
The Role of Carbonate Formation during CO2 Hydrogenation over MgO-Supported Catalysts: A Review on Methane and Methanol Synthesis



Institute of Chemical Engineering and Environmental Technology, Graz University of Technology, NAWI Graz, Inffeldgasse 25C, 8010 Graz, Austria
*
Author to whom correspondence should be addressed.
Energies 2023, 16(7), 2973; https://doi.org/10.3390/en16072973
Submission received: 5 March 2023
/
Revised: 20 March 2023
/
Accepted: 22 March 2023
/
Published: 24 March 2023
(This article belongs to the Special Issue Advances in Carbon Capture and Utilization)
Abstract
:Methane and methanol are promising products for CO2 hydrogenation for carbon capture and utilization concepts. In the search for effective, robust, easy-to-manufacture and stable catalysts, supported metal-based catalysts have proven advantageous. Whereas nickel for methane synthesis and copper for methanol synthesis stand out as efficient and cost-effective catalytically active metals, the best choice of support material is still a matter of ongoing debate. This review discusses the potential of the alkaline earth metal oxide MgO as support material for CO2 hydrogenation catalysts. Due to its basicity, it gives access to bifunctional catalysts as it shows pronounced CO2 adsorption capacity. Whereas carbonate formation seems to be beneficial in CO2 methanation, it may even have an adverse effect in methanol synthesis from CO2.
1. Introduction
The utilization of carbon dioxide (CO2) from industrial waste sources is highly promoted, aiming at mitigation of greenhouse gas emissions while exploiting industrial exhaust gas streams. Fossil resources like coal, oil and gas have been used as main fuels for transportation and as feedstock material in the chemical, power and energy sector for a long time. Their use is regarded as being the main driver for climate change due to their high CO2 emissions. In 2021, 36.3 Gt of CO2 were emitted from energy combustion and industrial processes, which was the highest annual level ever reached [1]. This value means that the average annual CO2 concentration in the atmosphere was 50% higher than that at the time when the industrial revolution began. In order to limit the global temperature rise to 1.5 K in line with the Paris Agreement, global leaders approved diverse goals to achieve a renewable and sustainable future. Together with the option of long-term storage (carbon capture and storage, CCS) of anthropogenic CO2 emissions, the utilization of CO2 makes an important contribution to achieving these climate goals. The technologies associated are referred to as carbon capture and utilization (CCU) technologies. Among them, catalytic CO2 hydrogenation with sustainably derived hydrogen (H2) adopts a leading role, with methane (CH4) and methanol (CH3OH) being potential hydrogenation products. Methane and methanol production via CO2 hydrogenation could make an important contribution to future sustainable energy supply by chemically storing renewably derived hydrogen while processing abundant, locally available industrial carbon sources.
CO2 hydrogenation to methane, the so-called Sabatier reaction or CO2 methanation, has already been discovered in 1913 by Paul Sabatier. It combines the endothermic reversed water–gas shift reaction (RWGS, Equation (1)) with the exothermic methanation of CO (Equation (2)) in one process step. Overall, CO2 hydrogenation to methane (Equation (3)) is thermodynamically favored (ΔRG0298 K = −130.8 kJ mol−1) but kinetically hindered. This makes the use of catalysts inevitable [2]. The reaction is generally carried out over nickel-, rhodium- or ruthenium-based catalysts [3]. Among them, supported nickel-based catalysts stand out by showing excellent catalytic performance and low cost, and have thus received great attention in recent years.
CO2 + H2 ⇌ CO + H2O ΔRH0298 K = 41.0 kJ mol−1
CO + 3 H2 ⇌ CH4 + H2O ΔRH0298 K = −206 kJ mol−1
CO2 + 4 H2 ⇌ CH4 + 2 H2O ΔRH0298 K = −164.9 kJ mol−1
Whereas direct CO2 hydrogenation to methane is thermodynamically favored, methanol synthesis from CO2 (Equation (4)) suffers from unfavorable chemical thermodynamics (ΔRG0298 K = 3.5 kJ mol−1); it requires stringent reaction conditions with high pressure to achieve sufficient CO2 conversion. Low temperatures would be desirable with regard to the thermodynamic equilibrium but are not feasible from a kinetic point of view. Among others, copper-based catalysts are used for methanol synthesis [4,5]. In this respect, water inhibition is reported to become an issue, especially at lower temperatures (<473 K). It is mentioned that a few kPa of water partial pressure are sufficient to eliminate the majority of the catalytic activity of copper-based catalysts. However, water formation is unavoidable during direct hydrogenation of CO2 to methanol [6].
CO2 + 3 H2 ⇌ CH3OH + H2O ΔRH0298 K = −49.4 kJ mol−1
Thus, different process concepts are currently under investigation. Whereas methanol synthesis from syngas (CO/CO2/H2) over Cu/ZnO/Al2O3 catalysts is an elaborate, major industrial process, the best concept for methanol synthesis from CO2 is controversial. While some researchers claim that a two-step process where the CO2/H2 feed is first partially shifted toward CO/CO2/H2 and subsequently, after the formed water has been removed, converted to methanol is beneficial [6,7,8], direct CO2 hydrogenation to methanol also has its advantages due to the simplified execution of the synthesis in one process step.
In this context, the design of tailor-made supported metal-based catalysts that are robust, highly active and selective for CO2 hydrogenation toward the respective reaction product is crucial. In catalysis with metal-based heterogeneous catalysts, not only the catalytically active metal species but also the carrier (support) material and the presence of potential promoters play a crucial role. To develop catalysts with high catalytic activity and product selectivity, the composition of the catalyst needs to be fine-tuned. Thus, the properties of the catalytically active metal species, their interactions with the carrier material and their individual role in the course of the reaction need to be known. While Ni for the synthesis of methane and Cu for the synthesis of methanol have emerged as suitable catalytically active materials, the role of the carrier material is still a subject of stimulated research and discussion [9,10]. Until now, several aspects remain controversial. This limits rational catalyst design.
Basic oxides, among them magnesium oxide (MgO), are regarded as promising carrier materials for a series of reactions, such as dry reforming, dehydrohalogenation, decomposition of CCl4, oxidative coupling of methane, hydrodesulfurization, methane dimerization, water–gas shift reaction [11,12] and also CO2 hydrogenation to methane and methanol [13,14,15]. MgO as a catalyst carrier material modifies the electronic state of the overall catalytic performance by electron transfer between the native catalytically active species and MgO as support. This influence is dedicated to the alteration of acid-base properties. However, the chemical composition of MgO, the preparation method and the preparation conditions are important factors that affect its surface and supporting properties as a catalyst carrier material. MgO with a high surface area and a nanocrystalline structure has encouraging applications. As a carrier material for CO2 hydrogenation catalysts, MgO offers an abundant source with the advantage of easy recycling of spent catalysts when the CO2 hydrogenation step is integrated into a compound process, for instance, in metal production, since MgO is an important slag former in the converter [14]. Whereas for methane synthesis, spent Ni/MgO catalysts can easily be recycled in a steel converter after their lifetime; for methanol synthesis, spent Cu/MgO catalysts can be recycled in a copper converter.
Apart from its role as a carrier material, the use of MgO is also propagated for CO2 adsorption as it readily adsorbs CO2 under the formation of carbonate. Thus, it has to be assumed that carbonate formation also has a decisive influence on the activity and selectivity of MgO-supported catalysts in CO2 hydrogenation. The purpose of this paper is to elucidate the role of carbonate formation during CO2 hydrogenation to methane and methanol over Ni- and Cu-based catalysts on MgO carrier material with the aim of giving access to tailor-made, customized catalyst design.
2. Magnesium Oxide—Occurrence, Production and Properties
2.1. Occurrence of MgO
Magnesium oxide, also known as magnesia, rarely naturally occurs as mineral MgO. In these rare cases, MgO is found as periclase, a cubic form of MgO that occurs in metamorphic rocks and dolomites [16]. However, in nature, it often forms a solid solution with wüstite (FeO), referred to as ferropericlase and magnesiowüstite, forming 20 vol% of the lower mantle of the earth. Occurring sites can be serpentine rocks and volcanic ejecta, and it can also be found in contact metamorphic limestone and dolomite. The reason why it generally does not form rocks or salt deposits is that it is readily converted to magnesium hydroxide (Mg(OH)2) by water vapor in the atmosphere [17]. The atomic stacking of MgO is characterized by a B1 cubic crystalline structure (space group Fm3m) known as the rock-salt (NaCl) structure. Its lattice parameter is about 4.21 Å at ambient conditions. Oxygen and magnesium atoms that are held together by ionic bonding make up its atomic structure that spreads by half the diagonal of the cube [18].
