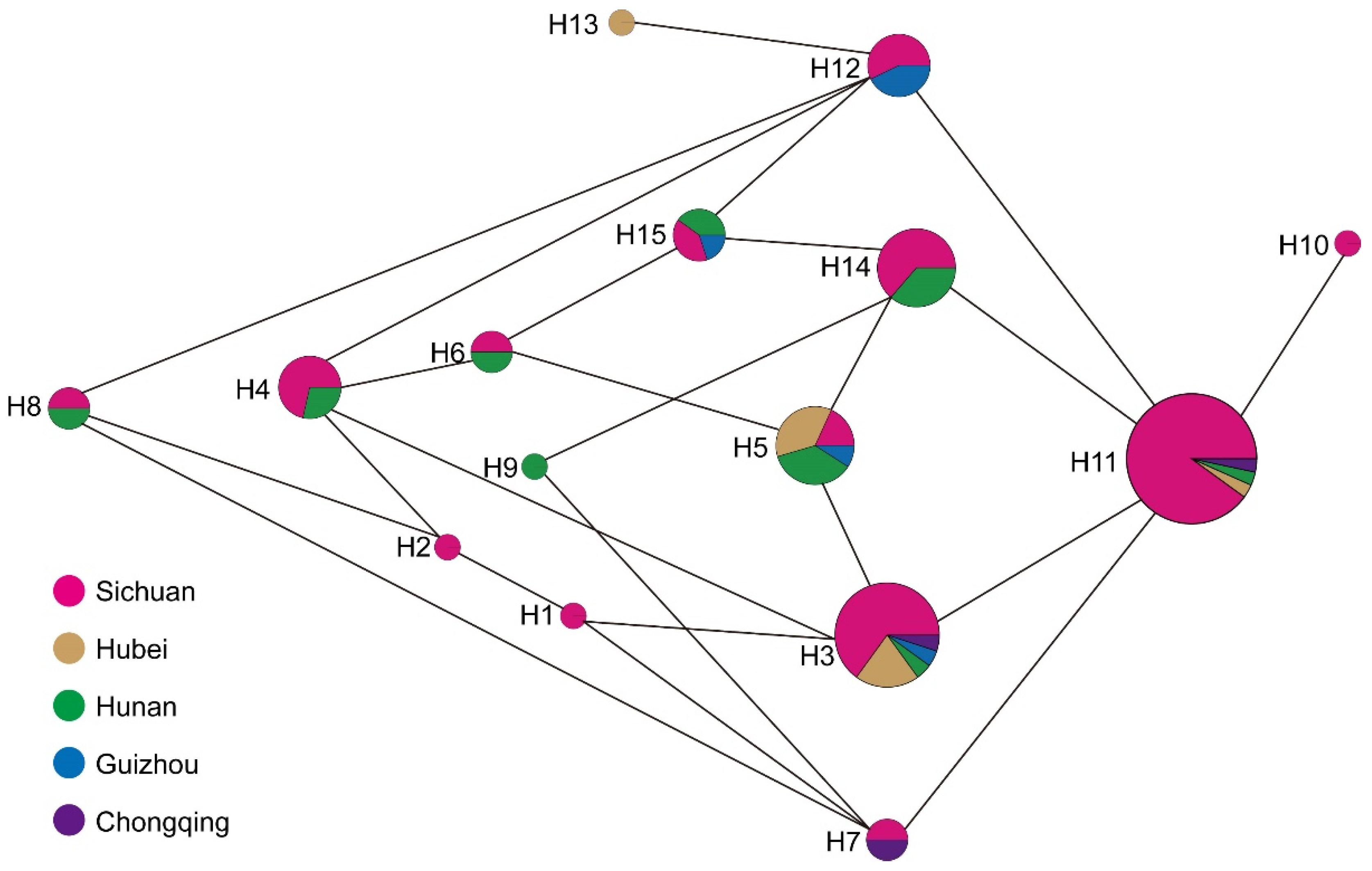
3.3. Chloroplast Haplotypes Variation
3.4. Phenotypic Diversity
3.5. Extraction of a Core Collection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yang, H.; Liu, Q.; Zhang, R.; Zhou, Z. Genetic diversity of second generation-parental germplasm of masson pine revealed by SSR markers and establishment of a core germplasm collection. Scand. J. For. Res. 2021, 36, 524–531. [Google Scholar] [CrossRef]
- Kopp, R.; Smart, L.; Maynard, C.; Tuskan, G.; Abrahamson, L. Predicting within-family variability in juvenile height growth of Salix based upon similarity among parental AFLP fingerprints. Theor. Appl. Genet. 2002, 105, 106–112. [Google Scholar] [CrossRef] [PubMed]
- White, T.L.; Adams, W.T.; Neale, D.B. Forest Genetics; CABI Publishing: Cambredge, MA, USA, 2007. [Google Scholar]
- Lv, J.; Li, C.; Zhou, C.; Chen, J.; Li, F.; Weng, Q.; Li, M.; Wang, Y.; Chen, S.; Chen, J.; et al. Genetic diversity analysis of a breeding population of Eucalyptus cloeziana F. Muell. (Myrtaceae) and extraction of a core germplasm collection using microsatellite markers. Ind. Crop. Prod. 2020, 145, 112157. [Google Scholar] [CrossRef]
- Scotti, I.; González-Martínez, S.C.; Budde, K.B.; Lalagüe, H. Fifty years of genetic studies: What to make of the large amounts of variation found within populations? Ann. For. Sci. 2016, 73, 69–75. [Google Scholar] [CrossRef]
- Gudeta, T.B. Molecular marker based genetic diversity in forest tree populations. For. Res. Eng. Int. J. 2018, 2, 176–182. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Che, Z.; Wang, L.; Wei, W.; Li, D. Genetic diversity assessment of sesame core collection in China by phenotype and molecular markers and extraction of a mini-core collection. BMC Genet. 2012, 13, 102. [Google Scholar] [CrossRef] [PubMed]
- Holden, J.H.W.; Williams, J.T. Crop Genetic Resources: Conservation & Evaluation; George Allen & Unwin: London, UK, 1984. [Google Scholar]
- Zhong, Y.; Wang, Y.; Sun, Z.; Niu, J.; Shi, Y.; Huang, K.; Chen, J.; Chen, J.; Luan, M. Genetic Diversity of a Natural Population of Akebia trifoliata (Thunb.) Koidz and Extraction of a Core Collection Using Simple Sequence Repeat Markers. Front. Genet. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Frankel, O.H. Genetic Manipulation Impact on Man & Society; Cambrdge University Press: Cambridge, UK, 1984. [Google Scholar]
- Xue, H.; Yu, X.; Fu, P.; Liu, B.; Zhang, S.; Li, J.; Zhai, W.; Lu, N.; Zhao, X.; Wang, J.; et al. Construction of the Core Collection of Catalpa fargesii f. duclouxii (Huangxinzimu) Based on Molecular Markers and Phenotypic Traits. Forests 2021, 12, 1518. [Google Scholar] [CrossRef]
