
Degradation by Electron Beam Irradiation of Some Elastomeric Composites Sulphur Vulcanized
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Sample Preparation
2.2. Sample Irradiation
2.3. Laboratory Tests
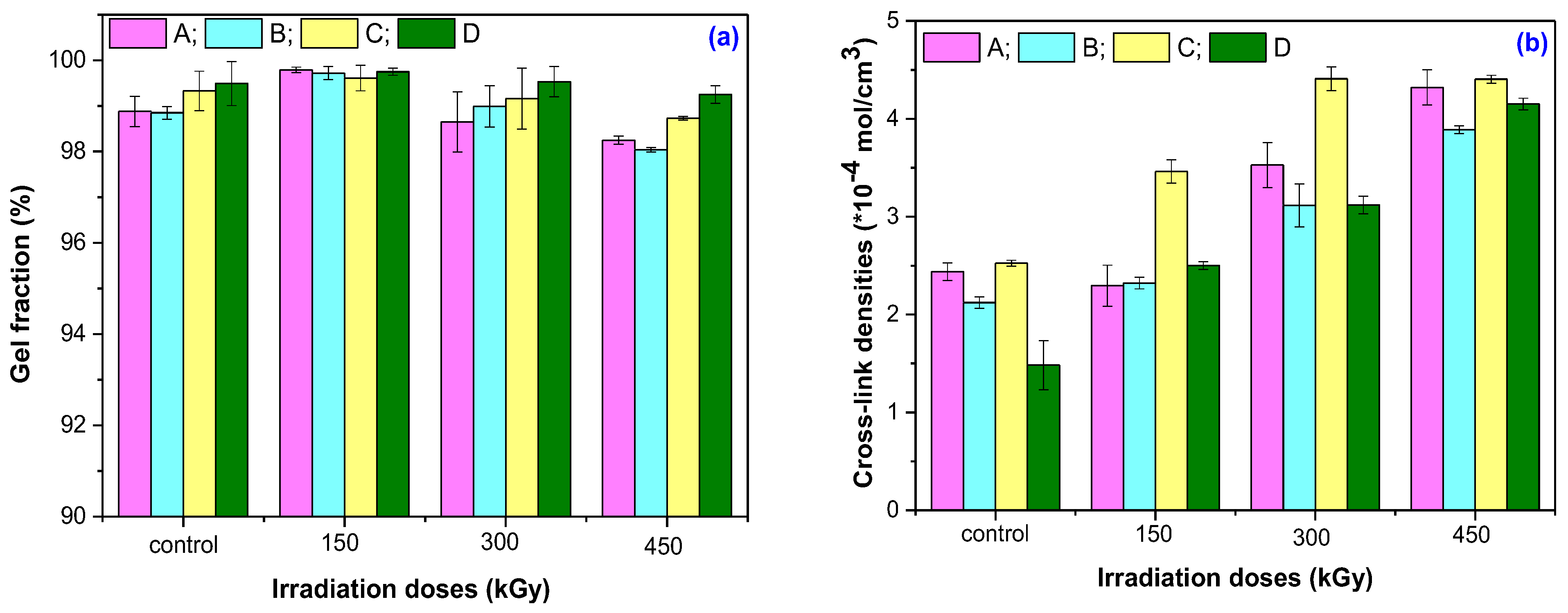
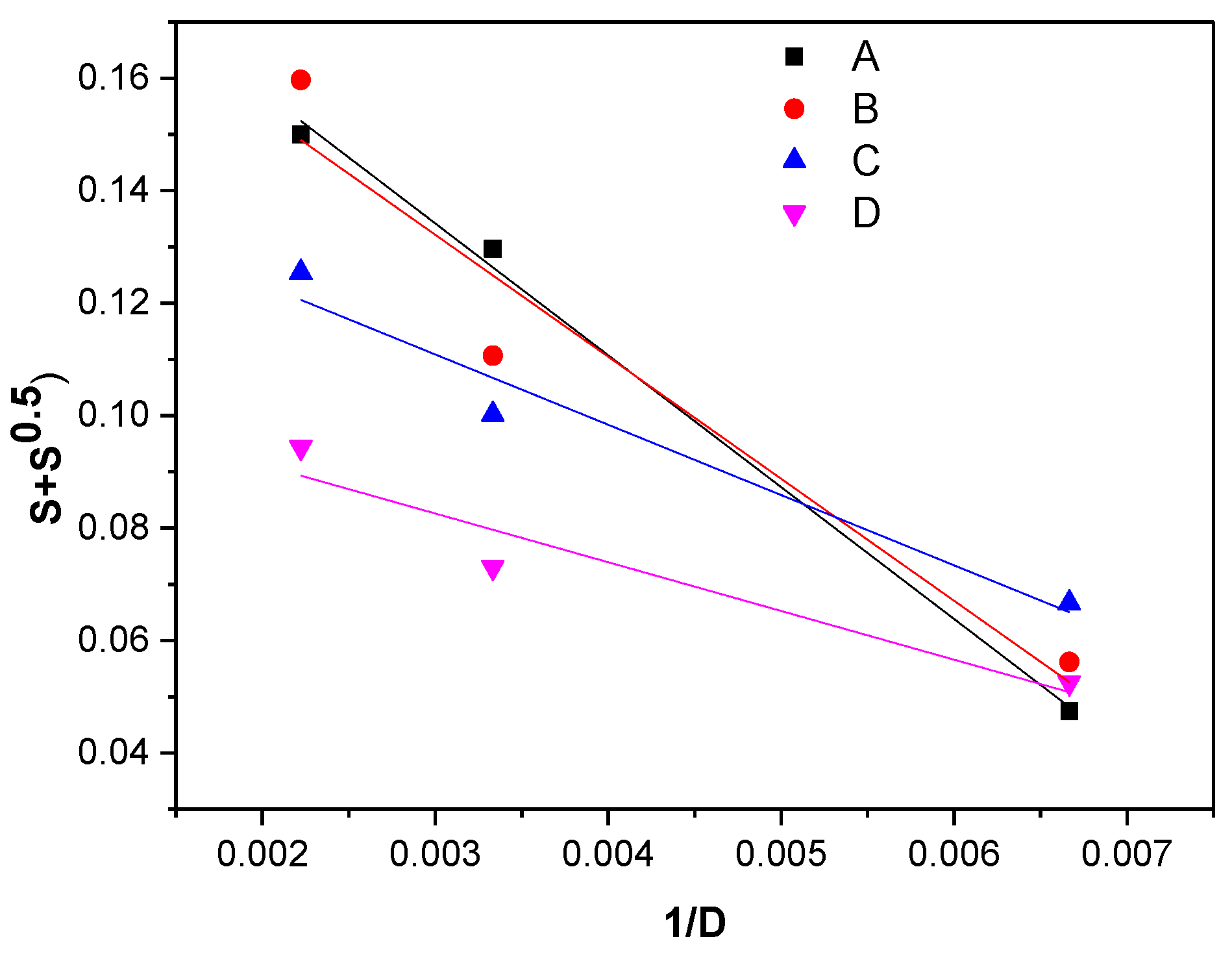
2.3.1. Gel Fraction and Cross-Link Density
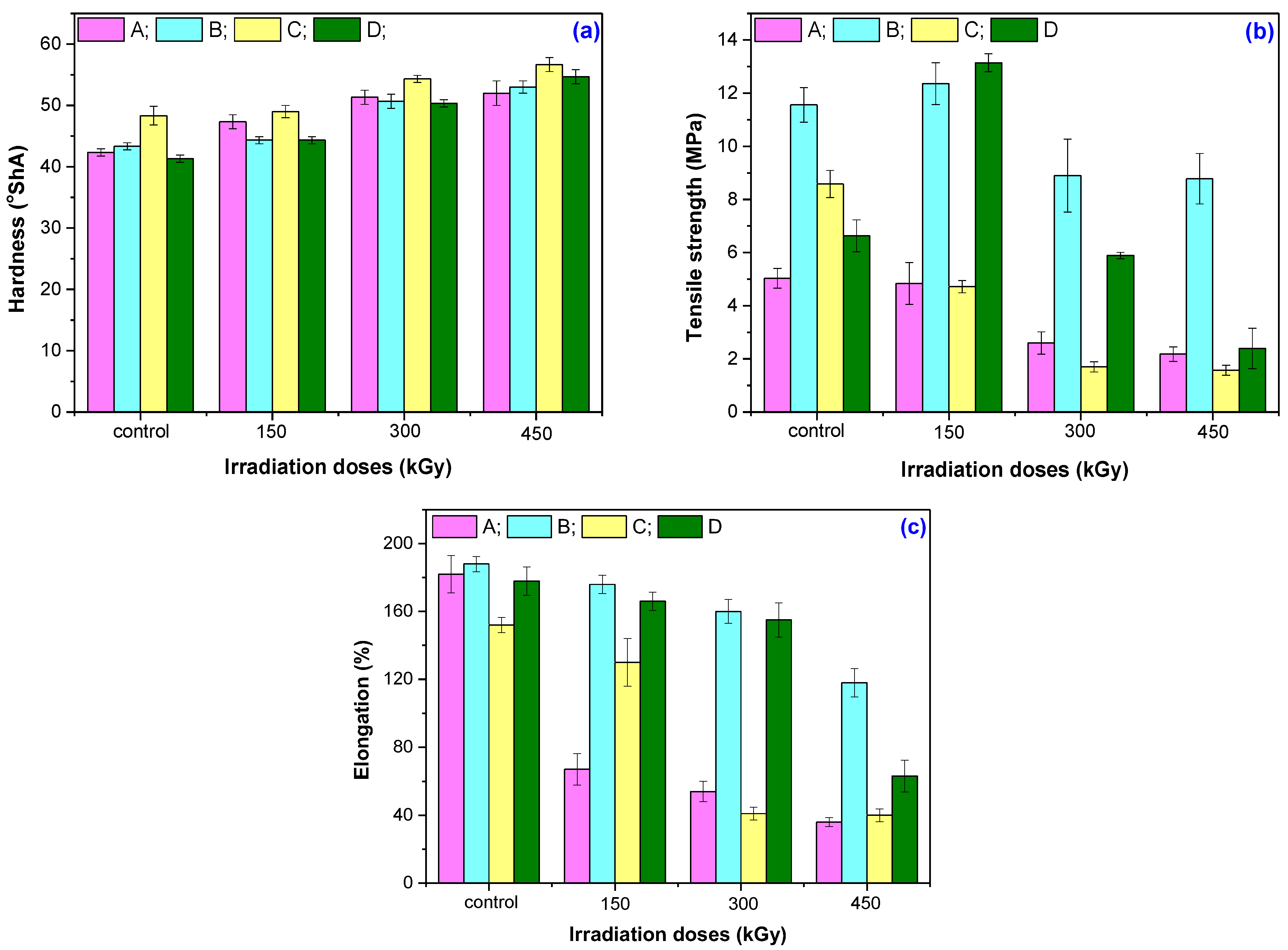