2.2. Production of MgO
MgO may be produced by two routes, beginning either with magnesium in sea water (wet process route) or with magnesite (MgCO3) ore (dry process route). The wet magnesia process from magnesium chloride (MgCl2) involves high-temperature hydrolysis and magnesium hydroxide precipitation. It is implemented in large scale in the United States and in England. With 600 m3 of seawater, one ton of MgO can be produced by addition of calcium hydroxide (Ca(OH)2) as a precipitating agent. Magnesium brines are another source for MgO production via the wet process route. They arise as byproduct in salt production from natural brines, mining or salt deposits. Magnesium brines mainly consist of MgCl2 and small amounts of MgSO4 and alkali chlorides, making sulfate removal essential for the precipitation of Mg(OH)2. MgO production from brines benefits from low amounts of bicarbonate and boron, which facilitate the complex removal process that is required for MgO production when seawater is used [17]. Use of the wet process route is in decline because it requires three times as much energy as the dry process route from magnesite ore. Therefore, the main process for producing magnesia is thermal processing of mined natural magnesite, a still energy-intensive process that was developed in Austria over 100 years ago [17]. It involves the firing of natural magnesite in a multiple hearth furnace, a shaft kiln or a rotary sintering kiln. The endothermic chemical reaction depends on high firing temperatures. The process starts at temperatures of around 823–1073 K, where magnesite is de-acidified, and CO2 is released, yielding caustic calcined magnesia causter as the reaction product. To produce sintered magnesia, causter is further processed at 1873–2473 K. The temperatures, along with the duration of the treatment, control the product quality, such as a high density and a well-crystallized final product [19].
Sommerbauer et al. (2016) suggested reductive calcination for the conversion of mineral magnesite to magnesium oxide. In a hydrogen atmosphere at ambient pressure and overpressure, methane, CO2 and CO are formed in the product gas without additional catalysts [20].
2.3. Properties of MgO
MgO as an alkaline earth metal oxide is a typical solid base. It has the lowest basic strength among the alkaline earth metal oxides: MgO < CaO < SrO < BaO. The surface areas increase in the reverse order with MgO giving access to high surface area materials (MgO > CaO > SrO > BaO) [21]. The generation, characterization and properties of the basic MgO sites were studied by Di Cosimo et al. (2014) [22]. The MgO samples showed surface sites of strong (low coordination O2− anions), medium (oxygen in Mg2+-O2− pairs) and weak (OH− groups) basicity. Increasing calcination temperature drastically decreased the density of strong basic sites and to a lesser extent that of weak OH− groups. The density of medium-strength basic sites was only slightly decreased. Wu and Goodman (1992) found that thermally generated defect sites on the surface exhibit a stronger basic character [23].
MgO may be a reactive substance, for instance, readily forming Mg(OH)2 in the presence of water or MgCO3 through adsorption of CO2, while under certain circumstances, it may be extremely temperature resistant. Its reactivity strongly depends on the temperature of the calcination process, meaning treatment at different temperatures produces MgO of different reactivity. Depending on the calcination temperature, MgO is classified into three grades—caustic, sintered and fused MgO:
- (i)
- Caustic MgO is formed when the solid minerals Mg(OH)2 or MgCO3 are slightly heated above their decomposition temperature. Caustic MgO can be divided into two types: light-burned and hard-burned MgO. Light-burned MgO, generally known as caustic calcined MgO, is formed at calcination temperatures of 1143–1273 K and is the most reactive form of MgO. Hard-burned MgO, which is calcined at temperatures of 1823–1923 K, has limited reactivity. The reactivity of caustic MgO decreases with increasing calcination temperature.
- (ii)
- Sintered or dead-burned MgO, also known as magnesia clinker, is produced at calcination temperatures of 1673–2273 K. It is an unreactive form of MgO with a high heat capacity and a high thermal conductivity, thus, generally being used as refractory material.
- (iii)
- Fused MgO is generally produced from naturally occurring MgCO3 in electric arc furnaces at 1273–1673 K (‘dead burned’) or by melting caustic magnesia. Fused magnesia is a crystalline substance with a melting point of 3073 K. When it is heated up to the melting point, no phase change takes place. Its tendency to undergo hydration is much lower than that of sintered or caustic calcined MgO, which makes it essentially stable toward the atmosphere. In a reducing atmosphere, the stability of fused magnesia is limited to 1973 K. Because it combines high electrical resistance and high thermal conductivity, it is mainly used as an insulating material [17].
Making use of the property of high temperature resistance, 87% of total MgO produced is used for refractory products: 65% in the steel industry, 15% in the cement industry and 7% for other refractory applications, such as non-ferrous metal or glass processing. Since global steel production is still increasing, the demand for magnesia-containing refractory products is also growing [19]. Other fields of application of MgO include the pharmaceutical sector, for instance, as antacid or dental filling, the food sector with use as an additive or anticaking agent, for example, and the agricultural industry as fertilizer or supplement for animal feed [17]. Moreover, MgO is used as a carrier material and catalyst support in the chemical industry [11].
3. CO2 Adsorption Behavior on MgO
3.1. Carbonate Formation—Thermodynamic Considerations
Metal oxides (MenO) readily react with CO2 forming metal carbonates (MenCO3) (Equation (5)). This reaction is known as carbonation. In the reverse reaction, known as calcination, CO2 is liberated from the metal carbonate at elevated temperatures, thereby regenerating the oxide. This reversible CO2 chemisorption process is exploited in CO2 capture technologies. Suitable oxides to capture CO2 are selected based on their CO2 capture capacity, adsorption rate, thermal stability, regeneration heat, costs and structural properties [24].
MenO + CO2 ⇋ MenCO3
The carbonation of metal oxides is exothermic with the extent of exothermics depending on the metal species. In the context of CO2 capture, it is noted that a highly exothermic metal oxide carbonation generally requires a large regeneration temperature for the oxide, entailing a high energy demand [24]. However, this characteristic renders metal carbonates to be suitable materials for thermochemical energy storage (TCES) processes [25,26,27]. The goal of TCES is to utilize waste heat through (thermal) energy storage by means of reversible chemical reactions, thereby enabling to balance the discrepancies between energy availability and demand. In TCES based on metal oxides/metal carbonates, the endothermic decomposition reaction (calcination/decarbonation of the metal carbonate) charges the storage material, whereas by recombination of the former decomposition products, namely the metal oxide and CO2, the exothermic reaction (carbonation) discharges the stored energy and restores the material. In general, one cycle consists of three steps: charging, storing and discharging [25]. Up to date, mainly CaCO3, PbCO3 and SrCO3 have been considered for TCES, predominantly for applications at medium or high temperatures [28,29]. Müller et al. (2019) showed that, in the presence of moisture, carbonate formation from CaO, MnO, ZnO, CoO and PbO can even be accomplished at temperatures below 373 K with an elevated CO2 pressure as the driving force. The selected metal oxides were treated with CO2 pressures of 1–55 bar (298 K) and 110 bar (333 K) [30]. Carbonation/decarbonation of MgO is also propagated for thermochemical energy storage. Shkatulov et al. (2022) investigated MgO promoted with the eutectic ternary mixture LiNO3-NaNO3-KNO3 for thermochemical storage of medium temperature heat [31]. Zhang et al. (2021) used MgO composites promoted with alkali metal nitrates [32]. In addition, MgO/Mg(OH)2 is currently getting attention as another potential system for TCES. Flegkas et al. (2019) investigated this system in fluidized bed reactors and conducted an economical study [33].