- Mao, X.; Zhu, S.; Li, S.; Hua, H.; Tian, S.; Zhong, W.; Dong, Y.; An, X. Core germplasm construction of Populus tomentosa based on the fluorescent SSR markers. J. Beijing For. Univ. 2020, 42, 40–47. [Google Scholar]
- Mohan, M.; Nair, S.; Bhagwat, A.; Krishna, T.G.; Yano, M.; Bhatia, C.R.; Sasaki, T. Genome mapping, molecular markers and marker-assisted selection in crop plants. Mol. Breed. 1997, 3, 87–103. [Google Scholar] [CrossRef]
- Luo, C.; Chen, D.; Cheng, X.; Liu, H.; Li, Y.; Huang, C. SSR Analysis of Genetic Relationship and Classification in Chrysanthemum Germplasm Collection. Hortic. Plant J. 2018, 4, 73–82. [Google Scholar] [CrossRef]
- Yang, X.-S.; Su, W.-J.; Wang, L.-J.; Lei, J.; Chai, S.-S.; Liu, Q.-C. Molecular diversity and genetic structure of 380 sweetpotato accessions as revealed by SSR markers. J. Integr. Agric. 2015, 14, 633–641. [Google Scholar] [CrossRef]
- Mahmoodi, R.; Dadpour, M.R.; Hassani, D.; Zeinalabedini, M.; Vendramin, E.; Leslie, C.A. Composite core set construction and diversity analysis of Iranian walnut germplasm using molecular markers and phenotypic traits. PLoS ONE 2021, 16, e0248623. [Google Scholar] [CrossRef]
- Ma, Q.; Ning, X.; Li, J.; Chen, H.; Yu, Y.; Lin, H. Mining elite sea-island cotton germplasm based on phenotyping and SSR markers. Cotton Sci. 2020, 32, 91–101. [Google Scholar]
- Di Guardo, M.; Scollo, F.; Ninot, A.; Rovira, M.; Hermoso, J.F.; Distefano, G.; La Malfa, S.; Batlle, I. Genetic structure analysis and selection of a core collection for carob tree germplasm conservation and management. Tree Genet. Genomes 2019, 15, 41. [Google Scholar] [CrossRef]
- Razi, S.; Soleimani, A.; Zeinalabedini, M.; Vazifeshenas, M.R.; Martínez-Gómez, P.; Mohsenzade Kermani, A.; Raiszadeh, A.R.; Tayari, M.; Martínez-García, P.J. Development of a Multipurpose Core Collection of New Promising Iranian Pomegranate (Punica granatum L.) Genotypes Based on Morphological and Pomological Traits. Horticulturae 2021, 7, 350. [Google Scholar] [CrossRef]
- Belaj, A.; del Carmen Dominguez-García, M.; Atienza, S.G.; Urdíroz, N.M.; De la Rosa, R.; Satovic, Z.; Martín, A.; Kilian, A.; Trujillo, I.; Valpuesta, V.; et al. Developing a core collection of olive (Olea europaea L.) based on molecular markers (DArTs, SSRs, SNPs) and agronomic traits. Tree Genet. Genomes 2012, 8, 365–378. [Google Scholar] [CrossRef]
- Kaviriri, D.K.; Zhang, Q.H.; Zhang, X.X.; Jiang, L.P.; Zhang, J.P.; Wang, J.Y.; Khasa, D.P.; You, X.L.; Zhao, X.Y. Phenotypic Variability and Genetic Diversity in a Pinus koraiensis Clonal Trial in Northeastern China. Genes 2020, 11, 673. [Google Scholar] [CrossRef]
