3.1. Kinematic Characterization of Pulverisette 6 Ball Mill
It is well known that the general outcome and efficiency of MA is strongly dependent on set process parameters. In order to give deeper insight into the basic physics involved in MA and to model the process effectively, kinematic equations describing the balls’ motion will now be presented. Modelling of MA was carried out to settle the salient factors affecting the process and to optimize it for a particular application. However, as the nature of MA is inherently stochastic [
25], realistic intensions of MA modelling described herein are restricted to estimate general trends rather than precisely predicting the outcomes.
The kinematic equations describing the ball movement inside the jar and consequent ball-to-powder energy transfer were derived by Burgio et al. [
26]. Magini and Iasonna [
27] implied that the prevailing energy transfer during milling is via collisions, at least when the filling charge of the vial is relatively low, and this assumption underlies subsequent calculations in this section. Although most researchers agree that collisions are assumed to be the primary energy transfer, incident, sliding, rolling and friction may also be relevant phenomena [
28]. Nevertheless, the kinematic and energetic relationships of the MA system used herein were designated on the basis of the Magini–Iasonna collision model, that is widely described in literature (e.g., [
26,
27,
29]).
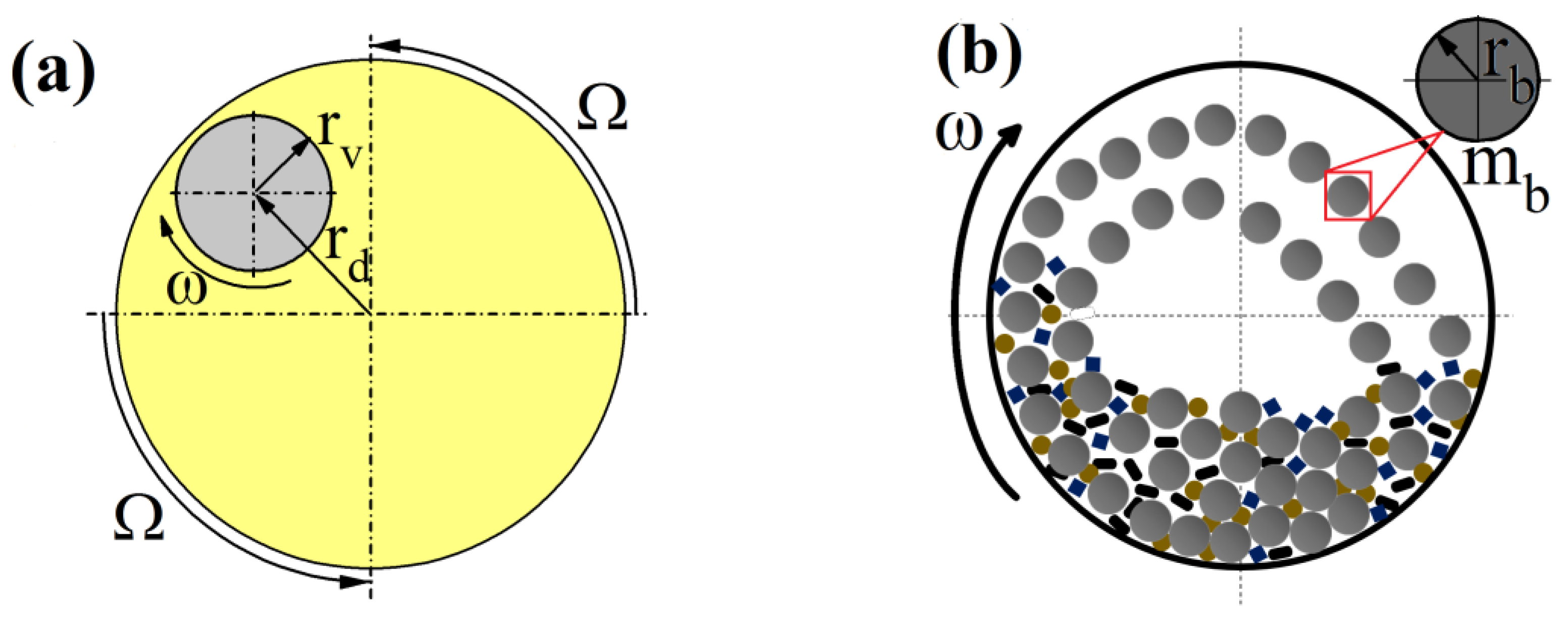
The geometrical dimensions and dependencies of the Pulverisette 6 (Fritsch, Idar-Oberstein, Germany) milling equipment are visualized in
Figure 2 and summarized in
Table 2. Accordingly,
,
and
are the main disc, vial and ball radii, respectively, h
v is the height of the jar,
is a ball’s mass and
is the ratio of vial’s rotational velocity (
has a negative value as it turns in opposite direction than main disc) with respect to main disc’s angular velocity (
).
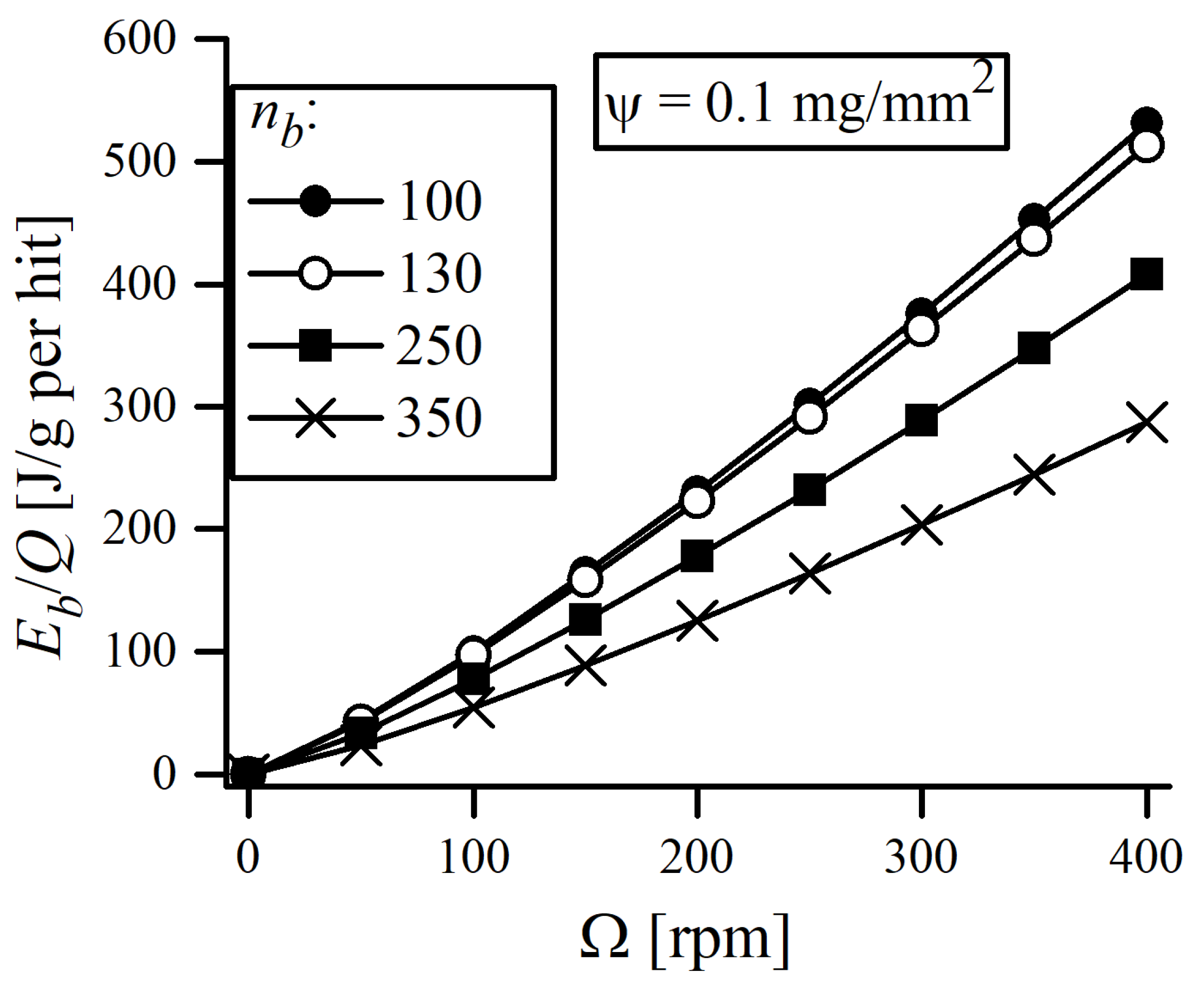
The energy transfer fundamental to the MA process is the one from the balls to the powder, which is only a fraction of the total energy output to the system. Therefore, it would certainly be more useful to specify the energy transfer going only to the powder, which can be calculated as a ratio of impact energy of balls
to the maximum quantity of material trapped between balls
, approximated by the powder adhering to the ball surface. Detailed derivation of the proper formulas can be followed in the literature ([
27,
29,
30]), so it will only be mentioned here briefly. After substituting
, where
is the density of milling balls, the equation considering energy input to the powder takes the form:
where
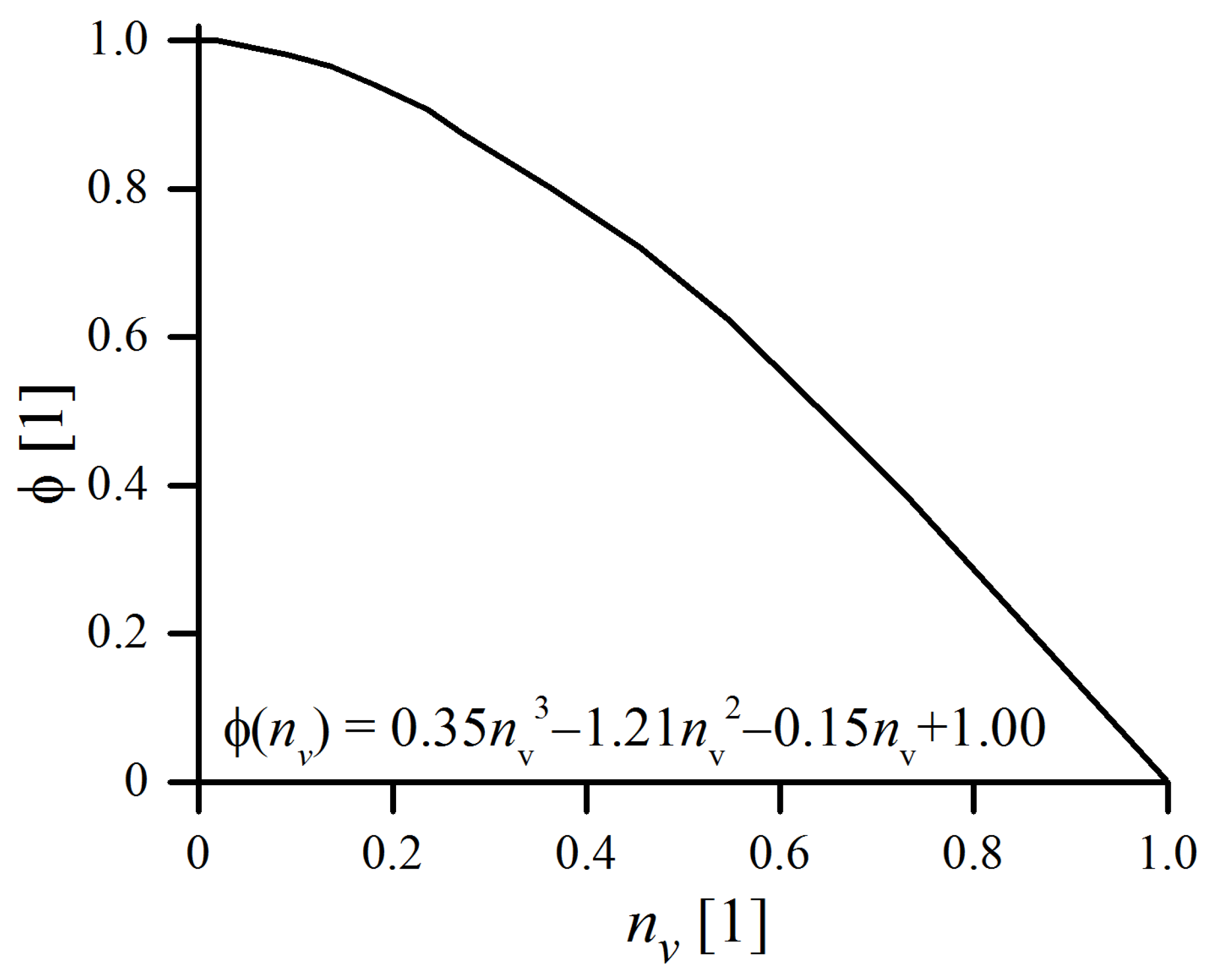
is the function related to the degree of filling of the jar,
is the relative impact velocity of a ball and
ψ is the surface density of the powder covering the balls. The
value with respect to the filling factor
is plotted in
Figure 3. It is equal to 0 in the case where the vial is completely full (no possible movement of balls) and
when the vial is nearly empty. When the degree of filling becomes greater, reciprocal collisions are no longer negligible, as the collisions themselves become less productive due to shorter free paths available for ball and, consequently, lower relative impact velocities [
29].