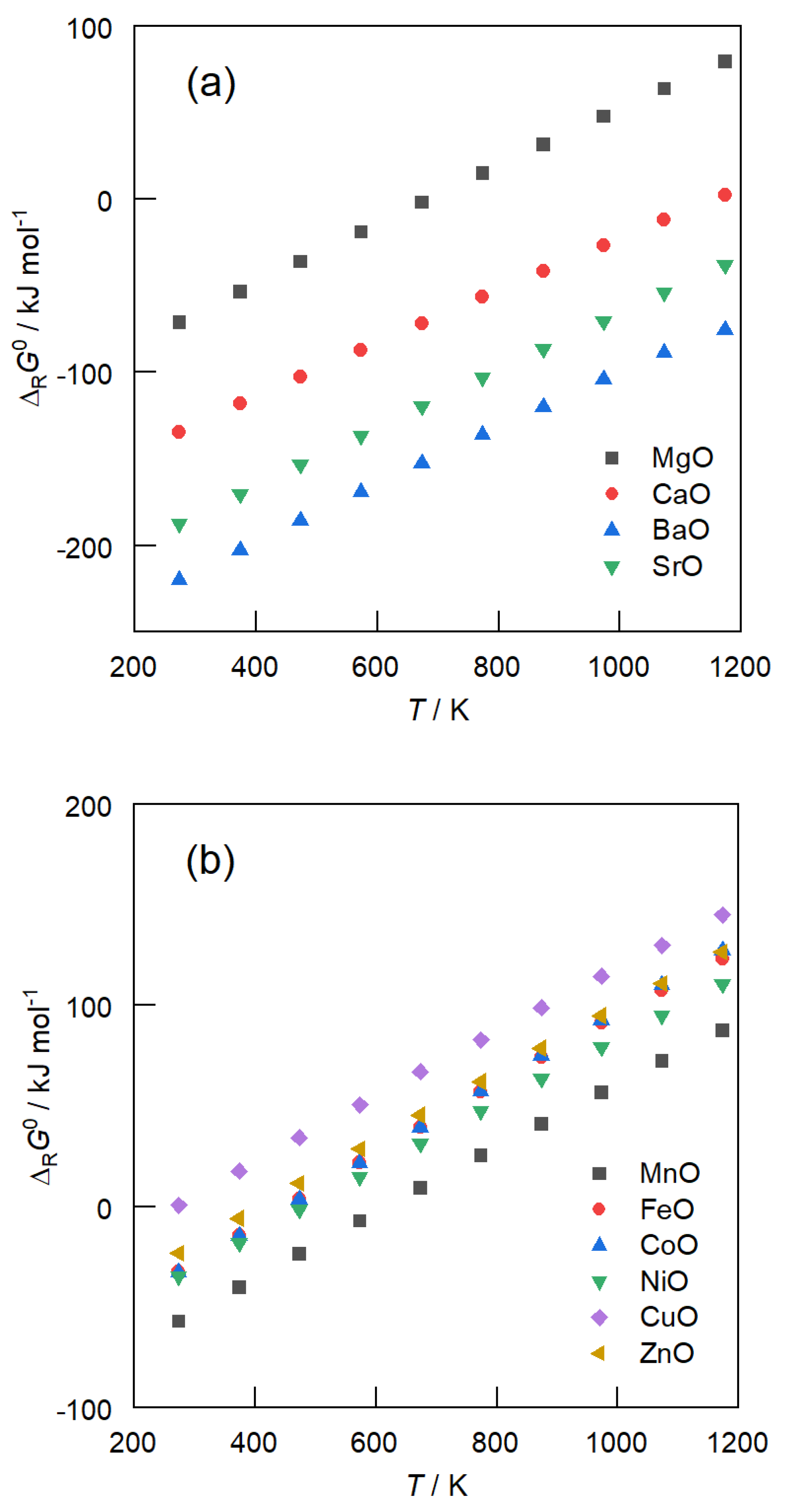
The temperature dependency of the standard free enthalpies of reaction ΔRG0 of carbonate formation from alkaline earth metal oxides (a) and transition-metal oxides (b) for temperatures between 300 and 1200 K at ambient pressure is depicted in Figure 1. Among the alkaline earth metal oxides, MgO has the highest ΔRG0 values. The standard free enthalpies of the reaction of transition-metal oxide carbonation are even further above. This means that, compared to CaO, BaO and SrO, MgO is more stable at comparably ‘lower’ temperatures. Above 670 K, carbonate formation from MgO is thermodynamically not favored any more with the chemical equilibrium lying on the educts’ side. Above 1073 K, MgO cannot react with CO2 at all, whereas BaO is still completely converted to its carbonate at this temperature. Thus, MgO is characterized by the fact that CO2 can be released from MgCO3 at comparably low temperatures, which renders it the most promising candidate for the carrier material in CO2 hydrogenation.
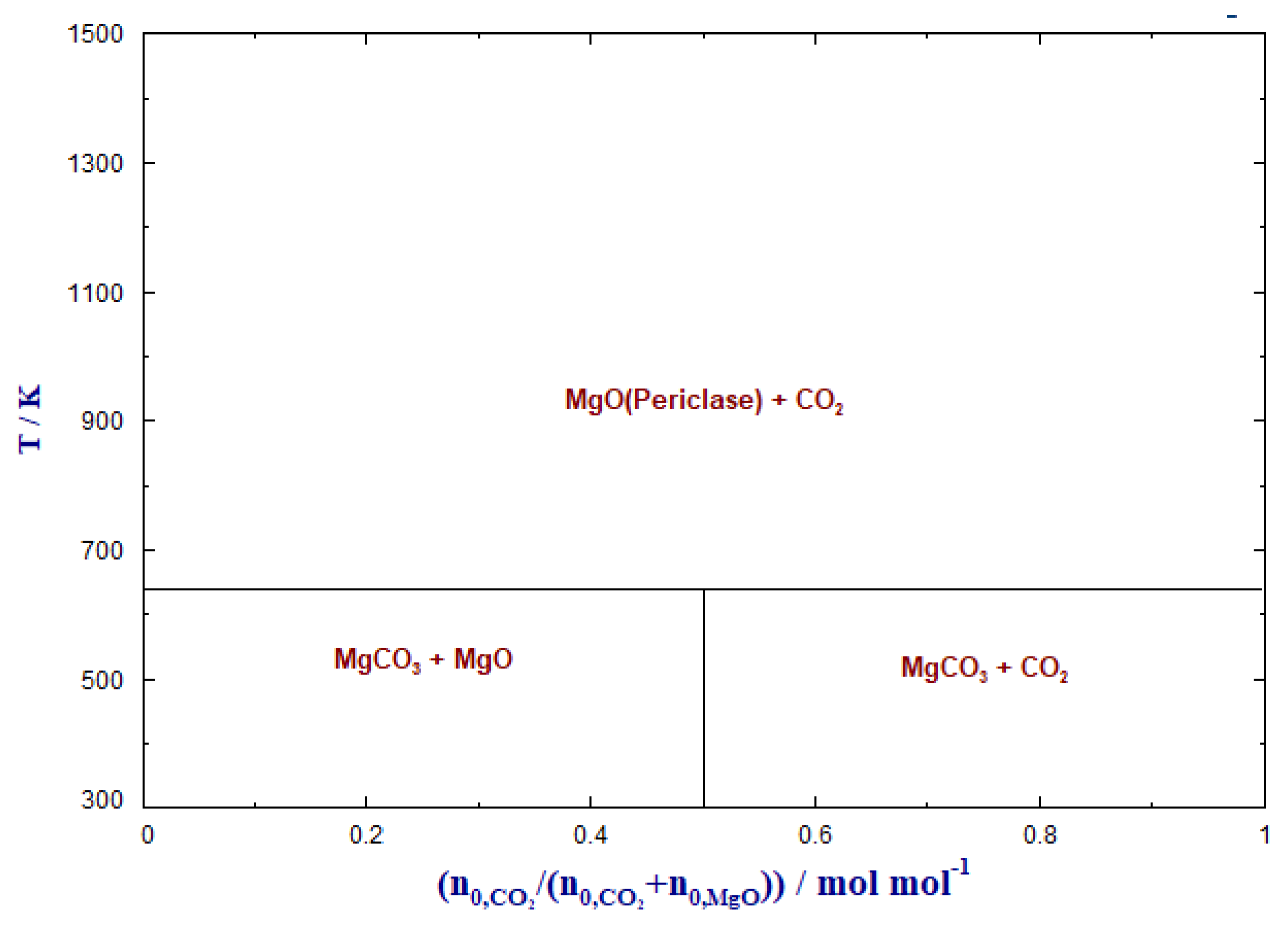
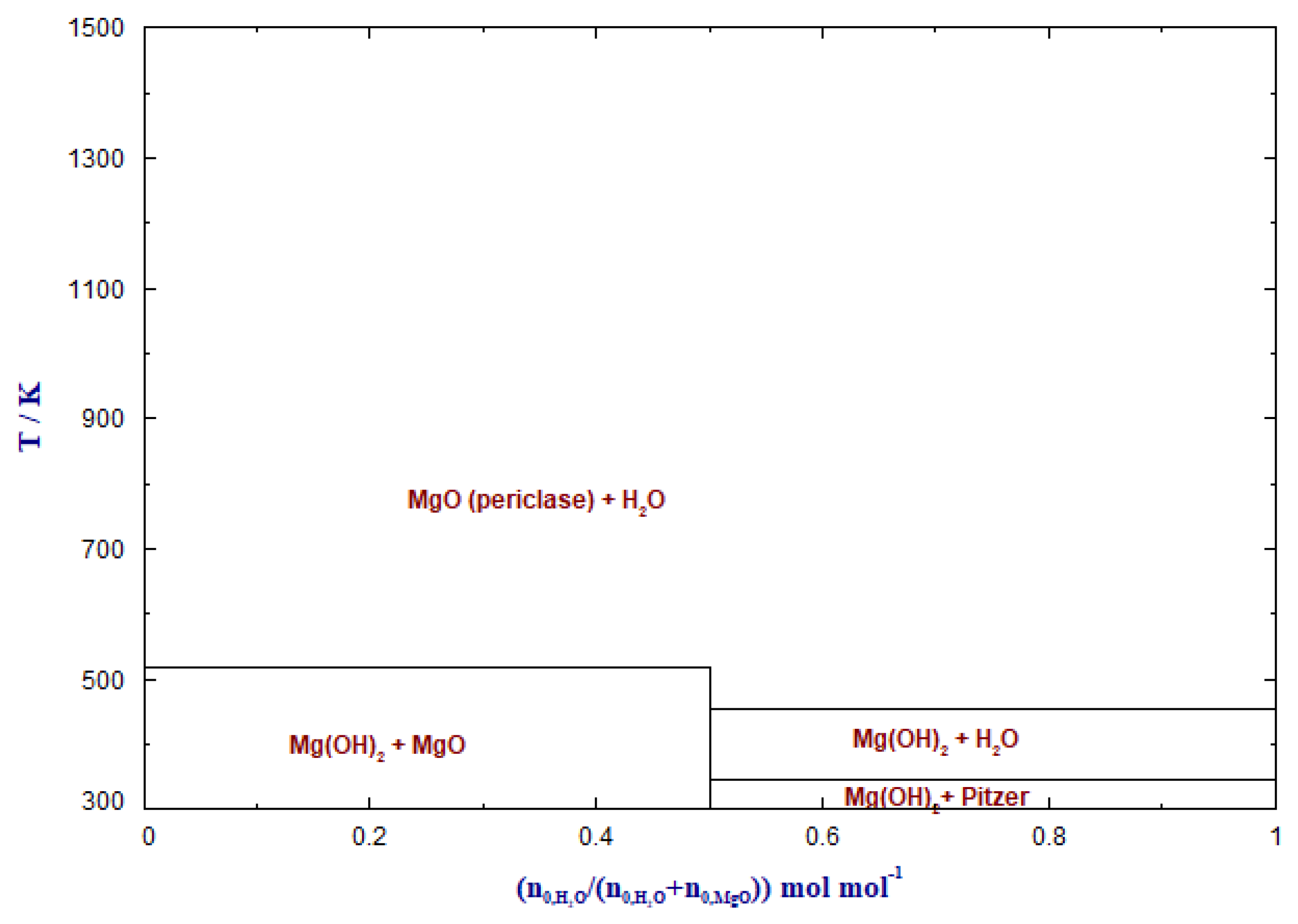
The phase transformation behavior of MgO in CO2 atmosphere is depicted in Figure 2. The solid phases at equilibrium were predicted as a function of the temperature and the feed CO2 (/(+ )), where n0 denotes the number of moles of the feed component.