- Gao, J.H.; Zhang, W.; Li, J.Y.; Long, H.L.; He, W.; Li, X.Q. Amplified fragment length polymorphism analysis of the population structure and genetic diversity of Phoebe zhennan (Lauraceae), a native species to China. Biochem. Syst. Ecol. 2016, 64, 149–155. [Google Scholar] [CrossRef]
- Li, X.W.; Li, J.; Huang, P.H.; Wei, F.N.; van der Werff, H. Lauraceae. In Flora of China; Wu, Z.Y., Raven, P.H., Hong, D.Y., Eds.; Science Press & St. Louis; Missorui Botanical Garden Press: Beijing, China, 2008; Volume 7, pp. 102–254. [Google Scholar]
- Xiao, J.-H.; Ding, X.; Li, L.; Ma, H.; Ci, X.-Q.; van der Merwe, M.; Conran, J.G.; Li, J. Miocene diversification of a golden-thread nanmu tree species (Phoebe zhennan, Lauraceae) around the Sichuan Basin shaped by the East Asian monsoon. Ecol. Evol. 2020, 10, 10543–10557. [Google Scholar] [CrossRef]
- Ding, X.; Xiao, J.H.; Li, L.; Conran, J.G.; Li, J. Congruent species delimitation of two controversial gold-thread nanmu tree species based on morphological and restriction site-associated DNA sequencing data. J. Syst. Evol. 2019, 57, 234–246. [Google Scholar] [CrossRef]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Phumichai, C.; Phumichai, T.; Wongkaew, A. Novel Chloroplast Microsatellite (cpSSR) Markers for Genetic Diversity Assessment of Cultivated and Wild Hevea Rubber. Plant Mol. Biol. Rep. 2015, 33, 1486–1498. [Google Scholar] [CrossRef]
- Petit, J.R.; Duminil, J.; Fineschi, S.; Hampe, A.; Salvini, D.; Vendramin, G.G. Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol. Ecol. 2005, 14, 689–701. [Google Scholar] [CrossRef]
- Shi, X.; Zhu, X.; Sheng, Y.; Zhuang, G.; Chen, F. Development of SSR markers based on transcriptome sequence of Phoebe zhennan. Sci. Silvae Sin. 2016, 52, 71–78. [Google Scholar] [CrossRef]
- Liu, D. Genetic Diversity of Germplasm Resources Revealed by SSR in Phoebe Bournei; Fujian Agriculture and Forestry University: Fuzhou, China, 2019. [Google Scholar]
- Li, Q.; Wan, J. SSRHunter: Development of a local searching software for SSR sites. Hereditas 2005, 27, 808–810. [Google Scholar]
- Chen, B.B.; Zeng, Y.; Li, C.L.; Xu, B.; Zhang, C.; Chen, W.; Li, S.L. Characterization of the complete chloroplast genome of the Phoebe zhennan S. Lee. Mitochondrial DNA Part B Resour. 2019, 4, 856–857. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.; von Holdt, B. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2011, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]