The ball impact velocity
depends on kinetic (
/
) and geometrical (
rd,
and
) relationships (
Table 2). Therefore, for each rotation speed of the main disc (
), the relative impact velocity can be simply obtained using the Equation (5):
For Pulverisette 6, after substituting the geometrical constants (
Table 2), and when using ø10 mm balls,
= 0.1658. It should be underlined that the value of
is valid only for the discussed MA setup, and would be different when changing the process conditions (e.g., different milling device or balls’ size).
Finally, after inserting the material (
Pa,
kg/m
3), and dimensional (
) constants for stainless steel, and using the previously derived relation that
(Equation (6)), the following formula is obtained:
As can be noticed, energy absorption of the powder for can be easily obtained using Equation (6) for the given rotational speed and the vial’s filling degree .
The crucial point in Equation (6) concerns the amount of powder subjected to each impact, which is assumed to be equal to that adhering to the balls’ surface, i.e., the surface density of the covering powder
. It is realistic to assume that the
value should represent the minimum quantity of trapped material, as some powder, not necessarily adhered to the balls, may also be involved in the collision event. Its value is probably dependent on many circumstances—the BPR, type and initial size of the powders used, type of balls, etc., and thus, is unreliable to presume arbitrarily. Moreover, the value of
certainly varies during the MA process, as most powders are more adherent during the early stages of the process, when the particles are relatively soft and have high affinity to weld together (welding predominance over fracturing) [
5]. Therefore,
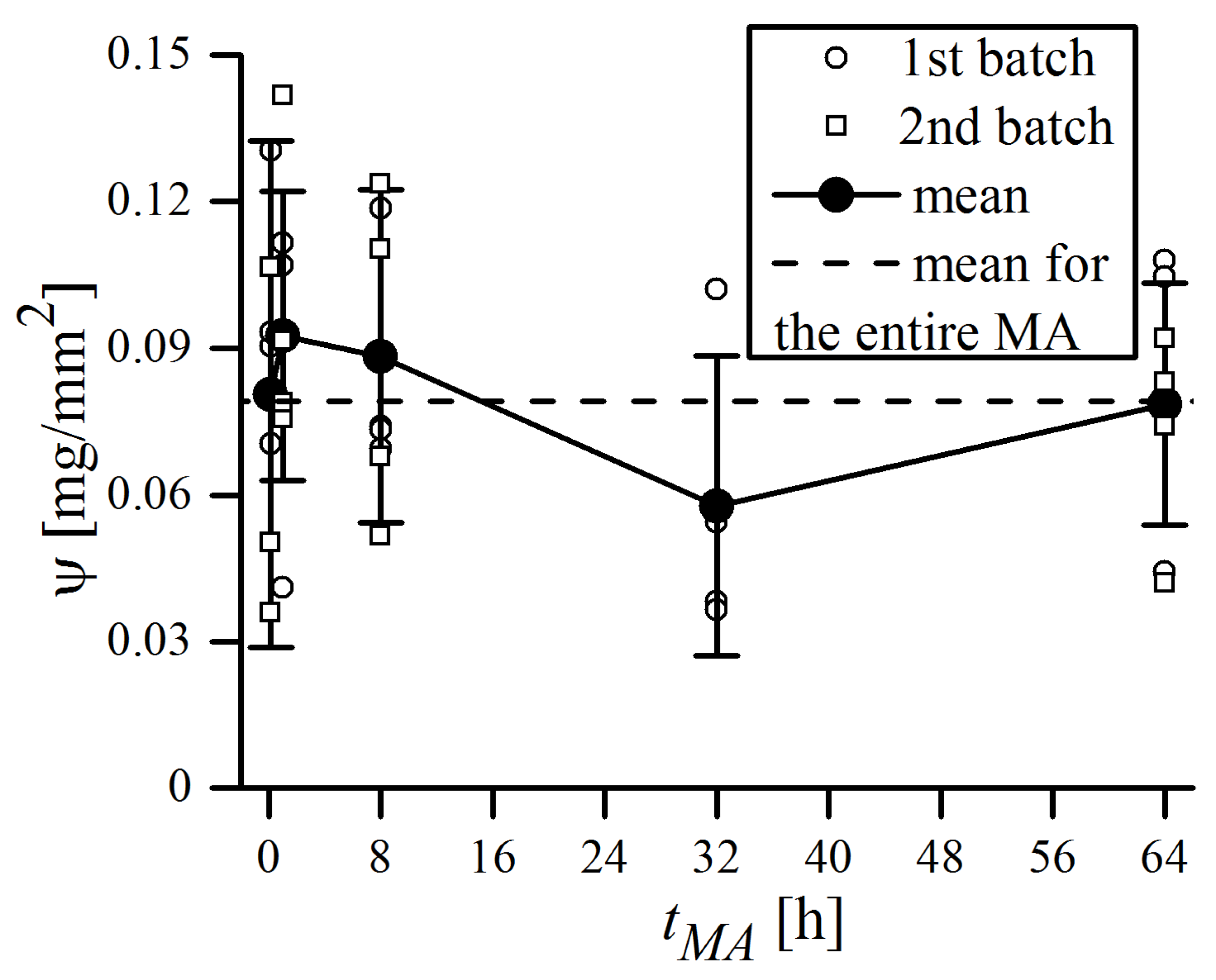
is apparently the major source of uncertainty in Equation (6), and should be obtained from direct weight measurements for the individual MA process. In this study, a few balls were carefully taken out from the vial after the specified MA time, each of them was placed in separate ceramic crucibles, and weighed altogether using a precise balance (Mettler Toledo XS205). Then, the balls and crucibles were cleaned of powder residues and weighted again. The resulting difference, i.e., the mass of the adhering powder, related to the ball surface area was defined as
. The
ψ values measured during MA are plotted in
Figure 4.
As can be seen, the mean
value averaged over the entire MA time is 0.079(25) mg/mm
2, and none of the single measurements ever exceeded 0.15 mg/mm
2. This is less than the typical values of
that can be found in the literature. Magini et al. [
27] reported a mean
= 0.3 mg/mm
2 for the Pd-Si system milled in Pulverisette 5, whereas Suryanarayana [
5] claimed that around 0.2 mg of powder is trapped during each collision (without reference to the surface area). The average measured value of
should constitute a minimum of the real quantity of material involved in single collision event, as the additional amount of powder, not necessarily adhering to the surface, may also be trapped between balls. This, plus the fact that single measurements, conducted even at the same batch and time of MA, may actually vary by a factor of ~4, makes
the primary source of uncertainty in the presented calculations. Therefore, it highlights the importance of measuring
for each milling system and type of powders used, as large discrepancies may arise when the literature values are assumed arbitrarily. The results of the energy transfer calculations are reported in
Figure 5, in form of an “energy map”, which evaluates the energy transferred per unit of powder mass.
The energy map can be used to estimate the required amount of energy to be transferred into the powder to obtain a solid solution of ODS alloy. According to
Figure 5, the most straightforward method for increasing the energy transfer to the powder is by simply increasing the rotational speed
. This is, however, accompanied with a rise in temperature inside the milling jar. In extreme cases, it may lead to the overheating the powder, causing its degradation and welding with the balls. Here, the rotational speed of 300 rpm was found to be the upper safe limit of
, as increasing the
to 350 rpm caused welding of particles to the balls. Although it is difficult to estimate the maximum local temperature (at the site of collision) inside vial, the temperature of the outer surface of the jar, measured by a pyrometer, was found to be 56 ± 4 °C when
= 300 rpm.
The energy map can be used to estimate the balance between maximizing the energy of impacting balls and the net yield of a MA batch. In this study, a yield of ~50 g of powder in a single batch was required for further processing and sintering by the spark plasma sintering (SPS) method. This, while preserving the typical 10:1 ball-to-powder ratio (BPR), required the use of 130 balls. As can be noticed, using 130 Ø10 mm balls had a negligible impact on energy transfer. The principal conditions of MA used in this study are presented in
Table 3. The process is usually performed in a protective atmosphere to prevent excessive oxidation of the batch. Ar can be perceived as a standard inert atmosphere and has been widely used, although some researchers found using He or H
2 to be more beneficial [
31,
32]. An important incentive to use a H
2 atmosphere is a lower oxidation degree of milled powders [
31]. During MA, H
2 can react with metal oxides (MO) and reduce them, accordingly to
, which may be a reasonable explanation of O reduction in powders milled in a H
2 atmosphere. As a consequence of elevated temperature inside the milling jar, the H
2O can turn into steam and further react with the C impurities contained in the jar:
. CO can also react with MO:
. Gaseous byproducts of the redox reaction, CO and CO
2, can be easily removed from the jar during purging. Bearing in mind the above statements, we decided to use H
2 as a protective MA atmosphere for the purposes of this work.
3.2. Characterization of Powders Prior to Mechanical Alloying
One of the biggest challenges of developing ODS steels for fusion applications is that the elements used in the alloy must fulfill certain criteria, so only a few of them can be considered. Most importantly, in case of ferritic (FS) and ferritic–martensitic (F/M) steels, these elements must form or stabilize the bcc structure and meet the low-activation requirement, ensuring that radioactivity from the material decays to low levels in less than 100 years. For the purpose of this study, ODS RAF alloys were manufactured using the commercially available, high purity Fe, Cr, W and Zr metallic elemental powders and Y
2O
3 nanoparticles. A brief description of the characteristics of the initial powders are listed in
Table 4.
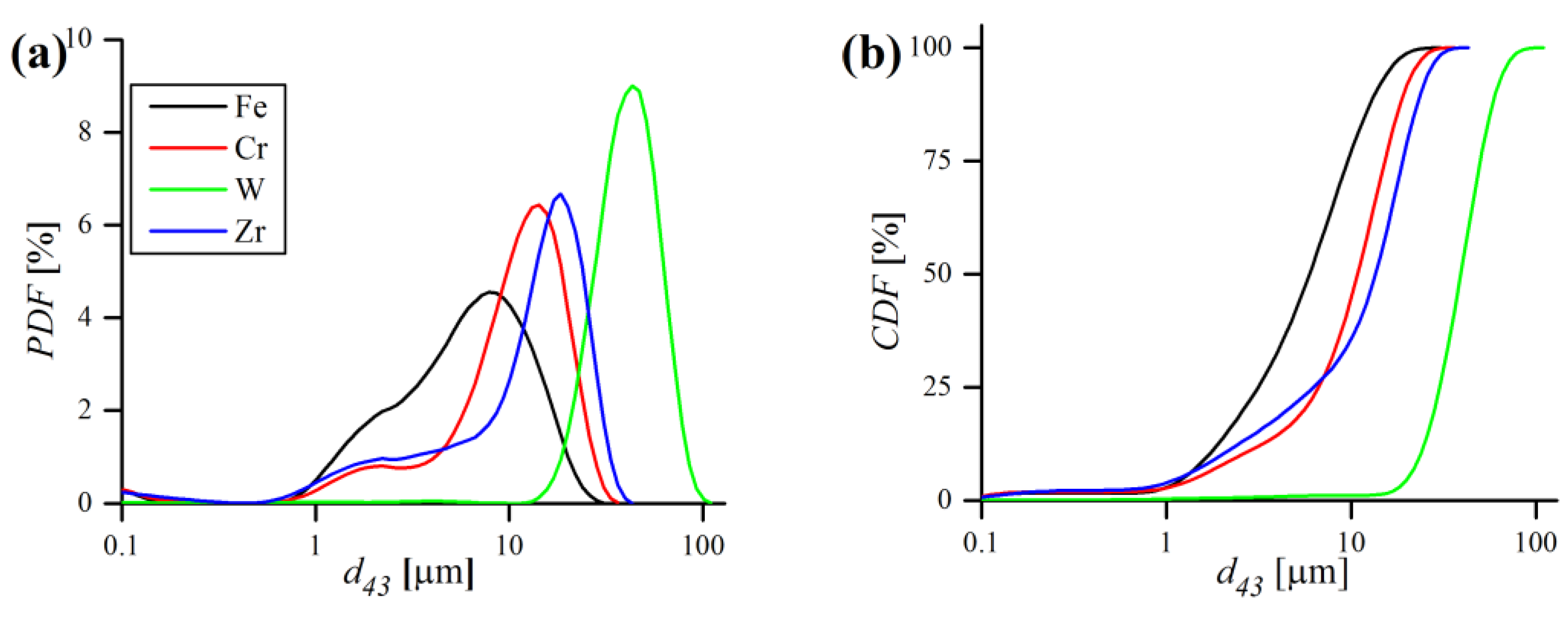
The substantial properties (size and morphology) of the initial powders prior to milling were determined by laser particle size measurements, XRD and SEM observations. Particle size measurements of starting elemental powders, in terms of probability (PDF) and cumulative density distribution functions (CDF) are presented in
Figure 6 and listed in
Table 4.