3.2. Mechanism of CO2 Adsorption on MgO
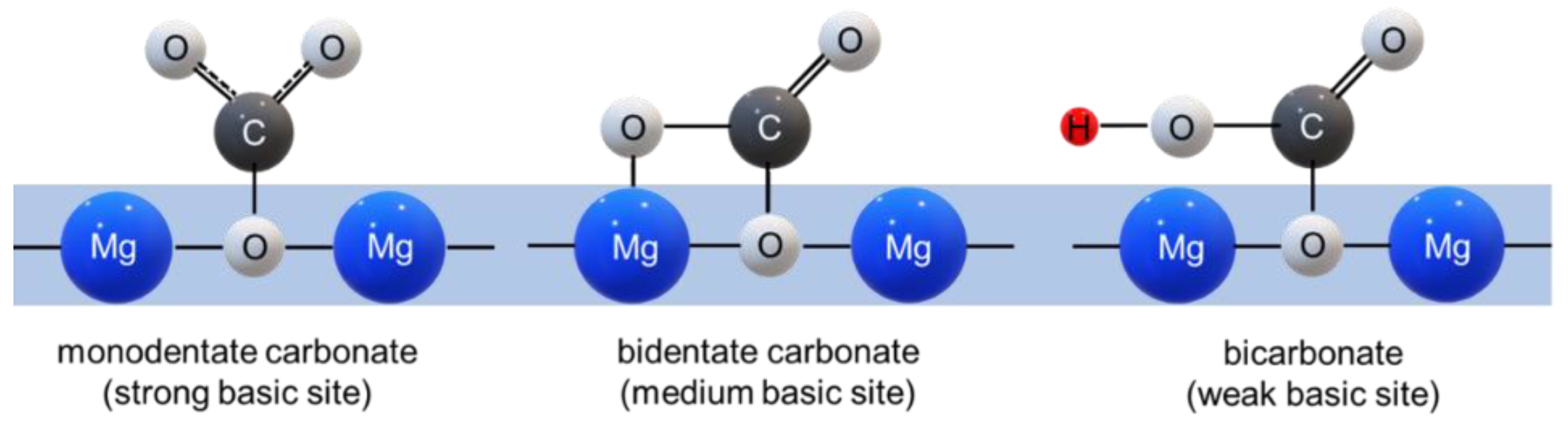
The adsorption of CO2 on MgO-based adsorbents occurs via the formation of carbonate species from acidic gaseous CO2 on basic Mg2+-O2− sites. Carbonate species may occur in the form of monodentate carbonate, bidentate carbonate and bicarbonate [12,34,35]. These are assigned to weak, medium and strong basic adsorption sites, respectively (Figure 3).
In the adsorption process, the number of basic sites is a key factor. Each adsorption site—weak, medium and strong—is classified according to the desorption temperature that is required. The desorption temperature of weak basic sites is lower than 473 K, the desorption temperature of medium basic site lies between 473 and 573 K, and the desorption temperature of strong basic sites is higher than 573 K [36]. The strong bonding that occurs between CO2 and Mg2+ and/or O2− is responsible for the formation of bidentate and monodentate carbonate with desorption temperatures > 573 K, while the interaction of hydroxyl groups (OH−) entails the formation of bicarbonate at desorption temperatures < 373 K [36]. It was concluded that the strength of the basic sites in MgO depends on the number of coordinated electronegative metal ions. Thus, this can be enhanced with a high surface area that facilitates an abundant number of O2− sites [37,38].
Yanagisawa et al. (1995) performed IR studies of CO2 adsorption on MgO powder from which they attributed the band observed to be monodentate carbonate species. Ab initio molecular modeling studies showed monodentate as the least stable carbonate species, being unstable at temperatures below 373 K, while it could be observed at a temperature of 773 K [39].
Huang et al. (2019) simulated a model for CO2 methanation on MgO (110) surfaces by three different pathways: (i) CO2 direct dissociation, (ii) a pathway via formate formation, and (iii) a carboxyl pathway. For CO2 adsorption on the MgO surface, they found that CO2 preferably adsorbs at the O-top site with an adsorption energy of −2.49 eV forming a carbonate (CO32−) structure that promotes activity and causes the strong basicity of MgO. The mechanism of CO2 methanation on MgO was illustrated by the reverse water–gas shift reaction followed by CO hydrogenation (RWGS + CO-hydro pathway): CO2*→ COOH*→ CO*→ HCO*→ H2CO*→ H2COH*→ CH2*→ CH3*→ CH4*→ CH4. In addition, they found that the MgO support is beneficial for OH removal which is responsible for hydration and water formation during CO2 methanation [40].
Kim et al. (2010) studied the CO2 methanation on MgO surfaces by using Kohn–Sham DFT calculations with a plane-wave VASP code and extensively discussed the effect of CO2 adsorption on MgO. The computational results showed monodentate carbonate formation on a MgO (110) surface over Pd-MgO/SiO2 catalysts. They reported that the carbonate draws electrons from nearby Mg atoms resulting in electron polarization of Mg atoms and a subsequent local anisotropic surface disordering of MgO (110) [41]. These findings confirm that not only stable species but also species with a lower stability (low-coordinated surface atoms, side edges, kinks) account for the activity of MgO [42,43,44]. In addition, Kim et al. (2010) suggested a mechanism for CO2 methanation on MgO surfaces that consisted of 9 steps: (i) CO2 adsorption on the MgO surface, (ii) hydrogen adsorption with CO2 and the formation of COOH on the MgO surface, (iii) COOH hydrogenation with a hydrogen atom and water formation as byproduct, (iv) hydrogen reaction with CO to form COH, (v) water formation as byproduct, (vi) hydrogen adsorption on MgO to form CH on the MgO surface, (vii) formation of CH* and transformation to CH2, (viii) hydrogen adsorption and the conversion of CH2 to CH3, and (ix) hydrogen adsorption resulting in the conversion of CH3 to CH4 [41].
3.3. CO2 Adsorption Studies on MgO
CO2 adsorption on MgO is extensively studied. Due to their abundant availability in nature (e.g., from magnesite ore), low cost and tunable physicochemical properties, MgO-based adsorbents are currently discussed as high-potential candidates for CO2 capture [36]. Thus, the CO2 adsorption performance of different types of MgO-based materials is well described in the literature over a wide range of temperatures. Compared to other metal-oxide-based adsorbents such as CaCO3 or Li2CO3, the application of MgO-based adsorbents is beneficial due to a comparably low regeneration temperature during desorption (<773 K). Their application is also promising in the intermediate temperature region of 473–673 K [34,45].
In general, potential CO2 adsorbents should feature a high surface area, large pore size and large pore volume, leading to a high number of active sites for adsorption. In addition to excellent morphologies, low diffusion resistance between CO2 and the adsorbent and a high surface basicity are crucial for a high CO2 uptake. Large emphasis is put on enhancing the adsorption performance of MgO-based adsorbents, their morphological properties and stability during repeated adsorption/regeneration cycles to foster industrial application. Measures include modification of the preparation method, variation of the type of magnesium precursor used and the addition of promoters, for instance, other metal oxides [36].