2.3.2. Mechanical Characteristics
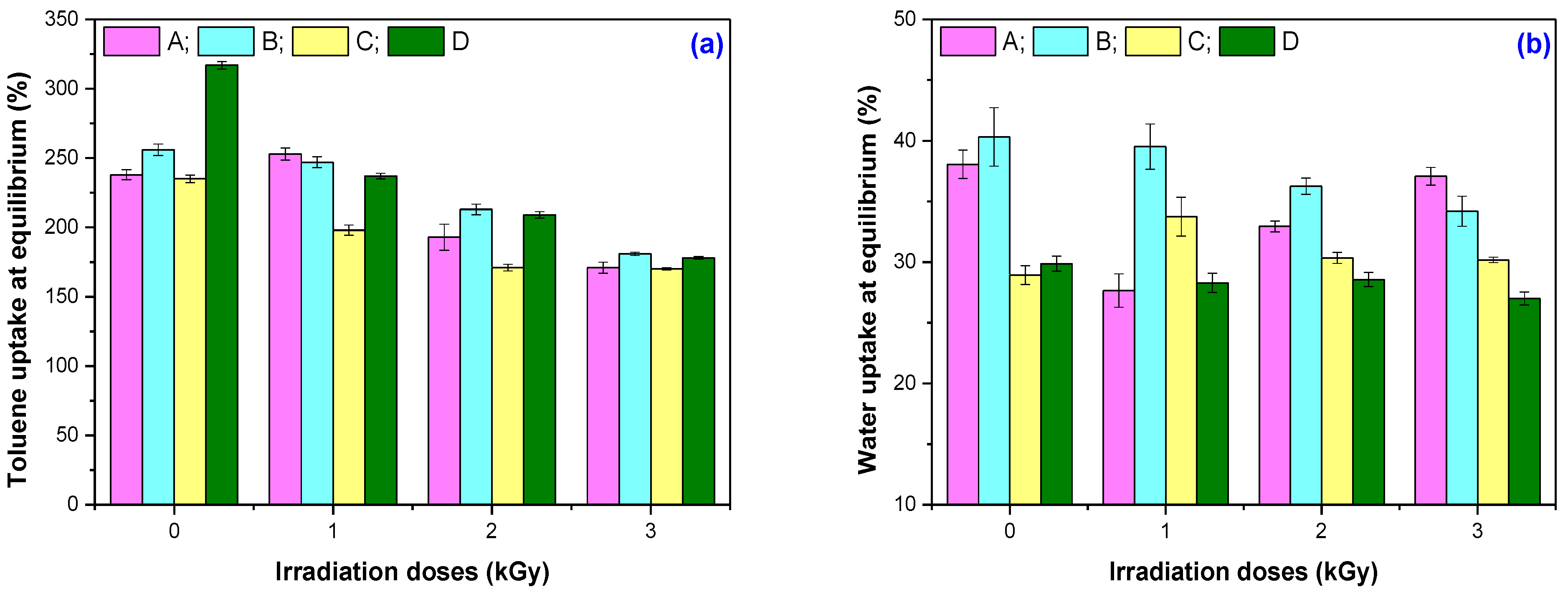
2.3.3. Water and Toluene Uptake and Weight Loss
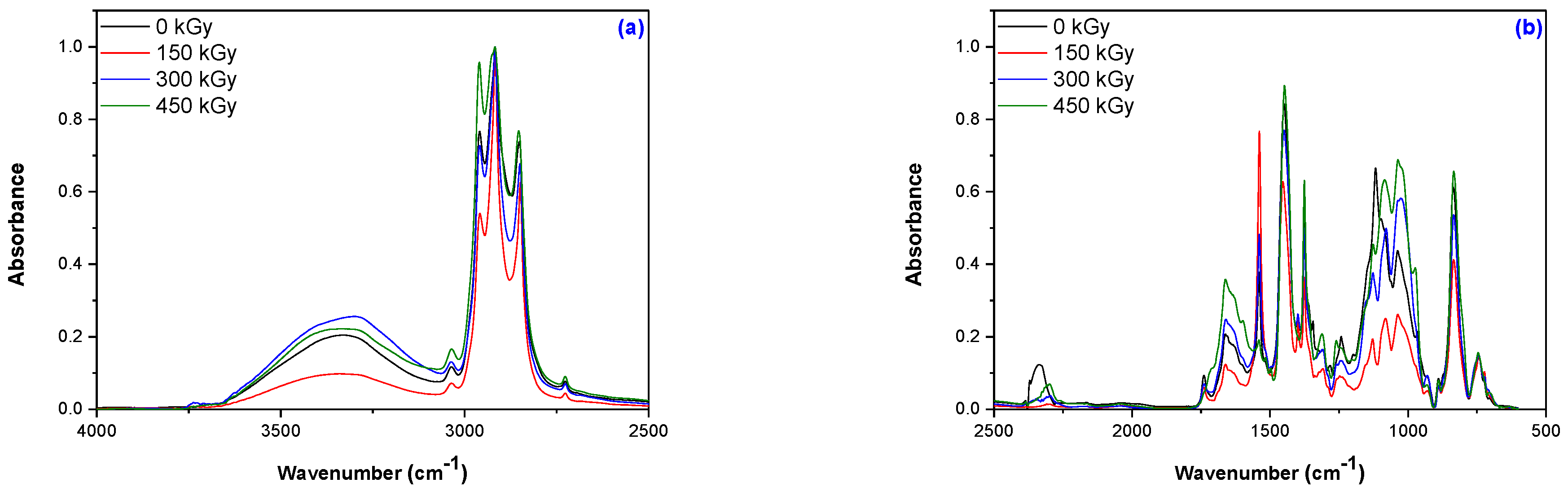
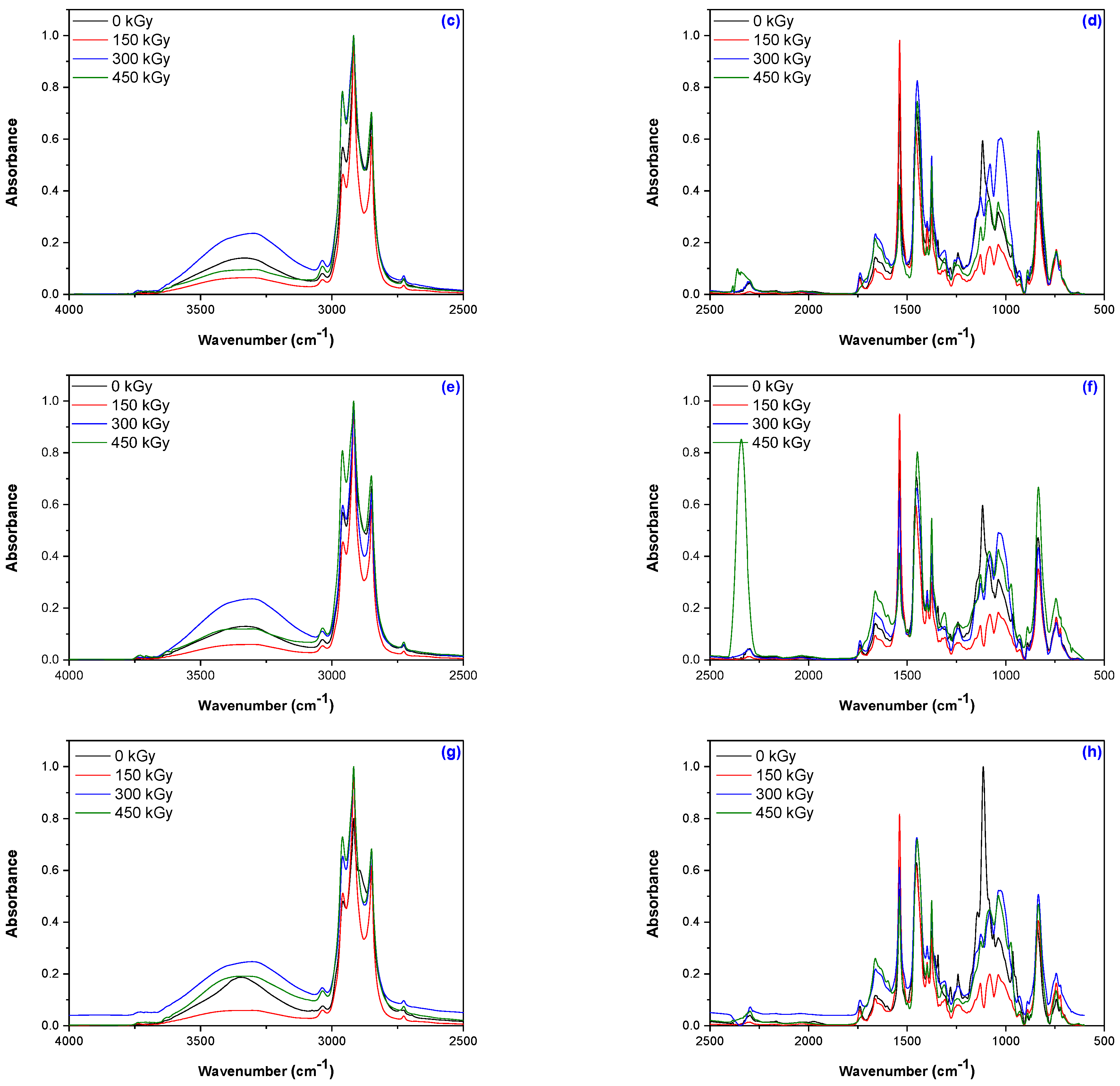
2.3.4. Structural Investigations by Fourier-Transform Infrared Spectroscopy
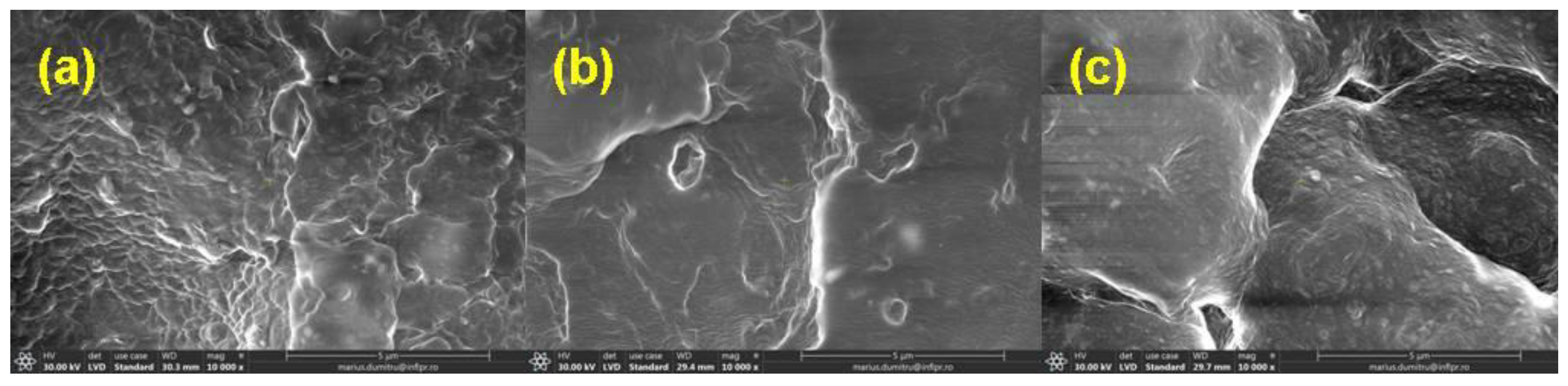
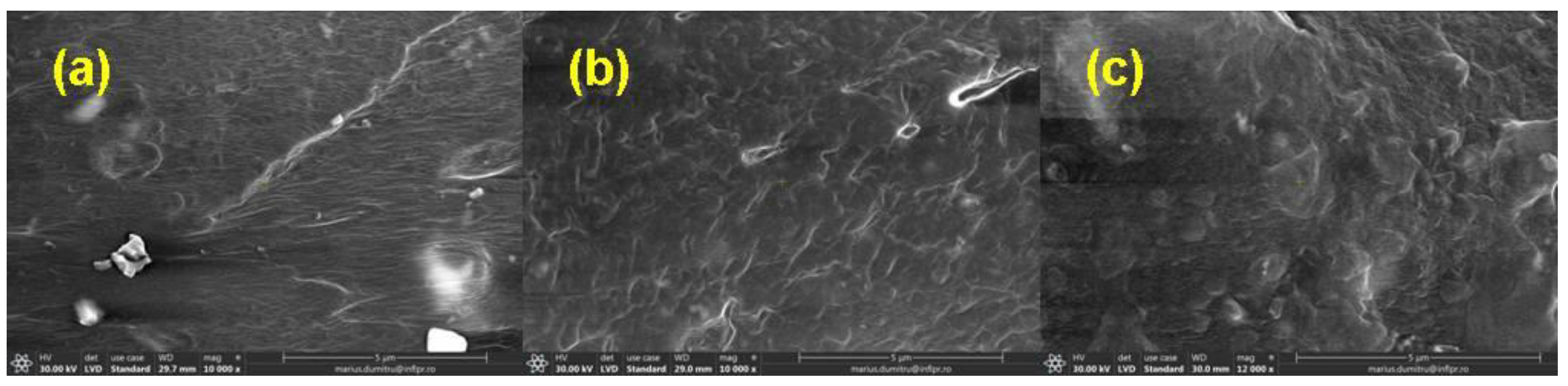
2.3.5. Morphological Investigations by Scanning Electron Microscopy
3. Results and Discussion
3.1. Gel Fraction and Cross-Link Density
3.2. Mechanical Characteristics
3.3. Solvent Uptake Tests and Weight Loss
3.4. Structural Investigations by Fourier-Transform Infrared Spectroscopy (FTIR) Technique
3.5. Morphological Investigations by Scanning Electron Microscopy (SEM) Technique
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Basik, A.A.; Sanglier, J.-J.; Yeo, C.T.; Sudesh, K. Microbial Degradation of Rubber: Actinobacteria. Polymers 2021, 13, 1989. [Google Scholar] [CrossRef]
- Available online: https://stacker.com/environment/how-long-it-takes-50-common-items-decompose (accessed on 31 January 2023).