According to the results, Fe, Cr and Zr powders are fine and have a similar
of ~10 µm, while W powder is considerably larger (~40 µm). The W powder size distribution is unimodal and symmetrical, whereas other powders PDFs are rather bimodal, with one major peak and a less pronounced minor one, in the very fine size range (
Figure 6a). The size of Y
2O
3 particles could not be measured on laser analyzer, as their size is smaller than minimal admissible size (0.08 µm) for this device. Therefore, the crystallite size of Y
2O
3 was calculated by the WPPM algorithm.
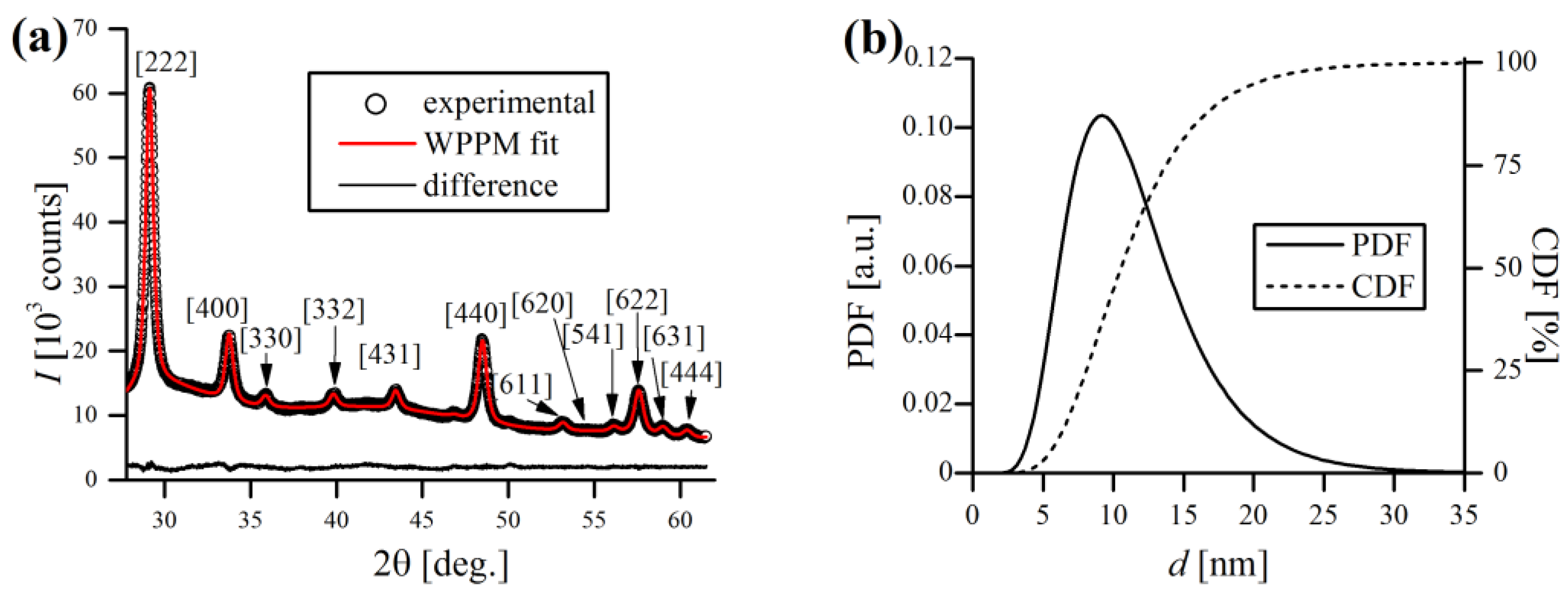
Figure 7a shows the diffractogram of pure Y
2O
3 with WPPM fit with lognormal domain size distribution (
Figure 7b). XRD revealed that used yttria has a cubic, corundum-like crystal system (Ia
space group), with a lattice parameter
= 10.6148(2) Å. As can be noticed in
Figure 7a, the Y
2O
3 peaks are rather broad, even before the mechanical treatment during MA, which indicates very small crystallite sizes. Indeed, the WPPM calculations confirmed the small mean domain size of Y
2O
3 of
= 11.5(4.7) nm (
Figure 7b), without any detectable strain broadening effects (
~0).
SEM investigation of the elemental powders revealed substantial differences in their shape. Even after a brief look, Fe particles are definitely the finest and are almost ideally spherical, having smooth, featureless lateral surfaces (
Figure 8a,b). The main fraction of Fe globes of ~5 μm diameter were surrounded with very fine (<1 μm) satellite spheres. Contrarily, Cr particles were considerably larger, flat and flaky, with rather rounded edges (
Figure 8c,d). W powders were spheroidal with multiple visible faces, which are similar to the shape of a polyhedron (
Figure 8e,f). Zr particles were spheroidal and have rounded edges, similar to pebble stones (
Figure 8g,h). Moreover, their lateral surfaces were clearly decorated with shallow, superficial holes. The particles of Y
2O
3 appeared on SEM micrographs as dendritic, feather-like structures with irregular, jagged boundaries (
Figure 8i,j).
Besides visual examination, more detailed image analysis was carried out using ImageJ software to mathematically determine the shape of the powders. Circularity
and roundness
are common shape indices used to characterize the grain form. In ImageJ,
and
are calculated according to the Cox [
33] and Pentland [
34] equations, respectively. Circularity is defined as the ratio of area to perimeter, with unity indicating a perfect circle. Lower values of
are characteristic for irregular and angular shapes. Similarly, roundness distinguishes particles with circular cross-sections from less circular ones (elliptical, etc.). As the values of dimensionless shape descriptors are in the range from naught to unity, the data was transformed using the logit function for the statistical analysis of data, using the following relation (Equation (7)) [
35]:
where
is the shape descriptor (herein
or
). Distribution of logit-transformed shape indicators was plotted as a histogram, using a bin size of 0.5, and was estimated using the normal distribution function. The obtained normal curves are demonstrated in
Figure 9.
It can be stated that the shape of Fe powders was most similar to a regular sphere, considering that the normal, logit-transformed mean values of
and
were the highest among the other powders (
Figure 9). On the other hand, Cr grains were most angular and irregular, and also deviated the most from the shape of a circle, with W and Zr powder shapes being somewhere between Fe and Cr extremes.
3.3. Microscopic Observations of Mechanically Alloyed Powder
As depicted in
Figure 10a,d, at early MA stages the particles form conglomerates consisting of multiple finer, elongated particles with clearly visible boundaries separating them from each other. Because the starting powders are relatively soft (proven later by microhardness measurements), the flattened layers tended to overlap and form cold welds. At short
, this lead to the formation of layered composite particles, consisting of a mixture of starting ingredients, which was recognized as a lamellar structure. In addition, the distinctive white sub-particles were clearly distinguished in these micrographs. As confirmed later by SEM EDS observations, these are W particles, which are not susceptible to etching by the reagent used.
As the MA process proceeds, the work hardening becomes more pronounced and the amount of defects introduced to the crystals seriously increased, providing short-circuit diffusion paths and facilitating the formation of an alloy [
5]. Moreover, the particles became harder and more susceptible to fracture. As shown in
Figure 10e,f, after 8 h of MA the individual constituents of lamellae were fine and hardly recognizable due to fragmentation of the fragile flakes. At this stage, the particles consisted of convoluted lamellae, a result of microstructure refinement and interdiffusion of the constituents.
At later stages of MA, the lamellae became even finer and more convoluted and no longer resolvable under the optical microscope (
Figure 10g,j). Eventually, a refined and homogenized microstructure was attained, with the absence of any conspicuous microstructural features. It should be noted that it is difficult to unambiguously estimate the structural features (e.g., grain size) of powders after MA due to lack of clarity in OM observations in such complicated microstructures. At the last stage of MA, featureless micrographs suggested a nano-sized microstructure of the powder.
In order to perform a more comprehensive analysis, subsequent SEM observations of etched cross-sections of the mechanically alloyed ODS powders were carried out. The results of these observations are presented in
Figure 11. Similar conclusions to the OM observations can be drawn in the case of short MA time—the particles were in form of agglomerates consisting of loosely bound sub-particles, with clearly visible boundaries (
Figure 11a–d). In addition, large, loose, single W particles, surrounded by much finer particles of the other chemical can be discerned (
Figure 11c). Despite the short time of the process, very fine (few hundred nm) sub-grains can be already observed at high magnification (
Figure 11d).
At the intermediate phase of MA (8 h), SEM revealed a very specific, fibrous microstructure of the alloyed powders (
Figure 11e,f). Some of the particles showed a mixed microstructure, consisting of fine (<1 µm), rather round, sub-particles in the interior and uniformly layered structure near the outer surface area (
Figure 11e). Characteristic, white and elongated W sub-particles, although much finer, could still be observed inside the grains.
The powders after MA (64 h) presented uniform microstructures without inhomogeneous inclusion or other characteristic features (
Figure 11g,h), suggesting that chemical composition of the individual particles approached that of the overall, nominal composition of the starting blend. Therefore, it indicated that alloy formation was finished. Additionally, at higher magnification, fine and elongated shapes, possibly representing sub-grains, can be distinguished (
Figure 11h). Generally, very similar SEM micrographs of ODS steel powders during MA were also obtained by other authors in [
36].