It is well known that different preparation methods and different preparation parameters for a respective preparation method result in different structural, morphological and textural properties. Preparation methods for MgO-based adsorbents include sol-gel, hydrothermal, flame aerogel, precipitation, calcination, urea hydrolysis and extrusion–spheronization methods, with various attempts to synthesize MgO-based adsorbents with a high surface area [36].
Apart from experimental studies, theoretical investigation of the MgO surface is of great interest as valuable information can be gained about the mechanism of catalysis. Computational studies enable differences in the properties of the bulk and surface defects to be identified and favorable positions for chemisorption to be found. Historically, it started with semi-empirical calculations followed by extensive ab initio calculations. Causa et al. (1987), for instance, studied the MgO (110) surface and CO adsorption thereon. They found that the fully ionic character of the chemical bond of bulk MgO and its (100) surface is also maintained at the (110) surface in spite of some redistribution of the electronic charge [46]. Usseinov et al. (2019) also highlighted the importance of understanding the atomistic interaction between defects and the active surface of solid adsorbents [47]. Kantorovich et al. (2000) provided an extensive review on MgO (001) surfaces implementing electronic structure methods based on density functional theory (DFT). They compared a perfect MgO (001) surface to surface irregularities such as steps and corners, as well as to point surface defects. Their theoretical results were also compared with experimental data [48].
3.3.1. Composite Promoters in MgO-Based Adsorbents
Introducing promoters such as alkali metal nitrates or carbonates (molten salt), metal oxides, and amines to MgO has a significant impact on MgO-based adsorbents, enhancing the CO2 uptake capacity and the performance stability. The introduction of promoters helps provide smooth mass transfer and improves the characteristics and properties of MgO as an adsorbent, facilitating CO2 adsorption. For instance, MgO composites exhibit increased basicity on the surface. This may be dedicated to the generation of oxygen vacancies and, thus, a reduction of the crystallite size. To modify MgO-based adsorbents with composite promoters in order to achieve a high CO2 uptake capacity, several factors need to be considered during the preparation procedure. These are the selection of the composite promoter, the amount of promoter introduced, and the calcination temperature. Care has to be taken as an excessive amount of the composite promoter reduces the surface area, the pore volume, and the theoretical adsorption capacity of MgO. Therefore, an optimum ratio/percentage of the introduced promoter needs to be found.
For instance, Yu et al. (2018) reported that the CO2 uptake of MgO-CeO2 is higher compared to MgO-Al2O3. Furthermore, MgO-CeO2 prepared by urea coprecipitation showed pronounced cyclic stability. The introduction of CeO2 increases the basicity of the MgO phase, which is a key factor for outstanding CO2 uptake capacity, although MgO-Al2O3 has a higher surface area and smaller MgO crystallites [49]. It is reported that an excessive amount of composite promoters has a negative effect on the pore volume, surface area, and adsorption capacity [50,51]. Therefore, to find the optimum molar ratio of promoters is an ongoing task. Jin et al. (2019) reported the highest CO2 uptake capacity and surface area for a molar ratio of Ce/Mg of 0.05, when MgO-CeO2 was prepared via a sol-gel combustion method at a decomposition temperature of 373 K. The addition of excess cerium significantly decreased the CO2 uptake capacity [50]. Gao et al. (2019) investigated molten salts MNO3/NO2 (M = Li, Na and K) as promoters. They found that NaNO3/NO2/MgO (10 mol% of NaNO3), prepared by wet impregnation, enhanced the CO2 uptake capacity at 625 K, and allowed multiple CO2 sorption–desorption cycles. However, the loading of 25% of NaNO3 reduced the CO2 uptake at various temperatures (550–650 K) [52]. Similarly, excessive amounts of FeO in FeO/MgO composite adsorbents negatively affected the CO2 uptake capacity. With FeO/MgO mass ratios higher than 3%, decreasing CO2 uptake capacity was reported [53]. It can be concluded that excessive amounts of metal/non-metal composites with MgO leads to low CO2 uptake capacities because of reduced adsorbent surface areas.
3.3.2. Effect of the Adsorption Temperature
The CO2 adsorption capacity decreases with increasing temperature due to the exothermic character of the adsorption process. Pure, pristine MgO suffers from poor CO2 adsorption performance due to a low surface area and slow adsorption kinetics [54]. Moreover, even though desorption temperatures for MgO-based adsorbents are low compared to CaCO3, thermal stability becomes an issue at elevated temperatures as sintering occurs, which reduces the surface area and deteriorates the CO2 adsorption performance and recyclability of the adsorbent [55,56].
Ito (1982) studied the initial sintering behavior of MgO that was freely dispersed in CO2 and discussed the effect on surface area diminution and crystallite growth. The sintering rate at 1123 K was found to be directly proportional to the CO2 pressure in a pressure range of 0.67–93.1 kPa. Sintering measurements in 18O-enriched CO2 revealed that most of the anions migrate only on the surface during sintering. It was concluded that sintering is enhanced by increased surface migration of O2−-ions being the slower moving ions, caused by repeated adsorption–desorption cycles of CO2 molecules regarded as anion-exchange mechanism [57].
Ho et al. (2017) studied the CO2 uptake capacity of MgO at various temperatures from 303 to 623 K. Their results confirmed that increasing temperature decreases the CO2 uptake capacity [58]. Vu et al. (2016) investigated the CO2 sorption behavior of a mesoporous MgO/Na2CO3/NaNO3 composite material in a temperature range of 623–723 K for its use in power plants. The simulated gas feed contained 10 vol% CO2 in N2. Increasing temperature resulted in an increase of the CO2 uptake capacity (48.7 to 51.6 wt%) at temperatures between 548 and 598 K. In a temperature range of 623–648 K, the CO2 uptake capacity decreased from 43.2 to 33.9 wt%. The lowest CO2 uptake capacity was found to be 12.8 wt% at 673 K [59]. Hiremath et al. (2017) found the highest CO2 uptake capacity of composite xMgO-TiO2 adsorbents at 473 K and encountered a decreasing value when the temperature reached 573 K and 673 K [60]. Tuan and Lee (2018) prepared MgO adsorbents by the precipitation method and found that the CO2 uptake capacity decreased for temperatures higher than 423 K [61]. On the contrary, Ho et al. (2017) [58] and Gao et al. (2018) [62] reported of good CO2 uptake capacities at temperatures of 303 K and 624 K using leaf-like, lamellar-shaped MgO. Chen et al. (2016) synthesized mesoporous MgO/carbon spheres by solid-state grinding synthesis and investigated the effect of temperature on the CO2 uptake [63]. The highest CO2 uptake capacity was obtained at 273 K. From these studies it is evident that both the adsorption temperature and the morphological structure of MgO affect the adsorption efficiency.
The adsorption temperature also has an effect on the formation of carbonate species. Park and McFarland (2009) reported that unidentate and bidentate carbonates form at room temperature on MgO adsorbents, with the unidentate carbonate changing to bidentate carbonate at a higher temperature of 513 K [64]. Both carbonate species disappear at 973 K. Fukuda and Tanabe (1973) studied the behavior of MgO as a support material by infrared (IR) spectroscopy analysis. They reported unidentate and bidentate carbonate formation on MgO after CO2 adsorption at room temperature. The results showed that the formation of bidentate carbonate increased with increasing outgassing temperature and decreased with increasing MgO loading [65].
3.4. CO2 Adsorption in the Presence of Water
For many CO2 capture technologies, the presence of water in the off gas is an issue due to lowering of the CO2 uptake capacity of the adsorbents. Normally, the emitted gases from coal-fired power plants contain water vapor in a range of 20–30% together with other components and 15–60% CO2 [66]. Water inhibition of metal-based heterogeneous catalysts may also become an issue [6]. Thus, the presence of water and its impact have a crucial role, both in CO2 capture and catalysis.
Regarding a deteriorating effect of water on CO2 adsorption, for MgO-based adsorbents, the contrary applies. This serves as the main argument for practical application: the presence of water has a beneficial effect on the CO2 uptake capacity. For instance, Vu et al. (2016) reported that the CO2 uptake capacity of wet gas mixtures containing 2.5% H2O and 10% CO2 in nitrogen is higher than that of the corresponding dry gas mixture in a temperature range of 548–627 K [59]. In addition, an 80% conversion of MgO into MgCO3 was reported in the presence of 10% H2O at 623 K [67]. Ding et al. (2016) found that the CO2 uptake capacity of MgO increased from 0.82 to 3.42 mol kg−1 when the relative humidity increased from 30 to 70%, respectively [68]. Ram Reddy et al. (2008) also showed the beneficial effect of water vapor that leads to more effective CO2 adsorption when using Mg–Al–CO3 layered double hydroxide (LDH) [69].