- Fainleib, A.; Vieira Pires, R.; Lucas, E.; Soares, B. Degradation of Non-vulcanized Natural Rubber—Renewable Resource for Fine Chemicals Used in Polymer Synthesis. Polimeros 2013, 23, 441–450. [Google Scholar] [CrossRef]
- Wisetkhamsai, K.; Patthaveekongka, W.; Arayapranee, W. Study on Degradation of Natural Rubber Latex Using Hydrogen Peroxide and Sodium Nitrite in the Presence of Formic Acid. Polymers 2023, 15, 1031. [Google Scholar] [CrossRef] [PubMed]
- Phinyocheep, P.; Phetphaisit, C.W.; Derouet, D.; Campistron, I.; Brosse, J.C. Chemical degradation of epoxidized natural rubber using periodic acid: Preparation of epoxidized liquid natural rubber. J. Appl. Polym. Sci. 2005, 95, 6–15. [Google Scholar] [CrossRef]
- Sadaka, F.; Campistron, I.; Laguerre, A.; Pilard, J.-F. Controlled chemical degradation of natural rubber using periodic acid: Application for recycling waste tyre rubber. Polym. Degrad. Stabil. 2012, 97, 816–828. [Google Scholar] [CrossRef]
- Curti, P.S.; Vidotti, G.J.; Rubira, A.F.; Muniz, E.C. Some kinetic parameters of the degradation of natural rubber induced by chloranil and iron (III) chloride, in solution. Polym. Degrad. Stabil. 2003, 79, 325–331. [Google Scholar] [CrossRef]
- Nariyoshi, K.; Shinzo, Y.; Yoshinori, F. Reclamation of Vulcanized Rubbers by Chemical Degradation. IX. Oxidative Degradation of cis-1,4-Polyisoprene by Phenylhydrazine-Iron(II) Chloride System. Bull. Chem. Soc. Jpn. 1978, 51, 625–628. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, S.; Kato, S.; Kawabata, N.; Okamoto, T. Reclamation of vulcanized rubbers by chemical degradation. VIII. Absorption of oxygen and degradation of cis-1,4-polyisoprene by phenylhydrazine–iron(II) chloride system. J. Appl. Polym. Sci. 1978, 22, 353–360. [Google Scholar] [CrossRef]
- Lewis, P.M. Effect of ozone on rubbers: Countermeasures and unsolved problems. Polym. Degrad. Stabil. 1986, 15, 33–36. [Google Scholar] [CrossRef]
- Banan, A.; Mehdipour, H. Controlled degradation and functionalization of natural rubber by ozonolysis in organic solvent. J. Polym. Res. 2021, 28, 333. [Google Scholar] [CrossRef]
- Nijpanich, S.; Nimpaiboon, A.; Rojruthai, P.; Sakdapipanich, J. Hydroxyl-Terminated Saponified Natural Rubber Based on the H2O2/P25-TiO2 Powder/UVC-Irradiation System. Polymers 2021, 13, 1319. [Google Scholar] [CrossRef] [PubMed]
- Valdes, C.; Hernandez, C.; Morales-Vera, R.; Andler, R. Desulfurization of Vulcanized Rubber Particles Using Biological and Couple Microwave-Chemical Methods. Front. Environ. Sci. 2021, 9, 633165. [Google Scholar] [CrossRef]
- Attallah, O.A.; Azeem, M.; Nikolaivits, E.; Topakas, E.; Fournet, M.B. Progressing Ultragreen, Energy-Efficient Biobased Depolymerization of Poly(ethylene terephthalate) via MicrowaveAssisted Green Deep Eutectic Solvent and Enzymatic Treatment. Polymers 2022, 14, 109. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, G.; Alessi, S.; Dispenza, C. Ionizing radiation-induced crosslinking and degradation of polymers. In Applications of Ionizing Radiation in Materials Processing, 1st ed.; Yongxia, S., Chmielewski, A.G., Eds.; Institute of Nuclear Chemistry and Technology: Warszawa, Poland, 2017; Volume 1, pp. 167–182. [Google Scholar]
- Ashfaq, A.; Clochard, M.C.; Coqueret, X.; Dispenza, C.; Driscoll, M.; Ulanski, P.; Al-Sheikhly, M. Polymerization Reactions and Modifications of Polymers by Ionizing Radiation. Polymers 2020, 12, 2877. [Google Scholar] [CrossRef] [PubMed]
- Adamus-Wlodarczyk, A.; Wach, R.; Ulanski, P.; Rosiak, J.; Socka, M.; Tsinas, Z.; Al-Sheikhly, M. On the Mechanisms of the Effects of Ionizing Radiation on Diblock and Random Copolymers of Poly(Lactic Acid) and Poly(Trimethylene Carbonate). Polymers 2018, 10, 672. [Google Scholar] [CrossRef] [Green Version]
- Volintiru, T.; Ivan, G. Bazele Tehnologice Ale Prelucrarii Elastomerilor; Ed Tehnica: Bucuresti, Romania, 1974; pp. 301–357. [Google Scholar]
- Available online: http://www.nocil.com/Downloadfile/DTechnicalNote-Vulcanization-Dec10.pdf (accessed on 3 February 2023).
- Stelescu, M.D.; Manaila, E.; Craciun, G.; Sonmez, M.; Georgescu, M.; Vilsan Nituica, M. Influence of crosslinking method on the properties of natural rubber mixtures. In Proceedings of the 6th International Conference on Advanced Materials and Systems, Bucharest, Romania, 20–22 October 2016. [Google Scholar]
- Stelescu, M.D.; Manaila, E.; Sonmez, M.; Nituica, M. Characteristics of polymer composites based on natural rubber. Leather Footwear J. 2017, 17, 147–154. [Google Scholar] [CrossRef]
- Manaila, E.; Craciun, G.; Ighigeanu, D.; Lungu, I.B.; Dumitru, M.; Stelescu, M.D. Electron Beam Irradiation: A Method for Degradation of Composites Based on Natural Rubber and Plasticized Starch. Polymers 2021, 13, 1950. [Google Scholar] [CrossRef]
- Lopez-Manchado, M.A.; Herrero, B.; Arroyo, M. Preparation and characterization of organoclay nanocomposites based on natural rubber. Polym. Int. 2003, 52, 1070–1077. [Google Scholar] [CrossRef]
- Chenal, J.M.; Chazeau, L.; Guy, L.; Bomal, Y.; Gauthier, C. Molecular weight between physical entanglements in natural rubber: A critical parameter during strain-induced crystallization. Polymer 2007, 48, 1042–1046. [Google Scholar] [CrossRef]
- Arroyo, M.; Lopez-Manchado, M.A.; Herrero, B. Organo-montmorillonite as substitute of carbon black in natural rubber compounds. Polymer 2003, 44, 2447–2453. [Google Scholar] [CrossRef]
- Available online: https://joss.nl/product_tag/accelerators-activators-for-rubber-compounding/ (accessed on 3 February 2023).