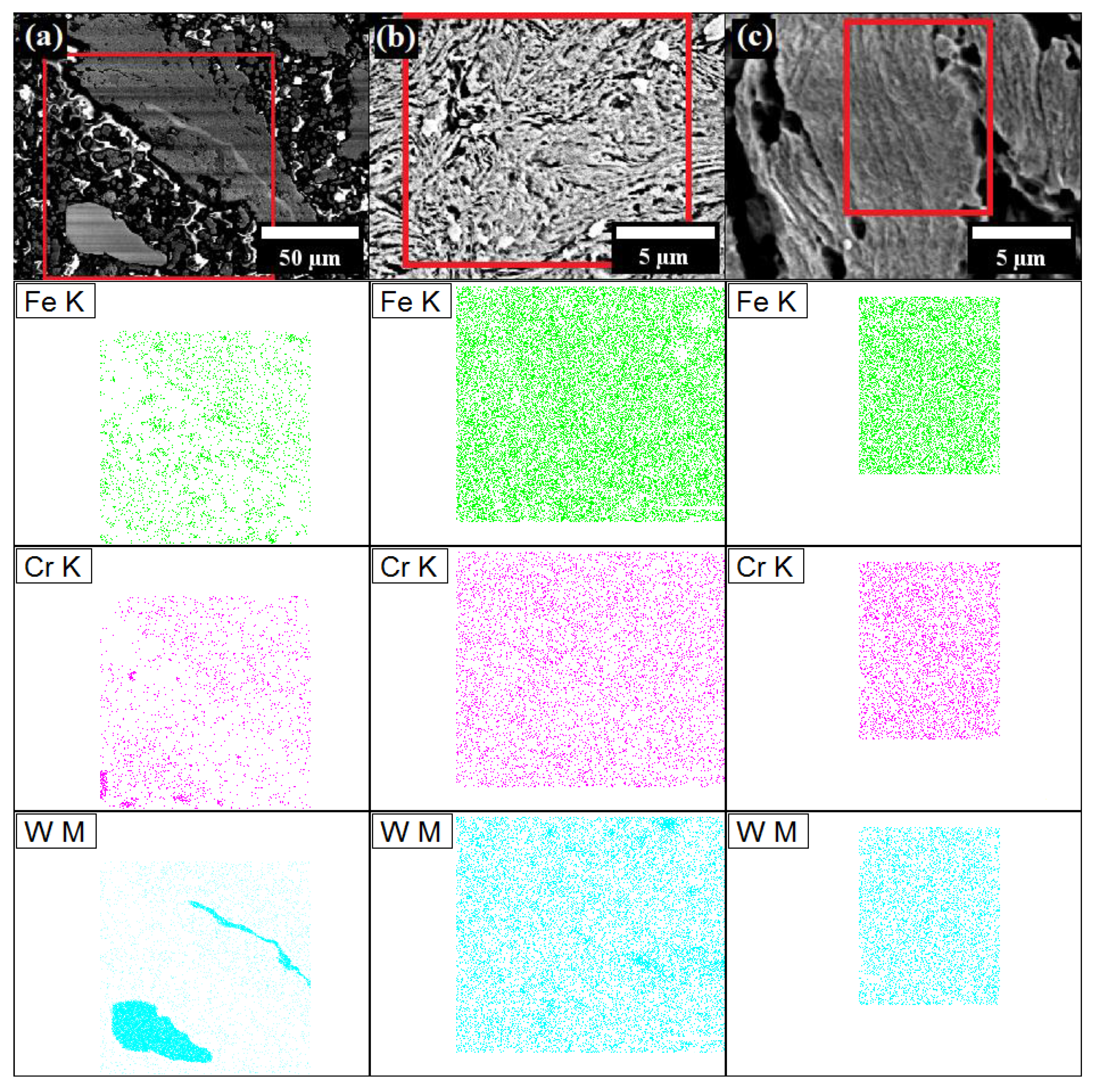
The results of SEM energy-dispersive X-ray spectroscopy (EDS), in terms of chemical mapping of Fe, Cr and W elements is presented in
Figure 12. Zr and Y
2O
3 were not detected by EDS, possibly mainly due to their very small mass fraction. Other theories to explain this phenomenon can be proposed as follows: first, Zr is added as a small fraction and, thus, is very rapidly dissolved in Fe, which was also confirmed by XRD. Second, nanometric Y
2O
3 particles are so small that are below the limit of detection by the EDS detector.
As expected, the results demonstrated that Fe, Cr and W elements are clearly segregated after a short MA time (
Figure 12a). In addition, it proved that the larger, white particles are W. After 8 h of MA, the chemical composition of the particles was much more uniform, although, the larger aggregates of W, however, were still detected (
Figure 12b). After 64 h, the chemical maps were entirely stochastic, without any larger agglomerates of any element, consequently manifesting that proper solid solution of the elements was achieved (
Figure 12c).
3.4. Particle Size Evolution during Mechanical Alloying
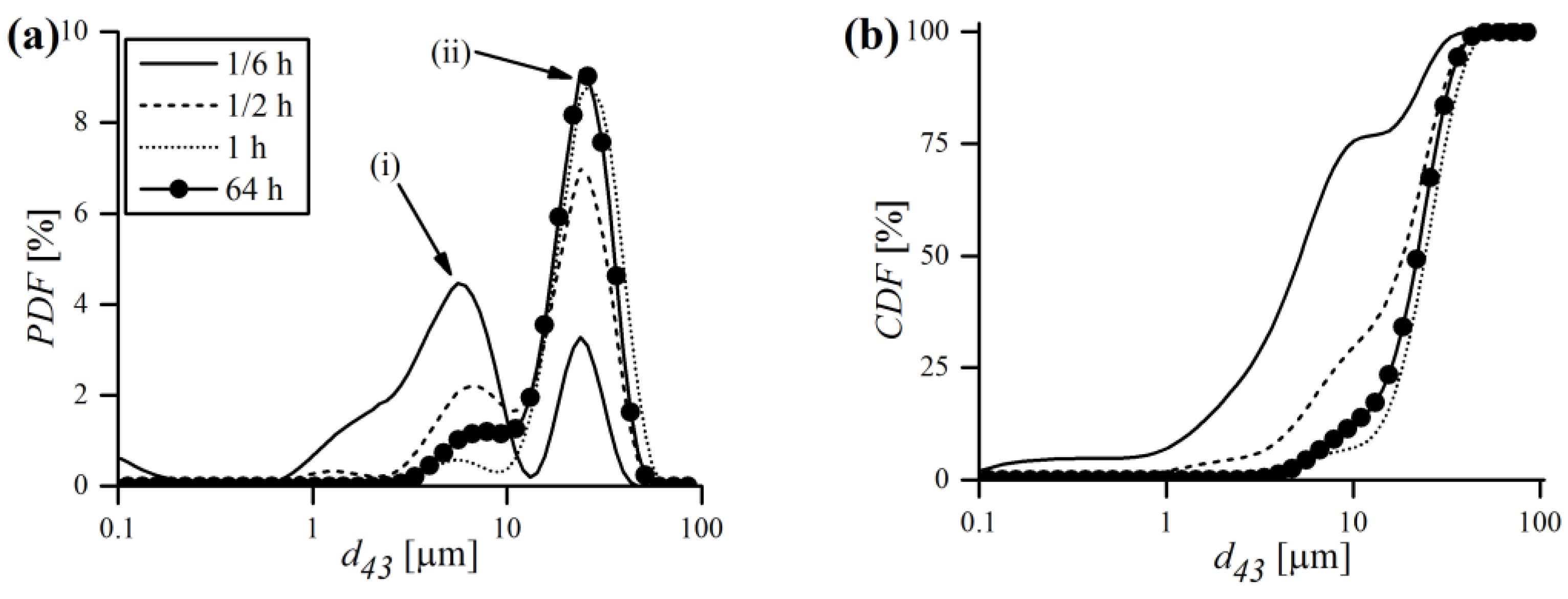
Particle size evolution during MA, in terms of probability and cumulative density functions, was obtained by a laser particle sizer and is shown in
Figure 13. As can be noticed, the probability distribution is bimodal, with two separate peaks labeled as (i) and (ii) (
Figure 13a). At the very beginning of MA (1/6 h), the finer (i) fraction prevailed over the coarser one (ii), and the vast majority (around 75%) of volumetric particle diameters (
) was in the 0.1–10 μm range (
Figure 13b). However, just after 1/2 h of milling, most of the (i) fraction decayed, which is accompanied with a mutual increase in the coarser (ii) fraction. It was also manifested by the shift of the cumulative curve towards higher
values. After 1 h of MA, the (i) fraction almost completely disappeared and the PDF became close to unimodal. At the end of MA, a recurrence of the smaller fraction was observed, manifested by a small rise of the (i) peak.
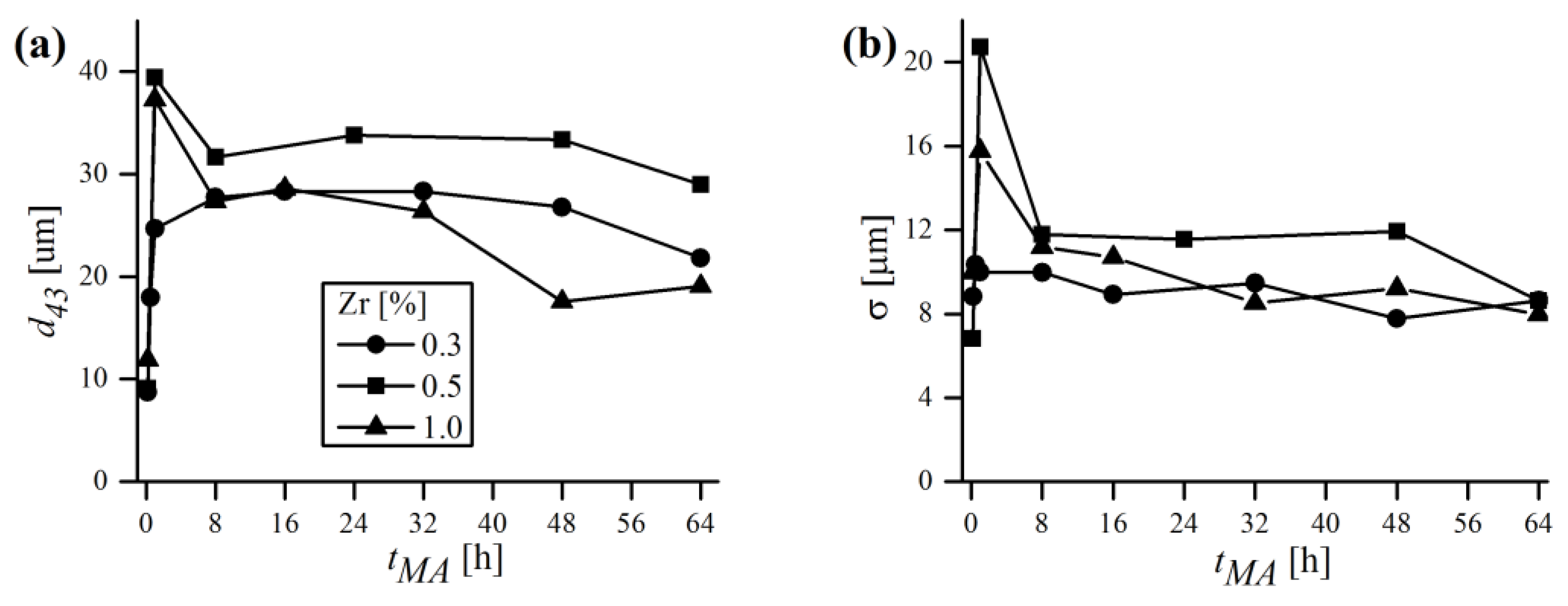
To better visualize the evolution of powder diameters during MA, the mean values of
and corresponding standard deviation
were plotted in
Figure 14 for three different chemical compositions. Generally, the trends of
and
were closely related and mimicked each other. At the beginning, the powders were very small (
~10 μm) and had a rather narrow distribution (
~8 μm). After 1 h of MA the
rose exceedingly, with an accompanying growth of
. As mentioned earlier, in the early stages of milling, particles were relatively soft and therefore more prone to weld together and form bigger particles. As a consequence, a broad range of particles developed, mostly larger than the starting ones, with some of them having a several times larger diameter. With continued deformation, the particles became so strongly work-hardened that they began to break into smaller pieces, which is manifested in
Figure 14 by the drop of
and
to lower values. It is worth noting, however, that even if at some MA stages the size of particles continued to be the same, their microstructure was still refined due to continued impact from the milling balls, which was supported by XRD. As the process continued, an equilibrium between the rate of welding (increasing the size) and fracturing (refining the size) was attained, and only slight deviations of
and
were observed. Finally, alloyed powder batches were obtained, characterized by fine size (
= 21.8; 29.0; 19.1 μm) and narrow distribution (
= 8.6; 8.6; 8.0 μm) in the case of 0.3, 0.5 and 1% Zr content, respectively. It is somehow puzzling that the final mean size of particles was higher than the starting one, despite relatively long milling times. Usually, especially in case of using single component or prealloyed powder, the final size after MA is lower than the starting size (e.g., [
37]). Herein, the evaluation of this phenomenon is more complicated because the chemical composition consists of multiple starting constituents, varying in size and shape. Therefore, it is plausible that the equilibrium particle size after MA of the powder mixture consisting of finer (Fe, Cr and Y
2O
3 in this case) and much coarser (W and Zr) starting powders can be higher than without MA, especially when the initial size is already very fine (~10 μm in this study). Another explanation might be the fact that during MA the powder is constantly contaminated with the material eroded from the vial and balls, which might be coarser than the batch and increase the particle size as a consequence. It is also worth noting that
may vary significantly even from batch to batch (difference in chemical composition is rather negligible to size evolution) despite using the same equipment and milling conditions, as demonstrated in
Figure 14.