The presence of water influences the reaction between MgO and CO2. Reactive MgO readily reacts with water to Mg(OH)2 (Equation (6)). During the carbonation process with CO2, both the initial adsorbent MgO and Mg(OH)2 are capable of forming MgCO3. Duan and Sorescu (2010) showed that the sorption temperature is lower than the transition temperature, and thus, carbonation of Mg(OH)2 (Equation (7)) dominates. When the sorption temperature is higher than the transition temperature, carbonation of MgO occurs [70].
MgO + H2O ⇋ Mg(OH)2
Mg(OH)2 + CO2 ⇋ MgCO3 + H2O
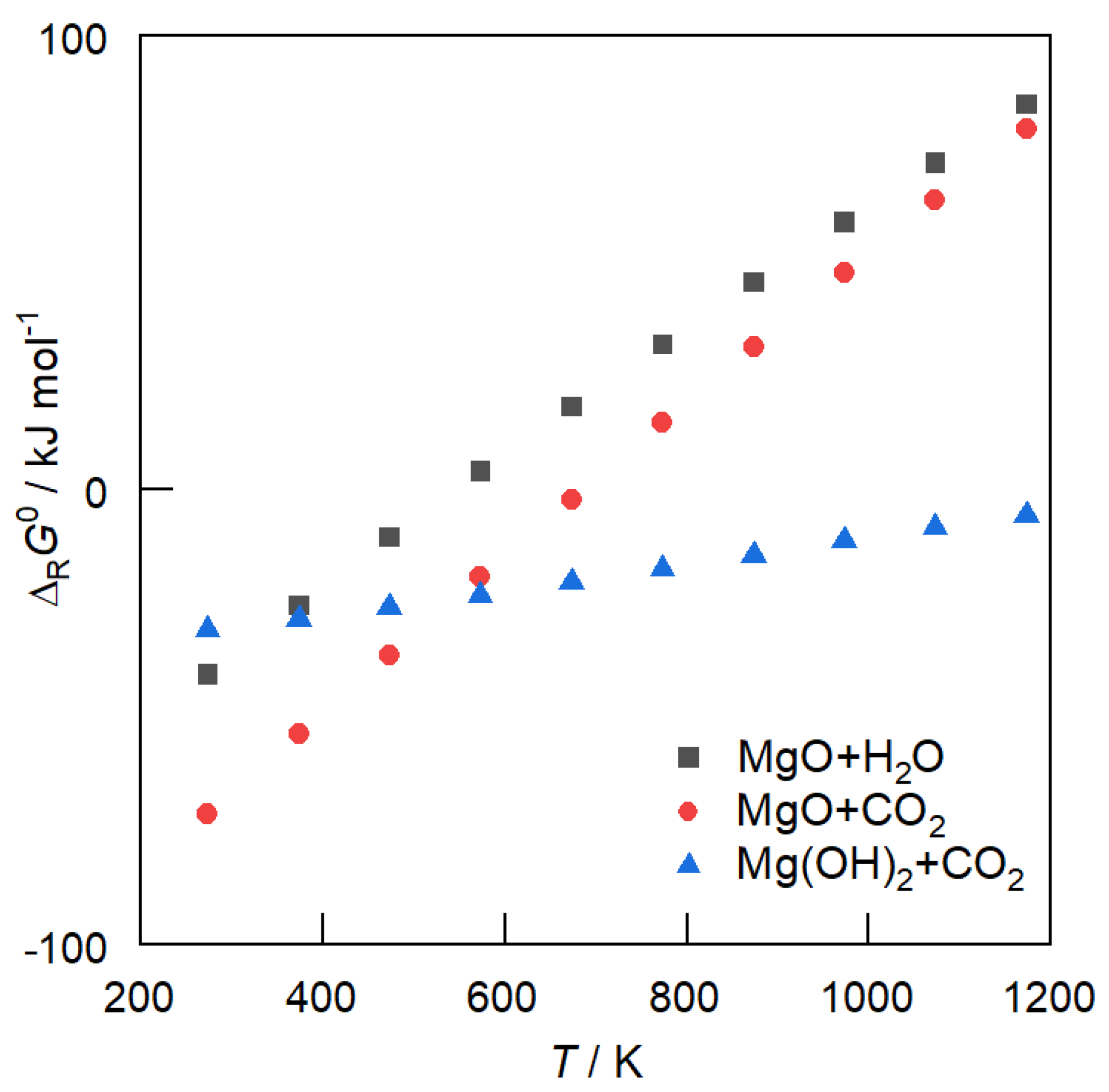
Figure 4 depicts the standard free enthalpies of reaction ΔRG0 for the reaction of CO2 with MgO (Equation (5), MgO + CO2 ⇋ MgCO3) and Mg(OH)2 (Equation (7)), and the reaction of H2O with MgO (Equation (6)) in a temperature range of 300 to 1200 K at ambient pressure. It can be seen that above 600 K, the reaction of CO2 with Mg(OH)2 is thermodynamically favored over MgO carbonation.
When water vapor is present at low system temperatures, CO2 is released from MgCO3, and Mg(OH)2 is formed instead of MgO during regeneration. Fagerlund et al. (2012) proposed a mechanism for MgO carbonation in the presence of water vapor. When MgO is surrounded by water, CO32− and H+ ions form. Free Mg2+ ions then react with CO32− forming MgCO3. It can thus be concluded that water vapor gives access to improved chemisorption interactions between MgO and CO2 [71]. However, for MgO-based adsorbents, a high water content in the sorbent system requires high energy due to the sensible heat. For MgO as a catalyst support, water may have a negative impact on the catalytically active metal species.
Wu and Goodman (1992) showed that water dissociates heterolytically on MgO surfaces [23].
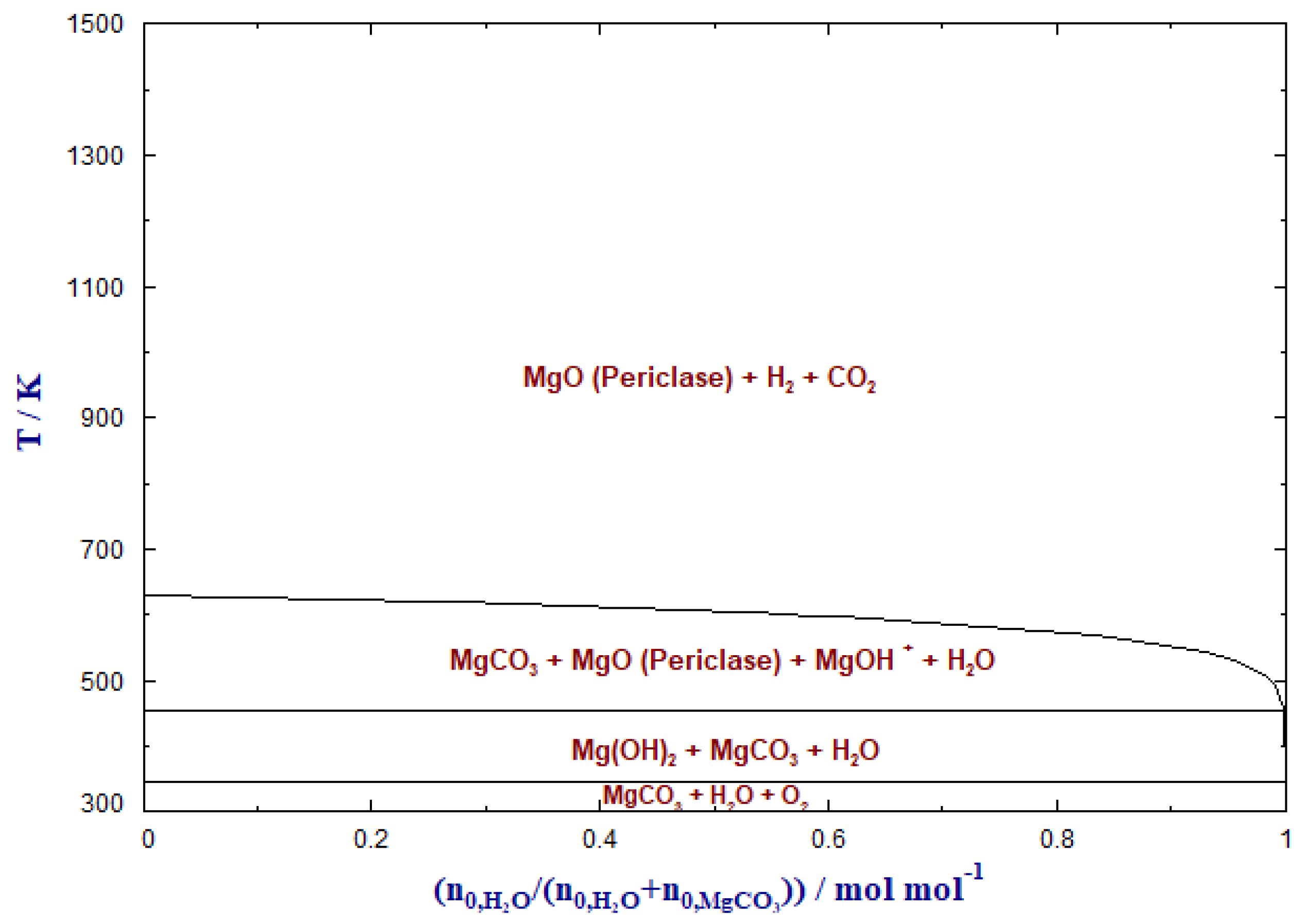
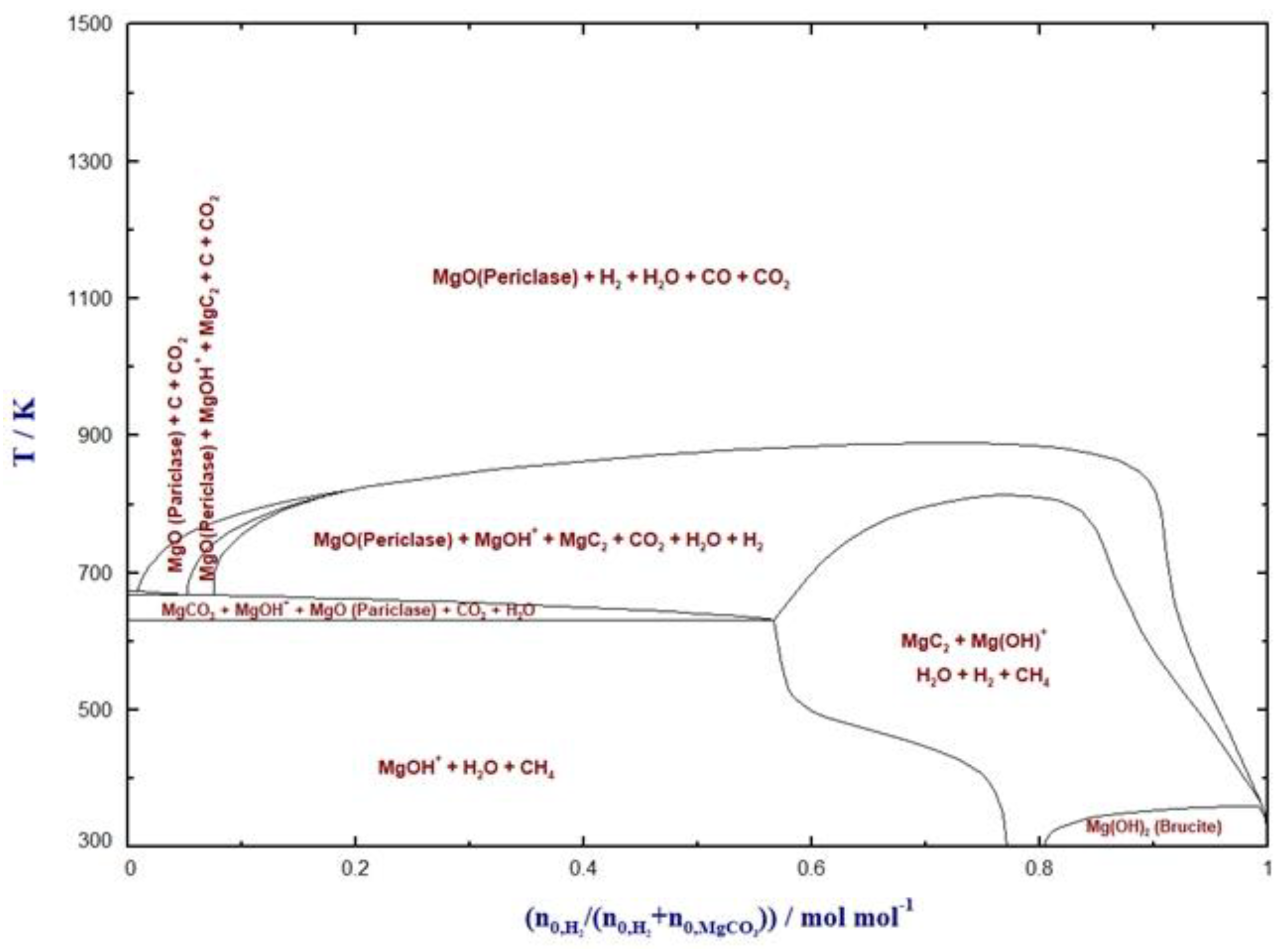
4. Hydrogenation of MgCO3
Baldauf-Sommerbauer et al. (2016) discussed the conversion of mineral magnesite to MgO, methane, CO2 and CO in a hydrogen atmosphere at ambient pressure and overpressure without additional catalysts. This process was termed reductive calcination. It was reported that low temperature and elevated pressure facilitate CH4 formation, whereas CO formation is enhanced at moderate to high temperature and low pressure. Methane is formed directly without any additional catalyst from mineral magnesite. It was stated that such reductively calcined MgO acts as a highly active reverse water–gas shift catalyst. This was confirmed by CO formation from gaseous CO2 and H2 at catalytically active, reductively calcined MgO. The reduction of CO to methane was not catalyzed by the reductively calcined MgO [20].
In another study from Baldauf-Sommerbauer et al. (2017), the reductive calcination of magnesite and dolomite to CO and methane was suggested as a means to reuse CO2 and produce value-added carbonaceous products in mineral processing. More than 70% of the CO2 emitted during the reductive calcination of the magnesite phase of a mixed magnesite/dolomite (1:1 mol/mol) sample could be converted to CO at moderate temperature without the admixture of catalysts [72].
Figure 7 depicts the phase transformation behavior of MgCO3 with H2.
5. Methane Synthesis over MgO-Supported Catalysts