- Rosenberg, N.A. Distruct: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Taylor, F.W. Rapid determination of southern pine specific gravity with a Pilodyn tester. For. Sci. 1981, 27, 59–61. [Google Scholar]
- Shannon, C.E. A mathematical theory of communication. ACM SIGMOBILE Mob. Comput. Commun. Rev. 2001, 5, 3–55. [Google Scholar] [CrossRef]
- Kim, K.-W.; Chung, H.-K.; Cho, G.-T.; Ma, K.-H.; Chandrabalan, D.; Gwag, J.-G.; Kim, T.-S.; Cho, E.-G.; Park, Y.-J. PowerCore: A program applying the advanced M strategy with a heuristic search for establishing core sets. Bioinformatics 2007, 23, 2155–2162. [Google Scholar] [CrossRef]
- Glaszmann, J.C.; Kilian, B.; Upadhyaya, H.D.; Varshney, R.K. Accessing genetic diversity for crop improvement. Curr. Opin. Plant Biol. 2010, 13, 167–173. [Google Scholar] [CrossRef]
- Li, X.; Tang, S.; Long, S.; Long, H.; Zhang, W. Timber physical and mechanical properties of Phoebe zhennan. J. Northeast For. Univ. 2013, 41, 77–79. [Google Scholar]
- Yang, J.; Ning, L.; Liu, M.; Liao, X.; Hu, R.; Zhang, M. Wood characteristicsand radial variations of Phoebe zhennan. J. Northwest AF Univ. (Nat. Sci. Ed.) 2017, 46, 7–14. [Google Scholar]
- Liu, D.; Liu, B.; Zeng, Q.; Chen, S.; Liu, B.; Li, Y. Genetic diversity of the superior geotypes of Phoebe bournei using SSR markers. J. For. Environ. 2019, 39, 449–453. [Google Scholar]
- Wright, S. The genetical structure of populations. Ann. Eugen. 1949, 15, 323–354. [Google Scholar] [CrossRef]
- Ohta, S.; Nishitani, C.; Yamamoto, T. Chloroplast microsatellites in Prunus, Rosaceae. Mol. Ecol. Notes 2005, 5, 837–840. [Google Scholar] [CrossRef]
- David, H.R.; Richard, F. How closely correlated are molecular and quantitative measures of genetic variation? A meta-analysis. Evolution 2001, 55, 1095–1103. [Google Scholar] [CrossRef]
- Duan, H.; Cao, S.; Zheng, H.; Hu, D.; Lin, J.; Cui, B.; Lin, H.; Hu, R.; Wu, B.; Sun, Y.; et al. Genetic Characterization of Chinese fir from Six Provinces in Southern China and Construction of a Core Collection. Sci. Rep. 2017, 7, 13814. [Google Scholar] [CrossRef]
- Brown, A. The case for core collections. In The Use of Plant Genetic Resources; Brown, A.H.D., Frankel, O.H., Marshall, D.R., Williams, J.T., Eds.; Cambridge University Press: Cambridge, UK, 1989. [Google Scholar]
- Li, Z.; Zhang, H.; Cao, Y.; Qiu, Z.; Wei, X.; Tang, S.; Yu, O.; Wang, X. Studies on the sampling strategy for primary core collection of Chinese indigence. Acta Agron. Sin. 2003, 29, 20–24. [Google Scholar]