3.5. X-ray Diffraction and Line Profile Analysis of Mechanically Alloyed Powders
Exemplary, general XRD patterns of the Fe-12Cr-2W-0.5Zr-0.3Y
2O
3 powder, taken at various
are presented in
Figure 15. As expected, the microstructure of an ODS alloy was purely bcc from the beginning to the end of MA, which was proven by five major ferritic peaks appearing in the graph, sequentially indexed with Miller indices (
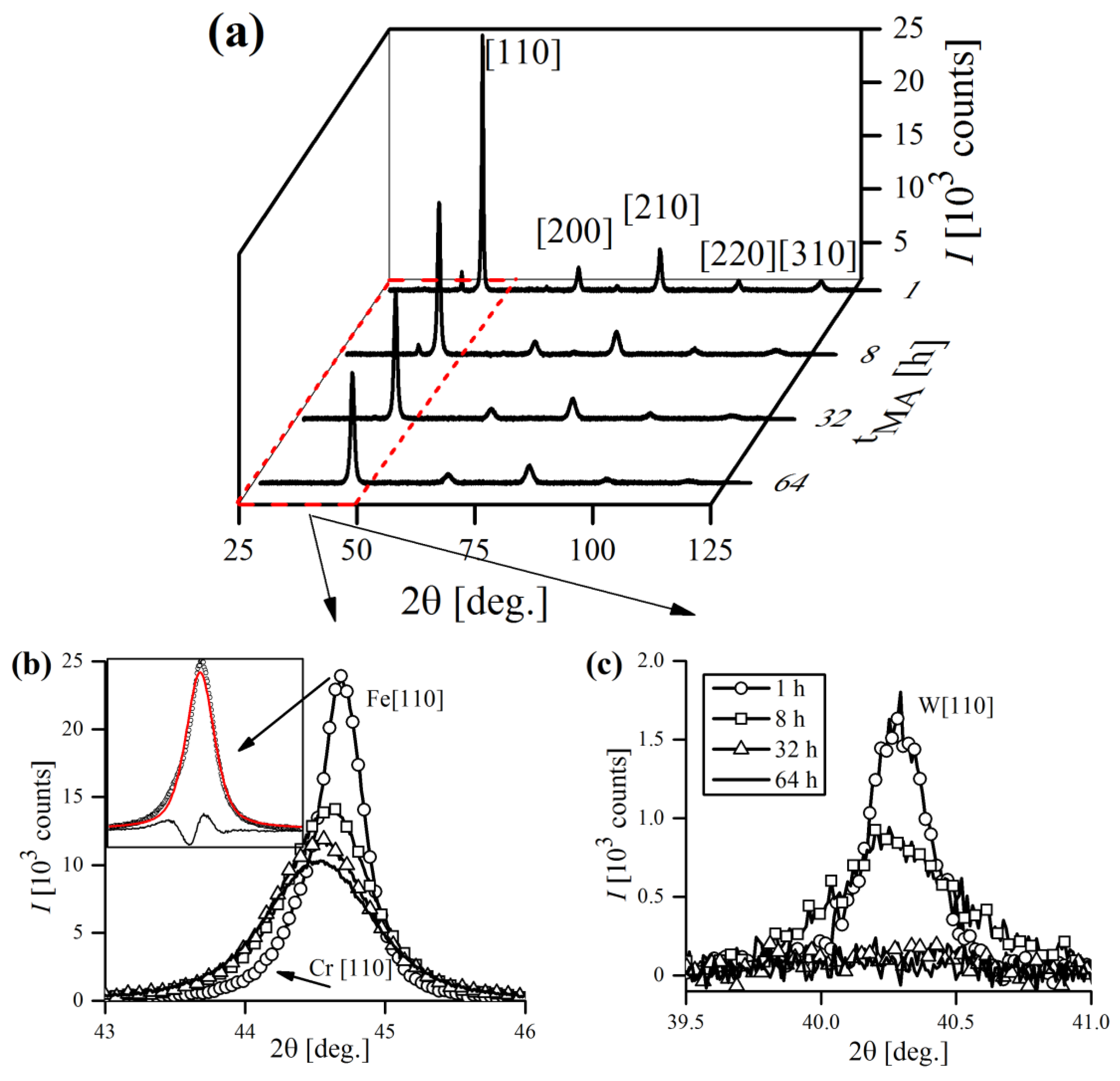
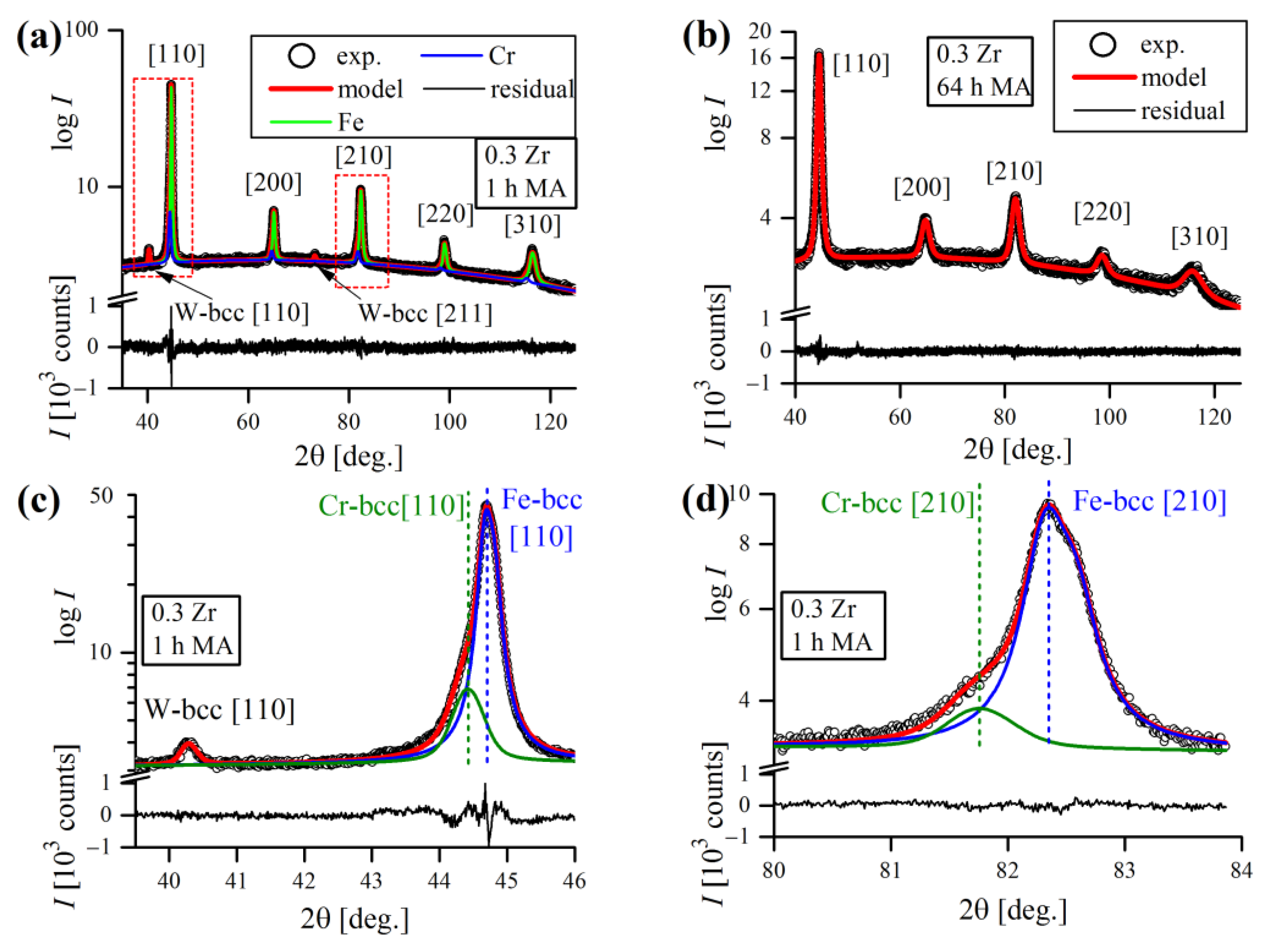
Figure 15a). Cr-bcc phase peaks were also evident, but not so obvious due to severe overlapping with Fe-bcc profiles. As a consequence, they were distinguishable only at the initial stages of MA, until Fe-bcc peaks became sufficiently broad, masking their presence.
Figure 15b,c presents the evolution of the most prominent Fe-bcc [110] and W-bcc [110] during the MA cycle. The inset in
Figure 15b focuses on Fe-bcc [110] reflection aberrated by the presence of overlapping Cr-bcc [110] peak, that is not obviously visible at first sight. However, the poor quality of the pseudo-Voigt fit clearly revealed the Cr-bcc contribution to the line profile.
Clearly, as the time of MA increased, the reflections were becoming broader, which was also accompanied with a drop in their intensity. The influence of mechanical treatment on line profiles was evident, as well as the anisotropic nature of the broadening (
Figure 15b,c). The origins of line broadening could be numerous, but certainly the most influential are size and microstrain factors [
38]. Besides broadening, the peaks also exhibited a shift towards lower
Bragg positions, suggesting an increase in lattice cell dimensions, which is consistent with Fe forming a solid solution with alloying additives. The deviation of XRD peak positions during MA has already been observed in analogous studies of ball-milled metals and is attributed to mechanisms related to severe plastic deformation [
39].
In general, classical MA is usually a lengthy process, as it requires dozens or even hundreds of hours to complete, depending on the alloy composition and equipment used. The situation is furtherly complicated by a multitude of factors related to MA—type of powders used, rotational speed, type of balls, etc., which most often differ from one study to another, making it impossible to impose a universal milling time. Thus, the process has to be controlled by XRD, and is usually considered finished when all of the alloying additives peaks disappear and only the reflections from matrix phase are left (Fe-bcc in this case). In this study, the MA was regarded as completed when the W [110] peak finally disappeared, which required 56–64 h. The intensity of W [110] reflection was greatly reduced after 32 h; it is still detectable, however, up until 56–64 h of MA (
Figure 15c).
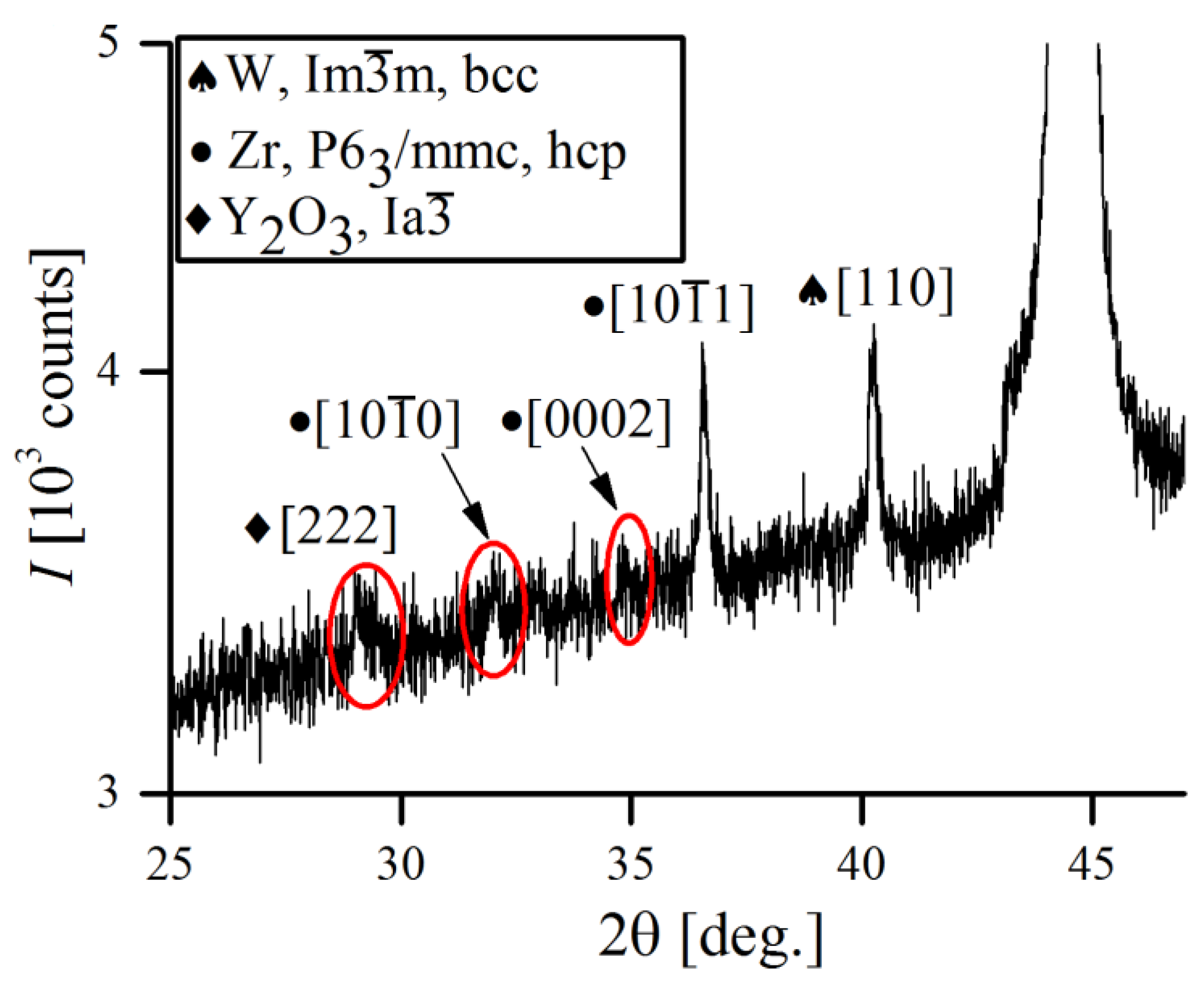
Besides those of the Fe-, Cr- and W-bcc phases, several reflections of minor alloying additives (Zr and Y
2O
3) were also detected. However, due to their small percentage in overall alloy composition (
1 wt.%), they were only detectable at the very beginning of the MA and rapidly vanished just after 1 h of processing, despite the solubility of Y
2O
3 in Fe being generally very low [
40]. Recent explanations state that severe fragmentation of Y
2O
3 particles drives them into amorphous sub-particles, making them undetectable to XRD [
41,
42]. Other authors also found that homogenization of Y
2O
3 in ODS steel during MA was achieved quickly, whereas for metallic components it took much more time [
43].