For methane synthesis via CO2 hydrogenation, a large number of studies has been conducted based on noble metal catalysts such as Ru [73,74], Rh [75,76,77,78] and Pd [79,80,81] that describe the high catalytic activity and excellent stability of these catalysts. However, the high price of noble metal catalysts is a limiting factor for industrial application. Non-noble, transition-metal catalysts such as Ni-based catalysts are a promising alternative; not only as they are more cost-effective compared to noble metal catalysts but also because of their improved catalytic activity at low temperatures. In CO2 methanation, low temperatures are favorable for high equilibrium conversions. As a drawback, Ni-based catalysts often suffer from deactivation due to the sintering of Ni particles, formation of mobile nickel sub-carbonyls or formation of carbon deposits [82].
In addition to the catalytically active metal species, CO2 methanation catalysts generally consist of metal oxides as carrier materials. These may also act as adsorbents. Thus, the support material provides a crucial factor that affects both the adsorption properties of the reactants and the catalytic activity of the catalyst. In general, a high surface area is required. Common support materials for Ni-based methanation catalysts include Al2O3, SiO2, TiO2, CeO2 and ZrO2, in addition to MgO [40]. MgO has the decisive advantage that it reduces catalyst deactivation such as sintering and carbon formation [41,83,84]. Therefore, several researchers have studied the use of MgO as a carrier material for Ni-based methanation catalysts, mainly in combination with other carrier materials and promoters.
5.1. Experimental Studies for CO2 Methanation over Ni/MgO Catalysts
In the literature, Ni/MgO catalysts were successfully used for CO2 methanation. Table 1 gives a compilation of the experimental studies that include Ni-based catalysts on MgO carrier material for CO2 methanation.
Takezawa et al. (1986) studied 13% Ni/MgO catalysts for CO2 methanation that were prepared by impregnation at various preparation conditions (calcination at 673 K, 773 K, 803 K, 873 K, and 973 K with air for 3 h and reduction at 773 K, 803 K, and 873 K with hydrogen at 50 mL min−1 for 13 h). The results showed that at a high calcination temperature (973 K), the Ni(II) ions are highly dispersed in the MgO carrier and become less reducible. In addition, the activity and selectivity of the Ni/MgO catalysts decrease considerably with increasing calcination temperature (973 K). The study presented a methane selectivity of 98% for calcination at 773 K and reduction at 873 K, a feed flow rate of 100 mL min−1 (CO2:H2 = 5:95), a pressure of 1 atm and a reaction temperature of 480 K [88].
Bette et al. (2016) studied the catalytic effect of 59 wt% nickel on (Mg,Al)Ox which they derived from a (Ni,Mg,Al)-hydrotalcite-like precursor on CO2 methanation in a fixed-bed tubular quartz reactor. The catalyst was calcined at 873 K in air, and metallic Ni particles supported on a spinel-type (Mg,Al)Ox matrix were obtained through reduction at 1173 K in a mixed flow of 10% H2 in N2. The reaction temperature was varied between 523 and 623 K at ambient pressure with a WHSV (weight hourly space velocity) of 1100 mL gcat−1 min−1 (H2:CO2:Ar:N2 = 18.5:4.6:12.8:64.1). The results showed a maximum CO2 conversion of 72–76% at reaction temperatures of 603–623 K. At a reaction temperature of 528 K, the catalyst provided good long-term stability under methanation conditions for up to 50 h on stream with a stable CO2 conversion of 17–23 % and a CH4 yield of 17–19 % [89].