- Faizal, N.A.S.; Samsudin, D.; Jalini, N.A.; Ahmad, Z.; Sarip, M.N.; Wahab, N.M.A.; Mustafa, M.S. The Effects of Devulcanize Microwave Radiation Time on Crosslink Density of EPDM Waste/Natural Rubber Blend. In Proceedings of the AIP Conference Proceedings, Proceedings of the 4th International Sciences, Technology & Engineering Conference (ISTEC), Penang, Malaysia, 8–10 April 2020; AIP Publishing LLC: Melville, NY, USA; pp. 100003-1–100003-6. [CrossRef]
- Kumnuantip, C.; Sombatsompop, N. Dynamic mechanical properties and swelling behaviour of NR/reclaimed rubber blends. Mater. Lett. 2003, 57, 3167–3174. [Google Scholar] [CrossRef]
- Nabil, H.; Ismail, H.; Azura, A.R. Compounding, mechanical and morphological properties of carbon-black-filled natural rubber/recycled ethylene-propylene-diene-monomer (NR/R-EPDM) blends. Polym. Test. 2013, 32, 385–393. [Google Scholar] [CrossRef]
- Dubey, K.A.; Bhardwaj, Y.K.; Chaudhari, C.V.; Kumar, V.; Goel, N.K.; Sabharwal, S. Radiation processed ethylene vinyl acetate-multiple walled carbon nanotube nano-composites: Effect of MWNT addition on the gel content and crosslinking density. Express Polym. Lett. 2009, 3, 492–500. [Google Scholar] [CrossRef]
- Charlesby, A.; Pinner, S.H. Analysis of the solubility behaviour of irradiated polyethylene and other polymers. Proc. R. Soc. Lond. Ser. A 1959, 249, 367–386. [Google Scholar] [CrossRef]
- Han, S.; Gu, B.; Kim, S.; Kim, S.; Mun, D.; Morita, K.; Kim, D.; Kim, W. Effect of Sulfur Variation on the Vulcanizate Structure of Silica-Filled Styrene-Butadiene Rubber Compounds with a Sulfide–Silane Coupling Agent. Polymers 2020, 12, 2815. [Google Scholar] [CrossRef]
- Han, S.; Kim, W.-S.; Mun, D.Y.; Ahn, B.; Kim, W. Effect of coupling agents on the vulcanizate structure of carbon black filled natural rubber. Compos. Interfaces 2020, 27, 355–370. [Google Scholar] [CrossRef]
- Waddell, W.; Evans, L. Use of nonblack fillers in tire compounds. Rubber Chem. Technol. 1996, 69, 377–423. [Google Scholar] [CrossRef]
- Bielinski, D.; Klajn, K.; Gozdek, T.; Kruszynski, R.; Swiatkowski, M. Influence of n-ZnO Morphology on Sulfur Crosslinking and Properties of Styrene-Butadiene Rubber Vulcanizates. Polymers 2021, 13, 1040. [Google Scholar] [CrossRef]
- Quirk, R.P. Overview of Curing and Cross-linking of Elastomers. Prog. Rubber Plast. Technol. 1988, 4, 31–45. [Google Scholar]
- Susamma, A.P. Studies of New Binary Accelerator Systems in Rubber Vulcanization. Doctoral Dissertation, Cochin University of Science and Technology, Kochi, India, 2002; pp. 4–5. [Google Scholar]
- Markl, E.; Lackner, M. Devulcanization Technologies for Recycling of Tire-Derived Rubber: A Review. Materials 2020, 13, 1246. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Isayev, A.I. Continuous ultrasonic devulcanization comparison of carbon black filled synthetic isoprene and natural rubbers. Rubber Chem. Technol. 2008, 81, 19–46. [Google Scholar] [CrossRef]
- Seghar, S.; Asaro, L.; Rolland-Monnet, M.; Hocine, N.A. Thermo-mechanical devulcanization and recycling of rubber industry waste. Resour. Conserv. Recycl. 2019, 144, 180–186. [Google Scholar] [CrossRef]
- Horikx, M.M. Chain Scissions in a Polymer Network. Rubber Chem. Technol. 1956, 29, 1166–1173. [Google Scholar] [CrossRef]
- Mathew, L.; Joseph, K.; Joseph, R. Swelling behaviour of isora/natural rubber composites in oils used in automobiles. Bull. Mater. Sci. 2006, 29, 91–99. [Google Scholar] [CrossRef]
- Valodkar, M.; Thakore, S. Thermal and Mechanical Properties of Natural Rubber and Starch Nanobiocomposites. Int. J. Polym. Anal. Charact. 2010, 15, 387–395. [Google Scholar] [CrossRef]
- Tangboriboon, N.; Phudkrachang, P.; Mulsow, L.O.; Kunchornsup, W.; Sirivat, A. Removal of water extractable proteins from concentrated natural rubber latex by eggshells. J. Elastom. Plast. 2012, 45, 253–269. [Google Scholar] [CrossRef]
- Nemtanu, M.; Brasoveanu, M.; Iovu, H. Degradation rate of some electron beam irradiated starches. Sci. Bull. Univ. Politeh. Buchar. Ser. B Chem. Mater. Sci. 2010, 72, 69–74. [Google Scholar]
- Bhat, R.; Karim, A.A. Impact of Radiation Processing on Starch. Compr. Rev. Food Sci. Food Saf. 2009, 8, 44–58. [Google Scholar] [CrossRef]
- Dinesh, B.; Squillaci, M.A.; Menard-Moyon, C.; Samori, P.; Bianco, A. Self-assembly of diphenylalanine backbone homologues and their combination with functionalized carbon nanotubes. Nanoscale 2015, 38, 12. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.; Yu, S. Fourier transform infrared spectroscopic analysis of protein secondary structures. Acta Biochim. Biophys. Sin. 2007, 39, 549–559. [Google Scholar] [CrossRef] [Green Version]
- Moretto, V.; Crisma, M.; Bonora, G.M.; Toniolo, C.; Balaram, H.; Balaram, P. Comparison of the effect of five guest residues on the .beta.-sheet conformation of host (L-val)n oligopeptides. Macromolecules 1989, 22, 2939–2944. [Google Scholar] [CrossRef]
- Toniolo, C.; Palumbo, M. Solid-state infrared absorption spectra and chain arrangement in some synthetic homooligopeptides in the intermolecularly hydrogen-bonded pleated-sheet β-conformation. Biopolymers 1977, 16, 219–224. Available online: https://scholar.google.com/citations?view_op=view_citation&hl=it&user=9F5vJRoAAAAJ&citation_for_view=9F5vJRoAAAAJ:bz8QjSJIRt4C (accessed on 1 March 2023). [CrossRef] [PubMed]
- Ferreira-Villadiego, J.; García-Echeverri, J.; Vidal, M.V.; Pasqualino, J.; Meza-Castellar, P.; Lambis-Miranda, H.A. Chemical Modification and Characterization of Starch Derived from Plantain (Musa paradisiaca) Peel Waste, as a Source of Biodegradable Material. Chem. Eng. Trans. 2018, 65, 763–768. [Google Scholar] [CrossRef]
- Munajad, A.; Subroto, C. Fourier Transform Infrared (FTIR) Spectroscopy Analysis of Transformer Paper in Mineral Oil-Paper Composite Insulation under Accelerated Thermal Aging. Energies 2018, 11, 364. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.S.; Lee, B.W.; Sohn, K.S.; Yoon, J.; Lee, J.H. Characterization of the UV Oxidation of Raw Natural Rubber Thin Film Using Image and FT-IR Analysis. Elastomers Compos. 2016, 51, 1–9. [Google Scholar] [CrossRef]
- Eng, A.H.; Tanaka, Y.; Gan, S.N. FTIR studies on amino groups in purified Hevea rubber. J. Nat. Rubb. Res. 1992, 7, 152–155. Available online: https://www.researchgate.net/publication/236170151_FT-IR_Studies_on_Amino_Groups_in_Purified_Hevea_Rubber (accessed on 3 February 2023).