Figure 16 shows the XRD pattern of the low intensity Zr and Y
2O
3 reflections obtained after initial mixing of powders (1 min of MA).
The quality of WPPM fit can be appreciated in
Figure 17, in terms of exemplary refined patterns of Fe-12Cr-W-0.3Zr-0.3Y
2O
3 after 1 h (
Figure 17a) and 64 h (
Figure 17b) of MA. A flat, almost featureless residual line (disagreement between experimental and modelled data) proves good quality of fit. Graphical analysis is generally the primary way to determine the quality of fit and to ensure that the model is correct and chemically plausible, which is particularly important in the case of multiphase patterns with overlapping reflections (
Figure 17c,d).
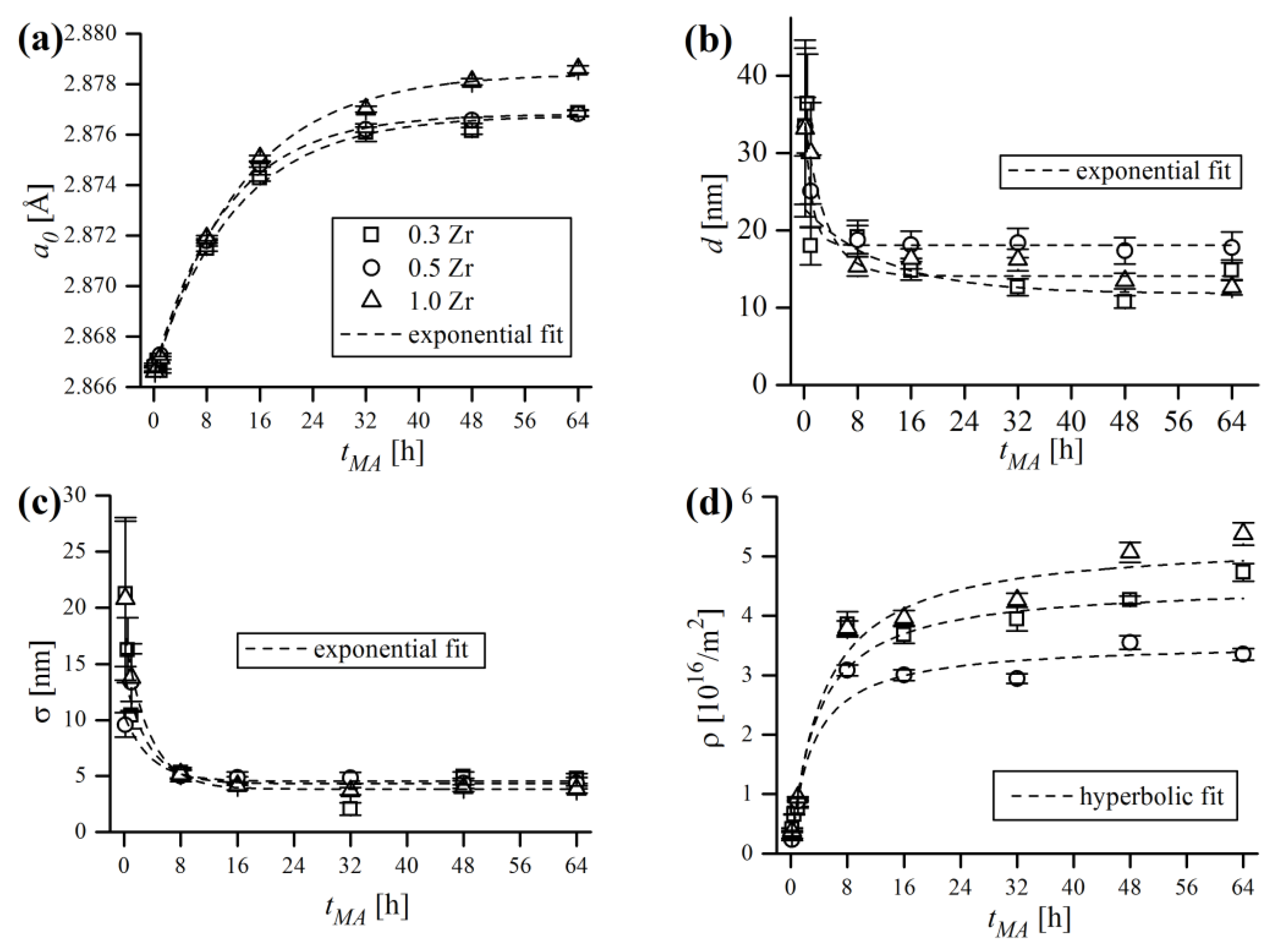
The principal WPPM results of mechanically alloyed powder compositions, in terms of bcc lattice parameter (
), lognormal mean domain size (
), lognormal standard deviation (
) and dislocation density (
) are revealed in
Figure 18. It is a common knowledge that MA greatly induces the expansion of the lattice cell due to the combined effects of severe plastic deformation and constant dissolution of alloying additives (Cr, W, Zr, Y
2O
3) and, also, due to various contaminants from the milling media into the ferritic matrix during mechanical treatment [
6,
44]. Volume expansion is further enhanced by fine crystallite size, which increases the solubility of vacancies and other crystal defects [
45].
Figure 18a shows the increase of the ferritic lattice constant
, which was particularly sharp at the early stage of MA (up to 16 h) and tended to saturate after 32 h of processing. The data points are almost identical in the cases of 0.3% and 0.5% Zr content, and correspond to 1.06% and 1.04% of the unit cell volume expansion
, respectively (for cubic lattice
). The mixture containing 1.0% Zr exhibited a slightly larger increase in
, corresponding to a 1.26% rise in cell volume, which is most probably caused by higher Zr content dissolved in the matrix, causing additional expansion of
.
The analysis of domain size trend (
Figure 18b) points out that the reduction of
is especially effective only at initial MA stages (up to 8 h), with only slight reductions being achieved afterwards. Lognormal standard deviation
(
Figure 18c) mimicked the trend of
, which showed that MA not only caused the reduction of mean domain size, but also caused the
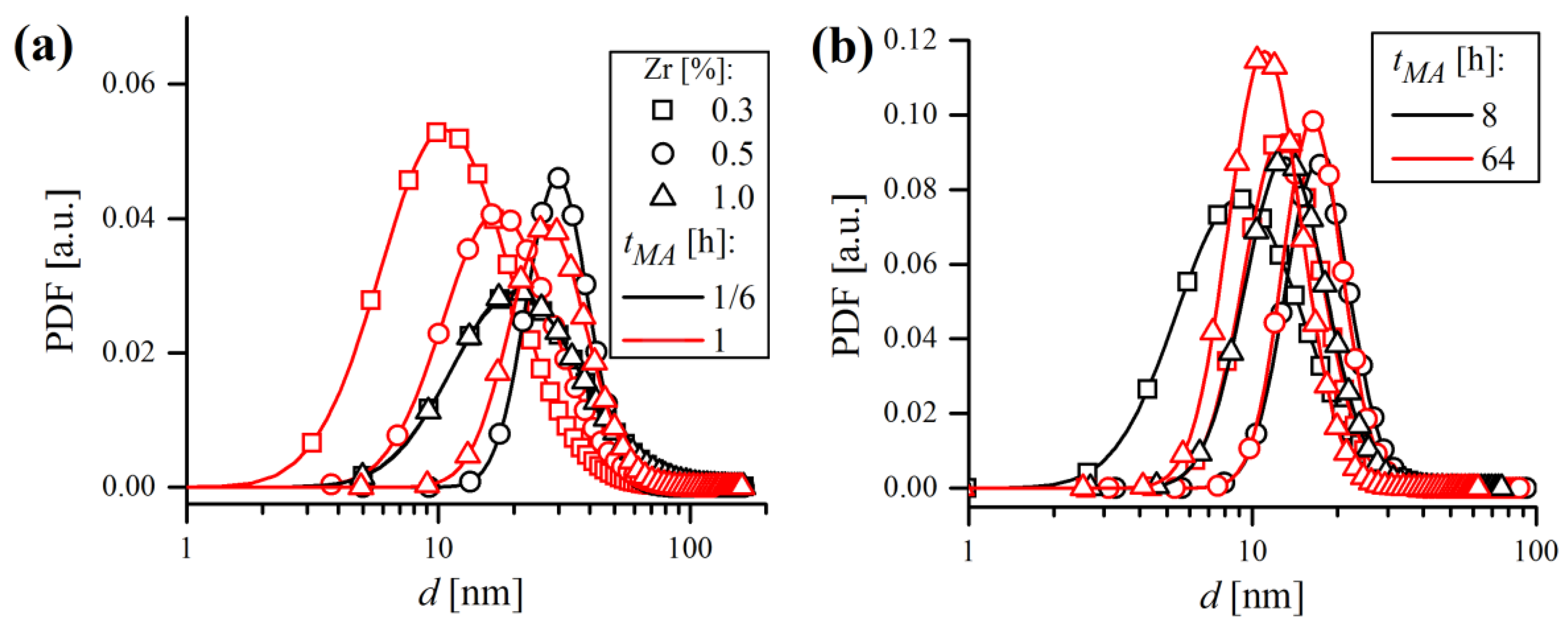
distribution to narrow with the milling time. Lognormal distributions of domain size, in terms of probability density functions (PDF), are shown in
Figure 19. Grain size distributions are roughly coincident in all samples at a given
tMA, regardless of the Zr content. It can be stated that during MA, the microstructure evolved towards a nanocrystalline state consisting of very fine (
~15 nm), narrowly dispersed (
~5 nm) crystalline domains.