- Jaradat, A.A. The dynamics of a core collection. In Core Collection of Plant Resouces; Hodgkin, T., Brown, A.D.H., van Hintum Th, J.L., Eds.; John Wiley and Sons: Chichester, UK, 1995. [Google Scholar]
- Silva-Junior, O.B.; Faria, D.A.; Grattapaglia, D. A flexible multi-species genome-wide 60KSNPchip developed from pooled resequencing of 240 Eucalyptus tree genomes across 12 species. New Phytol. 2015, 206, 1527–1540. [Google Scholar] [CrossRef]
- Goddard, M.E.; Hayes, B.J. Genomic selection. J. Anim. Breed. Genet. 2007, 124, 323–330. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Origin | Number of Germplasm | Germplasm Code | Latitude/° | Longitude/° |
|---|---|---|---|---|
| Sichuan province, China | 65 | ZNsc01-ZNsc65 | 30.8462 | 103.5600 |
| Hubei province, China | 11 | ZNhb01-ZNhb11 | 30.2669 | 108.9639 |
| Hunan province, China | 17 | ZNhn01-ZNhn17 | 29.3472 | 110.0844 |
| Chongqing province, China | 3 | ZNcq01-ZNcq03 | 29.3270 | 107.7538 |
| Guizhou province, China | 6 | ZNgz01-ZNgz06 | 27.1263 | 106.7335 |
| Locus | Na | Ne | I | Ho | He | uHe | F | h | uh | PIC | NPA |
|---|---|---|---|---|---|---|---|---|---|---|---|
| nSSR | |||||||||||
| ZiN-e8 | 10 | 1.476 | 0.761 | 0.048 | 0.325 | 0.325 | 0.852 | / | / | 0.306 | 8 |
| Unigene29601 | 8 | 2.894 | 1.354 | 0.228 | 0.658 | 0.658 | 0.651 | / | / | 0.617 | 4 |
| MN-e96 | 5 | 2.373 | 0.995 | 0.924 | 0.582 | 0.582 | −0.597 | / | / | 0.491 | 2 |
| MN-g30 | 33 | 11.276 | 2.885 | 1.000 | 0.916 | 0.916 | −0.097 | / | / | 0.906 | 17 |
| CL20730Contig1 | 2 | 1.160 | 0.265 | 0.149 | 0.139 | 0.139 | −0.080 | / | / | 0.128 | 0 |
| MN-g3 | 37 | 16.015 | 3.125 | 0.978 | 0.943 | 0.943 | −0.043 | / | / | 0.934 | 21 |
| MN-g5 | 34 | 14.387 | 3.029 | 0.278 | 0.936 | 0.936 | 0.701 | / | / | 0.926 | 21 |
| MN-g18 | 18 | 9.034 | 2.530 | 0.937 | 0.894 | 0.894 | −0.053 | / | / | 0.881 | 3 |
| CL4747Contig1 | 6 | 2.150 | 0.969 | 0.653 | 0.538 | 0.538 | −0.220 | / | / | 0.459 | 2 |
| Mean | 17 | 6.752 | 1.768 | 0.577 | 0.659 | 0.659 | 0.124 | / | / | 0.666 | 9 |
| cpSSR | |||||||||||
| PZmk03 | 2 | 1.940 | 0.677 | / | / | / | / | 0.484 | 0.489 | 0.328 | / |
| PZmf07 | 2 | 1.147 | 0.250 | / | / | / | / | 0.128 | 0.129 | 0.320 | / |
| PZmf05 | 2 | 1.710 | 0.606 | / | / | / | / | 0.415 | 0.419 | 0.375 | / |
| PZmf06 | 3 | 1.625 | 0.609 | / | / | / | / | 0.385 | 0.388 | 0.384 | / |
| PZmf10 | 2 | 1.020 | 0.055 | / | / | / | / | 0.019 | 0.020 | 0.109 | / |
| Mean | 2 | 1.488 | 0.440 | / | / | / | / | 0.286 | 0.289 | 0.303 | / |
| Haplotype | Count | PZmk03 | PZmf07 | PZmf05 | PZmf06 | PZmf10 |
|---|---|---|---|---|---|---|
| H1 | 1 | 179 | 267 | 238 | 204 | 222 |
| H2 | 1 | 179 | 267 | 238 | 205 | 222 |