- Riyajan, S.-A.; Sasithornsonti, Y.; Phinyocheep, P. Green natural rubber-g-modified starch for controlling urea release. Carbohyd. Polym. 2012, 89, 251–258. [Google Scholar] [CrossRef]
- Terpakova, E.; Kidalova, L.; Estokova, A.; Cigasova, J.; Stevulova, N. Chemical modification of hemp shives and their characterization. Procedia Eng. 2012, 42, 931–941. [Google Scholar] [CrossRef] [Green Version]
- Manaila, E.; Stelescu, M.D.; Doroftei, F. Polymeric composites based on natural rubber and hemp fibers. Iran Polym. J. 2015, 24, 135–148. Available online: https://www.researchgate.net/publication/273300630_Polymeric_composites_based_on_natural_rubber_and_hemp_fibers (accessed on 3 February 2023). [CrossRef]
- Ali, A.M.M.; Subban, R.H.Y.; Bahron, H.; Winie, T.; Latif, F.; Yahya, M.Z.A. Grafted natural rubber based polymer electrolytes: ATR-FTIR and conductivity studies. Ionics 2008, 14, 491–500. Available online: https://link.springer.com/article/10.1007/s11581-007-0199-3 (accessed on 3 February 2023). [CrossRef]
- Available online: https://www2.chemistry.msu.edu/faculty/reusch/virttxtjml/spectrpy/infrared/infrared.htm (accessed on 3 February 2023).
- Coates, J. Interpretation of Infrared Spectra, A Practical Approach. In Encyclopedia of Analytical Chemistry: Applications, Theory and Instrumentation, 1st ed.; Meyers, R.A., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2000; pp. 10815–10837. [Google Scholar]
- Manaila, E.; Stelescu, M.D.; Craciun, G. Degradation Studies Realized on Natural Rubber and Plasticized Potato Starch Based Eco-Composites Obtained by Peroxide Cross-Linking. Int. J. Mol. Sci. 2018, 19, 2862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaikumpollert, O.; Sae-Heng, K.; Wakisaka, O.; Mase, A.; Yamamoto, Y.; Kawahara, S. Low temperature degradation and characterization of natural rubber. Polym. Degrad. Stabil. 2011, 96, 1989–1995. [Google Scholar] [CrossRef]
- Nandiyanto, A.B.D.; Oktiani, R.; Ragadhita, R. How to Read and Interpret FTIR Spectroscope of Organic Material. Indones. J. Sci. Technol. 2019, 4, 97–118. Available online: https://www.researchgate.net/publication/332035024_How_to_Read_and_Interpret_FTIR_Spectroscope_of_Organic_Material (accessed on 3 February 2023). [CrossRef] [Green Version]
- Karabork, F.; Pehlivan, E.; Akdemir, A. Characterization of styrene butadiene rubber and microwave devulcanized ground tire rubber composites. J. Polym. Eng. 2014, 34, 543–554. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Gohs, U.; Müller, M.T.; Zschech, C.; Wießner, S. Evaluation of Electron Induced Crosslinking of Masticated Natural Rubber at Different Temperatures. Polymers 2019, 11, 1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahya, S.R.; Azura, A.R.; Ahmad, Z. Effect of Curing Systems on Thermal Degradation Behaviour of Natural Rubber (SMR CV 60). J. Phys. Sci. 2011, 22, 1–14. Available online: https://jps.usm.my/wp-content/uploads/2014/10/22.2.1.pdf (accessed on 1 March 2023).
- Haider, K.S. Rubber Soul. The Investigation of Rubber by Vibrational Spectroscopy. Master of Science Program in Polymer Science of the Freie Universitatat Berlin, Humboldt Universitatat zu Berlin, Technische Universitatat Berlin and Universitatat Potsdam. 2012, p. 55. Available online: https://incca.org/system/files/field_attachments/2012_ma_thesis_2012_rubber_analysis_latex.pdf (accessed on 1 March 2023).