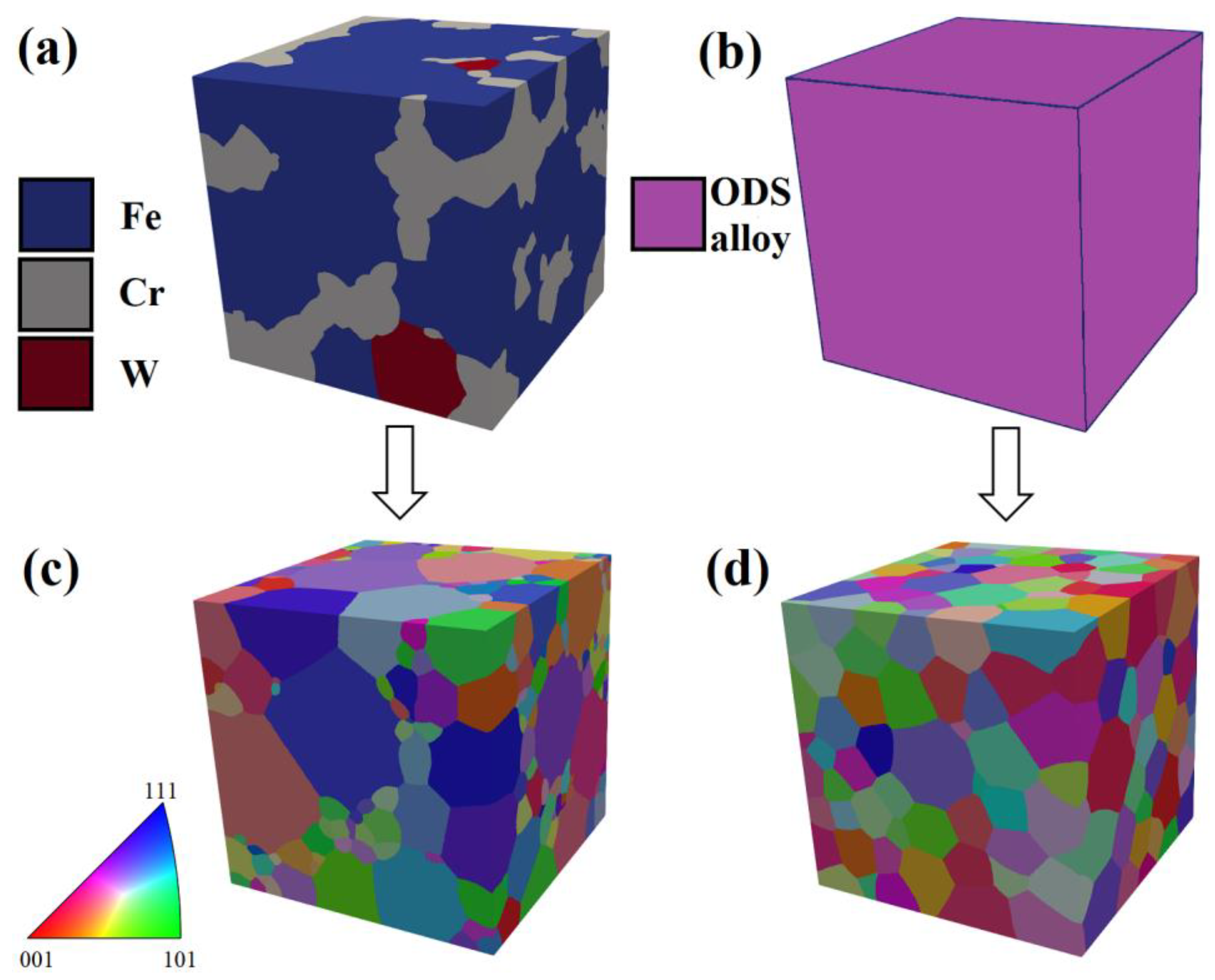
In an effort to visualize the evolution of microstructure in milled powders, a synthetic microstructure was created using domain size statistics obtained by WPPM for 0.5% Zr powder in the DREAM.3D software. According to the XRD analysis, after 1 h of MA, three distinctive phases (Fe, Cr, W) were detected, which later transformed into a single ferritic (bcc) ODS alloy phase after MA (
Figure 20a,b). The domain size distribution is visualized in
Figure 20c,d. The grains were assumed to be randomly oriented, which is expressed by their random color variation. It can be noticed that much finer domains existed in the gray fields, corresponding to the Cr phase, whereas Fe and W domains (blue and red, respectively) are coarser after 1 h of MA. After 64 h of MA, the visualized microstructure was much more homogenous, consisting of randomly oriented, equiaxed grains with little deviation in size.
Severe work hardening of the powder material during grinding was manifested by continuous accumulation of defects in crystals, mainly as dislocations. The evolution of dislocation density (
) calculated by WPPM analysis is presented in
Figure 18d, which has, in general, very similar trends as the lattice constant. Dislocation density grew during the first 16 h of MA, then the growth ratio slowed dramatically. The final obtained values of
are in range of a few times
—4.73(15)
, 3.35(10)
and 5.38(19)
for 0.3, 0.5 and 1.0% Zr, respectively, with an effective dislocation cut-off radius
of ~5 nm. Similar values were reported by other authors, such as Scardi and Leoni [
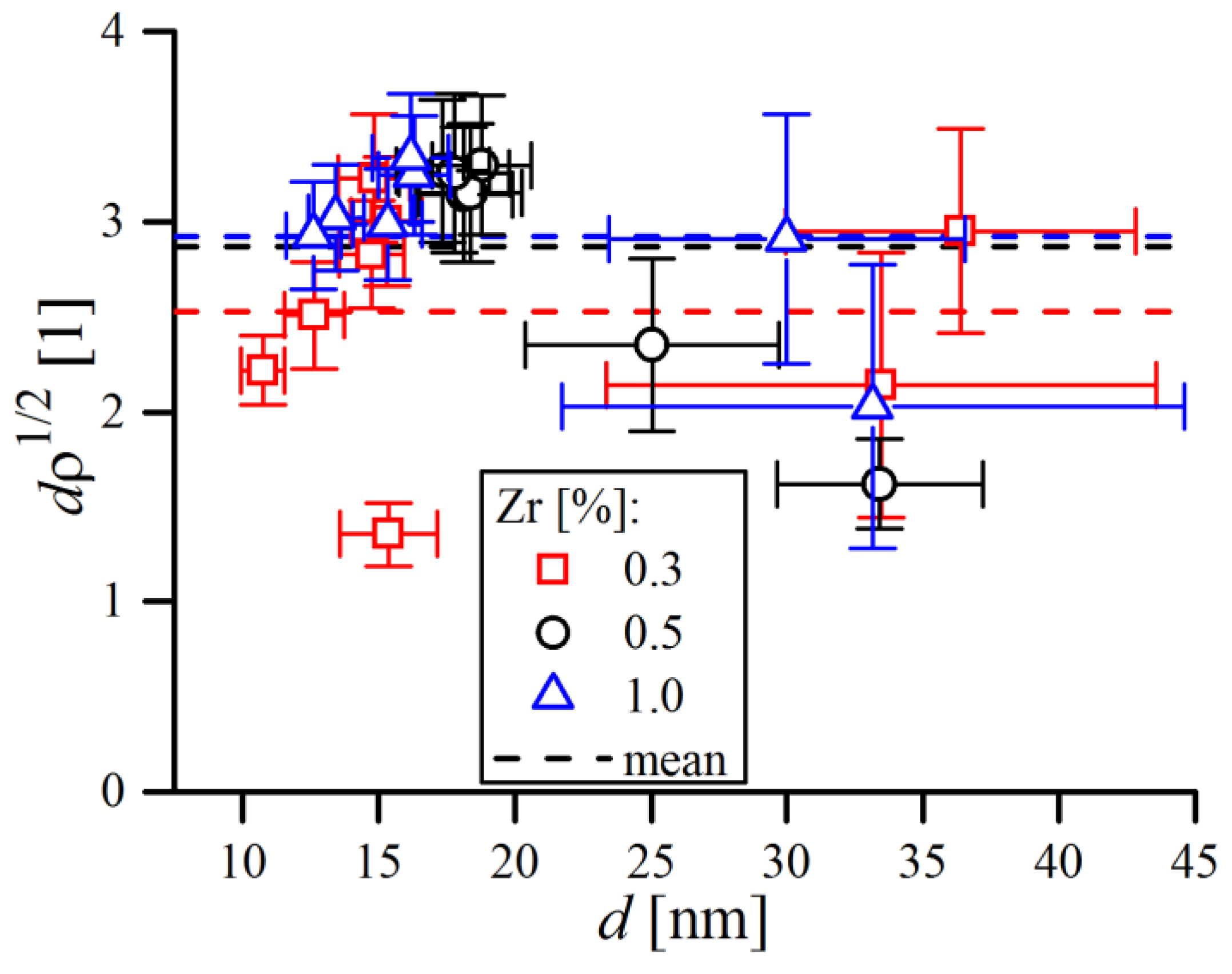
6], Kumar et al. [
22] and Rebuffi et al. [
24], in the case of FeMo, FeCrAl and Ni ball-milled powders. However, questions may arise about the actual meaning of
, as final
values were relatively high and seemed overestimated, roughly corresponding to around 2–3 dislocations per single grain (below grain sizes of ~20 nm, sub-grains do not form so crystalline domains can be perceived as grains [
46]). The average number of dislocations per grain was calculated as a ratio between the mean grain diameter
and mean dislocation distance
—i.e.,
, and the obtained data points are compiled in
Figure 21.
As shown, the average number of dislocations was always above unity, a condition certainly refuted by TEM observations [
24] as well as molecular dynamics simulations [
47]. Although some dislocation dipoles were found in the TEM observations of ball-milled materials, apparently many crystallites do not contain any dislocations, so the probably strain broadening observed in the XRD line profile has origins that are not solely due to dislocations [
48]. In fact, the quantity of dislocations in severely deformed, nanostructured metals is not so high, as dislocations generated during plastic deformation slip across the grains and merge into the grain boundary [
24]. Therefore, to avoid reporting
values which were clearly overestimated, referring to a more general term of ‘microstrain’
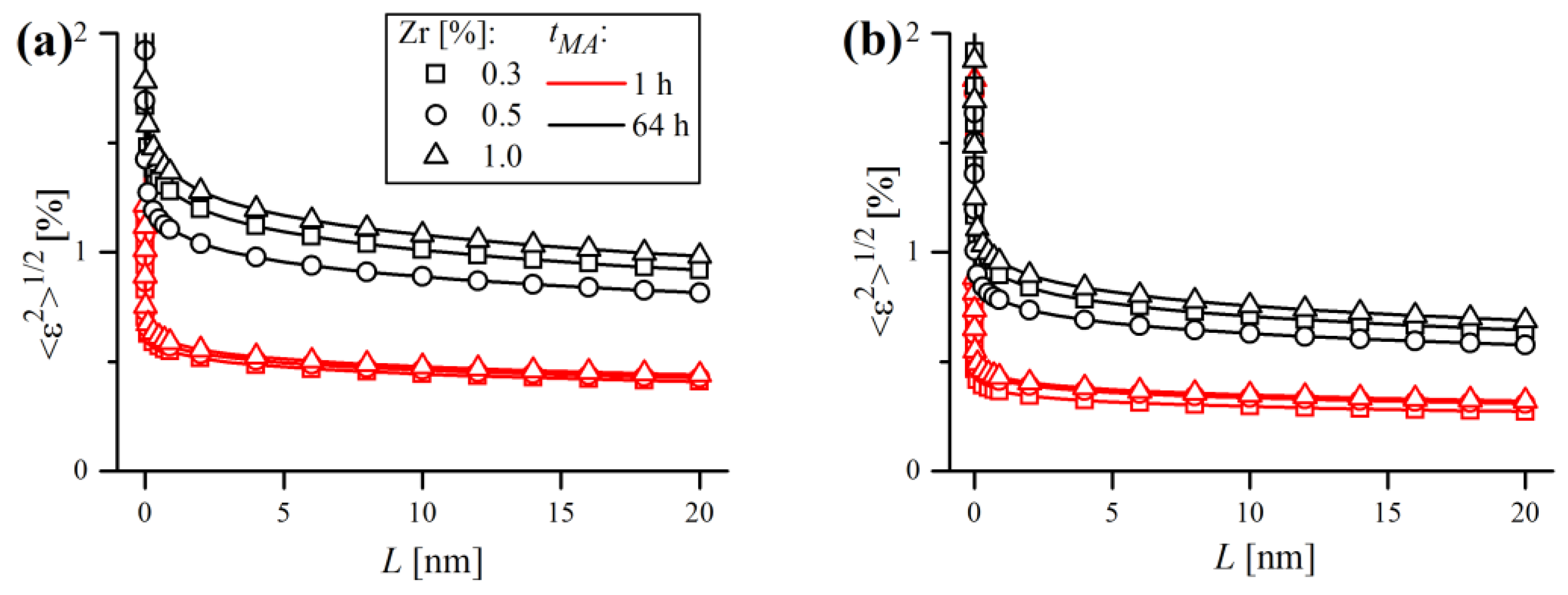
, i.e., root mean square elastic strain, can be more adequate to describe the strain broadening observed in diffraction lines. Microstrain can be simply calculated along any desired crystallographic direction by substitution of elastic strain parameters obtained by WPPM into the equation proposed by Wilkens (Equation (8)):
where
is the distance between pairs of atoms along the given crystallographic direction (correlation length),
is the effective dislocation cut-off radius and
is a monotonically, slowly decreasing logarithmic function, the so-called Wilkens function [
49]. Values of
were plotted in
Figure 22 for two crystallographic directions, [
h00] (a) and
(b), which are the softest and the stiffest crystallographic directions in Fe-bcc, respectively. As expected, microstrain values were higher towards the
direction compared to [
hhh], thus representing the upper and lower boundaries of the strain broadening.