| H3 | 20 | 179 | 268 | 238 | 204 | 222 |
| H4 | 7 | 179 | 268 | 238 | 205 | 222 |
| H5 | 11 | 179 | 268 | 239 | 204 | 222 |
| H6 | 2 | 179 | 268 | 239 | 205 | 222 |
| H7 | 2 | 180 | 267 | 238 | 204 | 222 |
| H8 | 2 | 180 | 267 | 238 | 205 | 222 |
| H9 | 1 | 180 | 267 | 239 | 204 | 222 |
| H10 | 1 | 180 | 268 | 238 | 203 | 222 |
| H11 | 30 | 180 | 268 | 238 | 204 | 222 |
| H12 | 7 | 180 | 268 | 238 | 205 | 222 |
| H13 | 1 | 180 | 268 | 238 | 205 | 223 |
| H14 | 11 | 180 | 268 | 239 | 204 | 222 |
| H15 | 5 | 180 | 268 | 239 | 205 | 222 |
| Traits | Core Collection | Whole Germplasms | Retention Rate/% | t Value | p Value |
|---|---|---|---|---|---|
| Phenotypic traits | |||||
| H | 5.1 | 5.3 | 96.3 | 0.532 | 0.597 |
| DBH | 5.3 | 5.5 | 95.7 | 1.023 | 0.311 |
| PSN | 2.3 | 2.4 | 96.5 | 0.533 | 0.596 |
| PEW | 2.4 | 2.4 | 97.3 | 0.494 | 0.623 |
| PV | 17.0 | 17.2 | 98.8 | 0.784 | 0.437 |
| FI | 8.9 | 8.7 | 101.4 | −0.204 | 0.839 |
| LW | 2.5 | 2.5 | 100.6 | −0.192 | 0.848 |
| PI | 0.8 | 0.8 | 101.4 | −0.212 | 0.833 |
| SVN | 21.5 | 21.5 | 99.9 | 0.450 | 0.655 |
| nSSR | |||||
| Na | 17 | 17 | 100.0 | 0.000 | 1.000 |
| Ne | 8.238 | 6.752 | 122.0 | 0.455 | 0.656 |
| I | 1.874 | 1.768 | 106.0 | 0.199 | 0.845 |
| Ho | 0.588 | 0.577 | 101.9 | 0.060 | 0.953 |
| He | 0.681 | 0.655 | 103.9 | 0.191 | 0.851 |
| uHe | 0.686 | 0.659 | 104.2 | 0.206 | 0.840 |
| cpSSR | |||||
| Na | 2.200 | 2.200 | 100.0 | 0.000 | 1.000 |
| Ne | 1.572 | 1.488 | 105.7 | 0.322 | 0.756 |
| I | 0.481 | 0.437 | 109.9 | 0.246 | 0.812 |
| h | 0.318 | 0.285 | 111.5 | 0.249 | 0.810 |
| uh | 0.323 | 0.288 | 112.2 | 0.263 | 0.800 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; An, W.; Peng, J.; Li, J.; Gu, Y.; Jiang, B.; Chen, L.; Zhu, P.; Yang, H. Genetic Diversity of Nanmu (Phoebe zhennan S. Lee. et F. N. Wei) Breeding Population and Extraction of Core Collection Using nSSR, cpSSR and Phenotypic Markers. Forests 2022, 13, 1320. https://doi.org/10.3390/f13081320
Zhu Y, An W, Peng J, Li J, Gu Y, Jiang B, Chen L, Zhu P, Yang H. Genetic Diversity of Nanmu (Phoebe zhennan S. Lee. et F. N. Wei) Breeding Population and Extraction of Core Collection Using nSSR, cpSSR and Phenotypic Markers. Forests. 2022; 13(8):1320. https://doi.org/10.3390/f13081320
Chicago/Turabian StyleZhu, Yan, Wenna An, Jian Peng, Jinwu Li, Yunjie Gu, Bo Jiang, Lianghua Chen, Peng Zhu, and Hanbo Yang. 2022. "Genetic Diversity of Nanmu (Phoebe zhennan S. Lee. et F. N. Wei) Breeding Population and Extraction of Core Collection Using nSSR, cpSSR and Phenotypic Markers" Forests 13, no. 8: 1320. https://doi.org/10.3390/f13081320