- Rose, K.; Steinbuchel, A. Biodegradation of Natural Rubber and Related Compounds: Recent Insights into a Hardly Understood Catabolic Capability of Microorganisms. Appl. Environ. Microb. 2005, 71, 2803–2812. [Google Scholar] [CrossRef] [Green Version]
- Afiq, M.M.; Azura, A.R. Effect of sago starch loadings on soil decomposition of natural rubber latex (NRL) composite films mechanical properties. Int. Biodeter. Biodegr. 2013, 85, 139–149. [Google Scholar] [CrossRef]
- Sansatsadeekul, J.; Sakdapipanich, J.; Rojruthai, P. Characterization of associated proteins and phospholipids in natural rubber latex. J. Biosci. Bioeng. 2011, 111, 628–634. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Li, J.; Hassan, A.A.; Wang, S. Accelerated liquefaction of vulcanized natural rubber by thermo-oxidative degradation. Polym. Bull. 2022, 79, 1767–1786. [Google Scholar] [CrossRef]
- Lomelí Ramírez, M.G.; Satyanarayana, K.; Iwakiri, S.; Bolzon de Muniz, G.; Tanobe, V.; Flores-Sahagun, T.S. Study of the properties of biocomposites. Part I. Cassava starch-green coir fibers from Brazil. Carbohyd. Polym. 2011, 86, 1712–1722. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.K.; Kim, D.S.; Won, J.S.; Jin, D.Y.; Lee, H.J.; Lee, S.G. Effects of Thermal and Humidity Aging on the Interfacial Adhesion of Polyketone Fiber Reinforced Natural Rubber Composites. Adv. Mater. Sci. Eng. 2016, 2016, 4159072. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accelerator | Group/Speed | Use | Chemical Structure | Property |
|---|---|---|---|---|
| MBT (2-Mercaptobenzothiazole) | Thiazoles/Scorchy. Ultra-fast | Primary accelerator |  | molecular weight: 167.2 melting point: 177–182 °C boiling point: 302 °C density: 1.42 g/cm3 |
| CBS (N-Cyclohexyl-2-benzothiazole sulfonamide) | Sulfenamides/Delayed action. Ultra-fast | Primary accelerator |  | molecular weight: 264.4 melting point: 97–105 °C density: 1.31 g/cm3 |
| DPG (Diphenyl guanidine) | Guanidines/Scorchy and slow cure rate | Primary and secondary accelerators |  | molecular weight: 211.27 melting point: 148 °C density: 1.15 g/cm3 |
| TMTD (Tetramethylthiuram disulfide) | Thiurams/Ultra-fast | Primary/secondary accelerator, and sulfur donor |  | molecular weight: 240.4 melting point: 155–158 °C boiling point: 129 °C density: 1.43 g/cm3 |
| Ingredients (phr) | Mixture Codes | |||
|---|---|---|---|---|
| A | B | C | D | |
| NR | 95 | 95 | 95 | 95 |
| Maleated NR (NR-g-MA) | 5 | 5 | 5 | 5 |
| Plasticized starch | 30.8 | 30.8 | 30.8 | 30.8 |
| ZnO | 5 | 5 | 5 | 5 |
| Stearic acid | 0.5 | 0.5 | 0.5 | 0.5 |
| PEG 4000 | 3 | 3 | 3 | 3 |
| Sulfur | 2.5 | 2.5 | 2.5 | 2.5 |
| Antioxidant 4010 (g) | 1 | 1 | 1 | 1 |
| DPG | - | 0.5 | 0.5 | - |
| MBT | 0.5 | 0.5 | 0.5 | 0.5 |
| TMTD | 0.5 | - | 0.5 | - |
| CBS | - | - | - | 0.5 |
| Doses (kGy) | Gel Fraction (%) | Cross-Link Density (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| A | B | C | D | A | B | C | D | |
| 150 | +0.92 | +0.87 | +0.28 | +0.26 | +5.85 | +9.50 | +37.09 | +68.55 |
| 300 | −0.23 | +0.14 | −0.17 | +0.05 | +44.66 | +46.80 | +74.66 | +110.28 |
| 450 | −0.64 | −0.82 | −0.60 | −0.24 | +77.20 | +83.33 | +74.45 | +179.76 |
| Mixture Type | p0/q0 Ratio |
|---|---|
| A (MBT + TMTD) | 0.2045 |
| B (MBT + DPG) | 0.1972 |
| C (MBT + DPG + TMTD) | 0.1484 |
| D (MBT + CBS) | 0.1036 |
| Irradiation Dose (kGy) | Mechanical Properties | |||
|---|---|---|---|---|
| A | B | C | D | |
| Hardness (%) | ||||
| 150 | +11.81 | +2.31 | +1.38 | +7.26 |
| 300 | +21.26 | +16.92 | +12.41 | +21.77 |
| 450 | +22.83 | +22.31 | +17.24 | +32.26 |
| Tensile strength (%) | ||||
| 150 | −3.74 | +6.06 | −44.96 | +98.26 |
| 300 | −48.36 | −23.65 | −80.13 | −11.07 |
| 450 | −56.67 | −24.66 | −81.75 | −63.99 |
| Elongation (%) | ||||
| 150 | −62.97 | −6.38 | −14.47 | −6.74 |
| 300 | −70.44 | −14.89 | −73.29 | −12.92 |
| 450 | −80.44 | −37.23 | −73.95 | −64.89 |
| Components (phr) and Vulcanizate Structures (%) | Vulcanization Systems | ||
|---|---|---|---|
| CV | Semi-EV | EV | |
| Component used | |||
| Sulfur (phr) | 2.0–3.5 | 1.0–1.7 | 0.4–0.8 |
| Accelerators (phr) | 1.2–0.4 | 2.5–1.2 | 5.0–2.0 |
| Accelerators/sulfur ratio | 0.1–0.6 | 0.7–2.5 | 2.5–12.5 |
| Cross-link type | |||
| Poly- and disulphidic cross-links (%) -C-Sx-C- and -C-S2-C- | 95 | 50 | 20 |
| Monosulphidic cross-links (%) -C-S-C- | 5 | 50 | 80 |
Cyclic sulphide concentration | high | medium | low |
| Irradiation Dose (kGy) | Mixtures | |||
|---|---|---|---|---|
| A | B | C | D | |
| Mass loss in toluene (%) | ||||
| 0 | 1.12 ± 0.01 | 1.15 ± 0.06 | 0.67 ± 0.02 | 0.51 ± 0.01 |
| 150 | 0.21 ± 0.06 | 0.28 ± 0.14 | 0.39 ± 0.28 | 0.25 ± 0.08 |
| 300 | 1.35 ± 0.60 | 1.01 ± 0.45 | 0.84 ± 0.67 | 0.47 ± 0.33 |
| 450 | 1.75 ± 0.09 | 1.96 ± 0.05 | 1.27 ± 0.04 | 0.75 ± 0.19 |
| Mass loss in water (%) | ||||
| 0 | 3.43 ± 0.06 | 2.78 ± 0.33 | 1.47 ± 0.09 | 1.45 ± 0.01 |
| 150 | 2.65 ± 0.21 | 3.65 ± 0.19 | 1.52 ± 0.30 | 1.76 ± 0.18 |
| 300 | 3.15 ± 0.24 | 3.79 ± 0.24 | 2.06 ± 0.12 | 1.81 ± 0.07 |
| 450 | 3.42 ± 0.13 | 4.38 ± 0.48 | 1.96 ± 0.02 | 2.10 ± 0.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manaila, E.; Craciun, G.; Lungu, I.B.; Dumitru Grivei, M.D.; Stelescu, M.D. Degradation by Electron Beam Irradiation of Some Elastomeric Composites Sulphur Vulcanized. Materials 2023, 16, 2152. https://doi.org/10.3390/ma16062152
Manaila E, Craciun G, Lungu IB, Dumitru Grivei MD, Stelescu MD. Degradation by Electron Beam Irradiation of Some Elastomeric Composites Sulphur Vulcanized. Materials. 2023; 16(6):2152. https://doi.org/10.3390/ma16062152
Chicago/Turabian StyleManaila, Elena, Gabriela Craciun, Ion Bogdan Lungu, Marius Daniel Dumitru Grivei, and Maria Daniela Stelescu. 2023. "Degradation by Electron Beam Irradiation of Some Elastomeric Composites Sulphur Vulcanized" Materials 16, no. 6: 2152. https://doi.org/10.3390/ma16062152