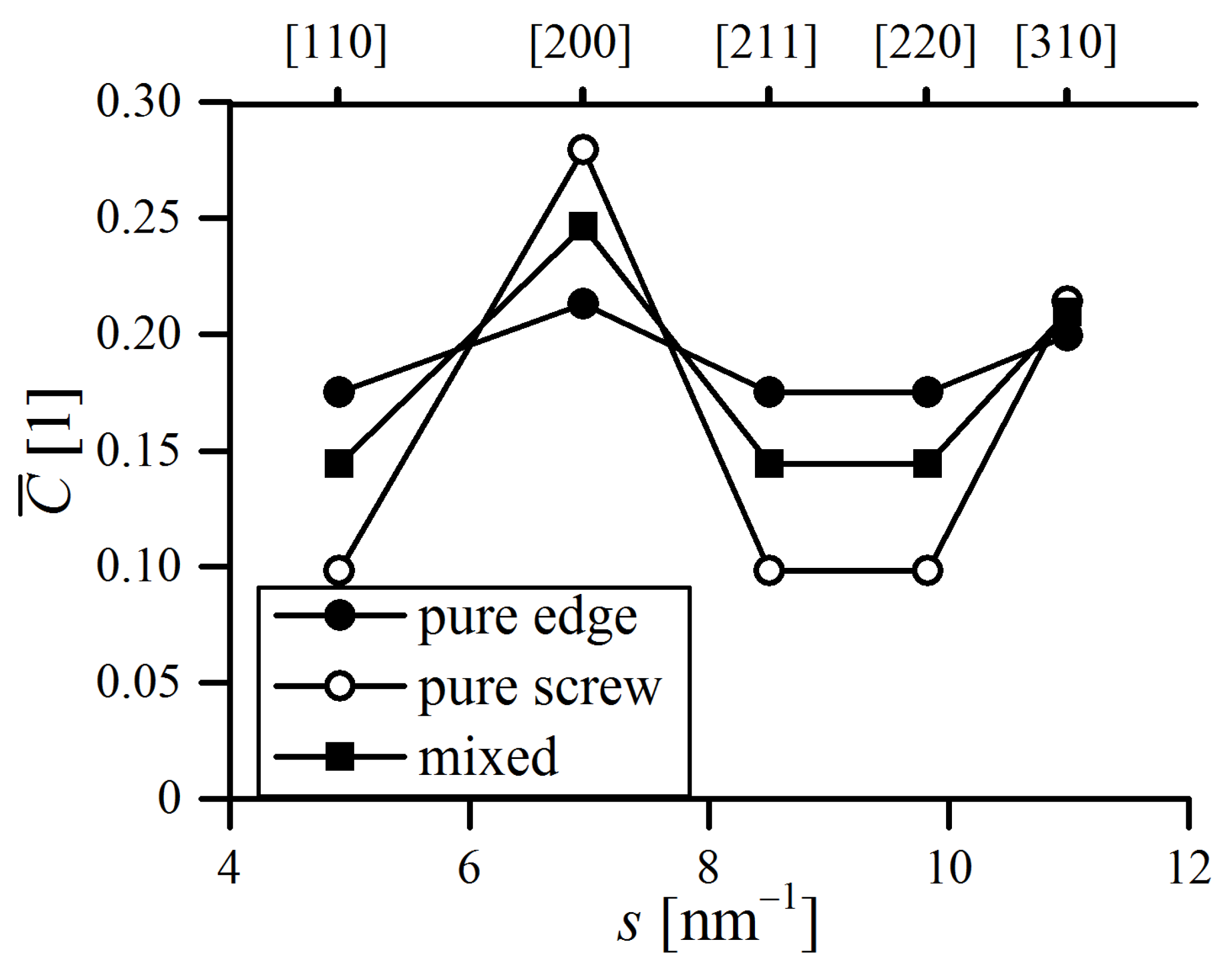
Furthermore, the dislocation character trend was plotted in the form of the edge dislocation fraction
, which is depicted in
Figure 23a. In general, the dislocation structure was in favor of screw dislocations over the whole MA process, as the
rarely exceeded 0.5, which might be caused by the rotational nature of the ball milling process, promoting the formation of twists in crystals [
50]. This is in line with other authors’ conclusions that edge dislocations are unstable in fine Fe domains [
24,
51].
The inspection of the Wilkens parameter
, was used to determine the correlation of dislocations in the material, i.e., to determine whether the arrangement of dislocations is random or strongly correlated. Its value is also directly correlated with the effective dislocation cut-off radius
, as
. It can be determined using XRD LPA, as the peak tail regions decay faster or slower, depending on the weak or strong character of the strain field. When dislocations are randomly distributed,
is large (
). On the contrary,
when dislocations are close to each other, i.e., when the strain field has the short-range character, known as the strong dipole configuration [
11]. In the present case (
Figure 23b),
values during MA oscillated around unity, finally reaching values in range of ~1.2–1.4 after 64 h of MA, supporting the hypothesis of strong interactions between dislocations, that were observed, for instance, in dislocation cell walls [
52]. This indicates that the dislocations were not randomly arranged in crystalline domains, but are rather densely accumulated at grain boundaries. Furthermore, ball milling caused microstructure evolution towards a nanocrystalline state, consisting of fine, narrowly dispersed crystalline domains with strongly correlated extrinsic dislocations forming non-equilibrium grain boundaries.
According to the work hardening theory of cold worked metals, the growth in defects during plastic deformation should be accompanied with corresponding improvements of mechanical properties, e.g., hardness. The Vickers microhardness of the powder samples was determined using a digital microhardness tester using two load values: 0.098 N and 0.245 N (10 and 25 g of force, respectively), and 15 s of dwelling time. It was necessary to apply two different load values due to substantial disparities in microhardness between samples tested after short and long MA times. A load of 0.098 N turned out to be too small in the case of harder particles, as the imprints produced were so small that they could not be properly measured. Contrarily, a 0.245 N load was too much for samples after ≤1 h of MA, causing the particles to break apart. As a consequence, powders after short MA times (≤1 h) were tested using a 0.098 N load, and all others used a 0.245 N load. At least 20 indentations were performed on each powder to calculate the mean experimental values of .
3.6. Microhardness Testing of Mechanically Alloyed Powders
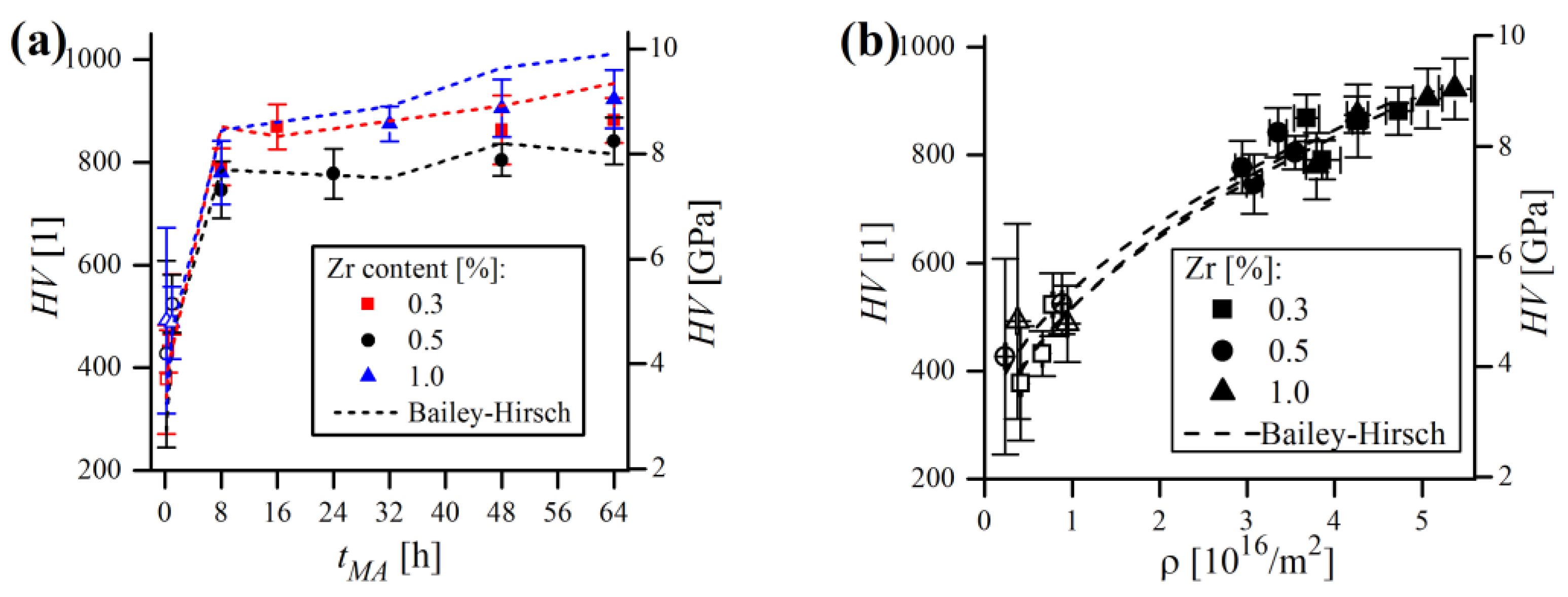
The results of microhardness testing of the mechanically alloyed powders are presented in
Figure 24. Powder containing 1% Zr exhibited the highest microhardness after MA (922(56) HV), while 881(44) and 841(46) HV were measured in the case of 0.3% and 0.5% Zr samples, respectively. As seen in
Figure 24a, all of them show very similar, exponential trends in HV, with microhardness values raising sharply during the first 8 h of MA, with little increase measured afterwards. At the beginning of MA, the microhardness exhibited the largest variation (expressed in the standard deviation), because the particles were far from being homogenous and were rather complex composites of the starting powders. At this stage, the chemical composition varied significantly from particle to particle and even within the single particle, causing strong dispersion of the obtained
values. As the process continued, the particles became more homogenous, resulting in lower scattering of measured microhardness.
As the trend of microhardness was, in general, very close to the
trend (
Figure 24b), and the increase in microhardness itself should result from the accumulation of defects in material, efforts were undertaken to find alleged
and
correlations. The formula relating the material’s strength and dislocation density is basically known as the Bailey–Hirsch relation, originally relating yield strength (
) and
[
53]. This model assumes that dislocation interaction is the main factor responsible for the hardness increase. However, considering that the hardness of a typical metal is generally 2.5–3 times its yield strength
−
), the following relation, obtained by Yin et al. [
54] will be utilized in this work:
where
is a constant (here
= 1) and
is a shear modulus (
). After grouping the constants by substituting
and
one obtains:
Genuinely, the above equation was derived for ball-milled, pure Fe (
GPa,
. In the case of the alloys considered in this work,
was estimated as the hardness of pure ferritic Fe, which is ~83.5 HV (0.819 GPa) [
55]. The shear modulus was calculated as
= 63 GPa, accordingly to elastic constants reported earlier (
Table 1), while
= 0.249 nm, as derived from XRD. After substituting these constants, the
relation can be written as:
or, alternatively, Vickers hardness units can be used, by multiplying the result obtained by a factor of 102 (1 GPa = 102 HV). The first summand in Equation (11) represents the hardness of pure Fe, while the second addend describes dislocation forest hardening. The experimental values of microhardness were compared to those obtained by the model (using
values calculated by WPPM) and plotted with relation to MA time in
Figure 24a. In any case,
values obtained from the model mimicked the experimental ones but did not reproduce them clearly. However, modelling the
(
relationship with Equation (11) (with free
and
parameters and the
exponent constrained to
) provided a reasonable fit (
in all cases), which is presented in
Figure 24b. Therefore, the converged
and
constants derived from trend lines were collated in
Table 5.
As shown,
values were rather far from the theoretical value of 0.819 GPa, and
, expected to be around
GPa × m tended to be slightly underestimated. The possible reasons for these discrepancies in experimental and modelled
might be various. First of all, as it was discussed earlier,
values obtained from WPPM analysis are probably overestimated. Moreover, the Bailey–Hirsch model might not necessarily describe the macroscopic microhardness evolution in ball-milled materials flawlessly, as it can result not solely from defect accumulation but rather from the combination of collateral effects involved in MA—plausibly related mainly to grain size refinement. As can be noticed in
Figure 24a, the theoretical results are understated during early stages of MA (≤1 h) and, in contrast, rather inflated afterwards. Presumably, the major factor contributing to the microhardness of short milled powder is the grain size refinement, which is not accounted for in the Bailey–Hirsch relation, instead of dislocation density, which was low at this stage, causing the theoretical values of
to be underestimated.































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}