Guo et al. (2014) studied the catalytic behavior of 10 wt% Ni/SiO2 that was modified with 1–4 wt% MgO prepared by co-impregnation and calcination at 723 K for 3 h and a cool down to room temperature by mixed 50% H2 in N2. The methanation reaction was carried out in a fixed-bed continuous flow quartz reactor in a temperature range of 473–773 K at atmospheric pressure and a feed flow rate of 50 mL min−1 (CO2: H2: N2 = 1:4:3). They found that modification with MgO increases the dispersion of Ni species and also suppresses sintering and oxidation of metallic Ni. Furthermore, catalyst (10 wt% Ni/SiO2) modification with MgO in a quantity of as low as 1 wt% features pronounced catalytic activity and stability at 623 K for 50 h with 66.5% CO2 conversion and 96.8% methane selectivity [83].
Nakayama et al. (1997) investigated Ni/MgO catalysts (10, 30, 50, 70 and 90% Ni) by using citric acid and calcination in air at 773 K for 3 h followed by reduction in pure H2 at 773 K for 2 h. A fixed-bed flow reactor was used for hydrogenation of CO2 with a feed flow rate of 180 mL min−1 (with varied feed ratio of CO2:H2 = 1:4 and 1:8) and a temperature of 553 K at atmospheric pressure. The CO2 conversion at 553 K with 30, 50, 70 and 90% Ni-catalysts were 62%, 79%, 85% and 79%, respectively, and the methane selectivity ranged at 99–100% for all catalysts with ≥30 wt% Ni. It was suggested that aggregation of metallic Ni particles was depressed by MgO that was dispersed in the Ni particles during the reduction of the NiO-MgO solid solution [87].
Varun et al. (2020) prepared NiO/MgO nanocomposite catalysts for CO2 hydrogenation via four preparation methods. The preparation methods included sonochemical, co-precipitation, impregnation, and solution combustion methods. The reaction temperature was 448–723 K at atmospheric pressure. The catalysts prepared by the sonochemical route gave the best catalytic performance with respect to CO2 conversion and methane selectivity. They reported the highest CO2 conversion of 85% with 98% methane selectivity at 673 K with NiO/MgO catalysts and concluded that the MgO carrier promotes CO2 adsorption, which provides more interaction time for H2 and CO2 that enables methane to be formed at low temperature. They also impregnated NiO/MgO catalysts with 2% Co, Cu and Fe, whereby impregnation with Co resulted in a significantly reduced activation energy for CO2 hydrogenation to methane [85].
Baldauf-Sommerbauer et al. (2018) tested two catalysts with a Ni loading of 11 and 17 wt% on MgO. The catalysts were prepared by wet impregnation and resulted in a Ni/MgO solid solution with a cubic lattice. They performed experiments with a controlled increase of the catalyst temperature to 773 K (due to the exothermicity of the reaction) and compared them to steady-state experiments. The two experimental procedures yielded comparable results in terms of CO2 conversion and methane selectivity. It was shown that at a moderate reaction temperature of 598 K and a feed composition of H2:CO2:N2 = 4:1:5 at a flow rate of 250 mLSTP min−1, CO2 conversion and methane selectivity were near the thermodynamic equilibrium. With reactor operation for several days at a reaction temperature of 603 K, the long-term stability of Ni/MgO (17 wt% Ni) catalysts was proven [15]. Loder et al. (2020) extended this study and investigated the effect of Ni loading (0 to 27 wt%) and MgO quality on the rate of methanation in a temperature range of 533–648 K. To investigate the effect of matrix elements, a mixed MgO/CaO carrier material was tested with 21 wt% nickel loading. The reaction kinetics of CO2 methanation was modeled with a Langmuir–Hinshelwood approach describing a bifunctional character of the catalyst. Both studies show that Ni/MgO catalysts act as robust, active and highly selective catalysts for CO2 methanation, with a CO2 conversion of 87% and a methane selectivity of 99% [13].
5.2. Reaction Mechanism for CO2 Methanation
In the literature, the reaction mechanism for CO2 hydrogenation to methane is generally classified according to the initial CO2 hydrogenation step in three typical pathways: (i) CO2 direct dissociation pathway, meaning CO2 direct dissociation into CO*, (ii) formate pathway, meaning C-terminal hydrogenation of CO2 to HCOO* species, and (iii) carboxyl pathway, meaning O-terminal hydrogenation of CO2 to COOH* species [90,91].
However, regardless of the pathway, initial adsorption and activation of the reactants, CO2 and H2 are the decisive steps for the reaction. Several researchers have discussed the importance of synergistic interactions at the metal/metal oxide interface of the catalyst. Moreover, Ni/MgO catalysts have proven to exhibit bifunctional catalytic activity during CO2 hydrogenation to methane; while Ni provides the adsorbent capacity for hydrogen, MgO activates CO2 through chemisorption. For Ni/MgO catalysts the crucial role of carbonate formation via CO2 adsorption has also been pointed out [40].
To elucidate the roles of Ni and MgO and any support effects on the reaction mechanism of CO2 hydrogenation to methane, Huang et al. (2019) [40] performed DFT calculations. They investigated CO2 hydrogenation on pure Ni (111), hydrogen-assisted MgO (110) and Ni/MgO catalysts. From their calculations, they confirmed that in Ni/MgO catalysts Ni is the catalytically active site, and MgO takes over the role as promoter for CO2 hydrogenation to methane. Ni is not only responsible for the dissociation of H2 to H atoms but also provides the active site for CO2 hydrogenation. Huang et al. also reported strong metal–support interactions between Ni and MgO in Ni/MgO catalysts compared to pure Ni surfaces. This distinctly improves the Ni reducibility of the Ni/MgO surface, leading to the fact that the terminal C of CO2 gains more electrons from the Ni/MgO surface. Thus, on the Ni/MgO surface, the formate pathway via H2COO* intermediates is promoted. This differs from the methane formation on Ni (111), which follows the formate pathway via HCOOH* intermediates. Moreover, H-spillover and strong OH adsorption on the MgO carrier favor OH removal and H2O formation during CO2 hydrogenation. Huang et al. also pointed out that on the MgO (110) surface, CO2 preferentially adsorbs at the O-top site of MgO by carbonate formation. On Ni/MgO, the CO2 molecules are adsorbed at the interface of Ni/MgO and those C atoms form Ni–C bonds with a Ni atom [40].
Loder et al. (2020) studied the reaction mechanism of so-called bifunctional Ni/MgO catalysts for CO2 methanation in a temperature range of 533–648 K and atmospheric pressure. To describe the reaction kinetics, a Langmuir–Hinshelwood reaction model was developed that considers CO2 and H2 adsorption on the catalyst [13]. This supports the bifunctional catalytic activity of Ni/MgO catalysts. The postulated reaction model was based on five steps: (i) dissociation of H2 and adsorption on Ni (Equation (8)), (ii) adsorption of CO2 on the Ni-MgO interface, whereby adsorbed CO2 is attached to two active Mg sites and one active nickel site, as shown by Huang et al. (2019) (Equation (9)), (iii) reaction of adsorbed CO2 with adsorbed hydrogen (Equation (10)) to produce methane and the byproduct H2O (Equation (11)), (iv) desorption of the reaction product methane (Equation (12)), and (v) desorption of the byproduct water (Equation (13)). In the literature, the adsorption of hydrogen on Ni is described as the rate-controlling step for Ni-based catalysts [92]. As with bifunctional Ni/MgO catalysts, hydrogen (on Ni) and CO2 (on the Ni-MgO interface) are adsorbed; the availability of active nickel sites is decisive. Hence, species adsorption on Ni in general was assumed to be rate-controlling by Loder et al. (2020). As their basic aim was to develop a simple rate law with adequate accuracy, hydrogen and CO2 adsorption were combined, and it was assumed that both were rate-controlling. As postulated by Huang et al. (2019), it was assumed that methane is adsorbed on one nickel site and water on one magnesium site [13].
H2 + 2 Ni ⇌ 2 HNi
CO2 + 2 Mg + Ni ⇌ CO2(Mg)2Ni
H2 + CO2 + 2 Mg + 3 Ni ⇌ 2 HNi + CO2(Mg)2Ni
CO2(Mg)2Ni + 8 HNi ⇌ CH4 Ni + 2 H2OMg + 8Ni
CH4Ni ⇌ CH4 + Ni
H2OMg ⇌ H2O + Mg
Several researchers have mentioned carbonate formation as an intermediate step during CO2 methanation over Ni-based catalysts on various carrier materials. Pan et al. (2014) investigated Ni-based catalysts on Ce0.5Zr0.5O2 and γ-Al2O3 using in situ FTIR spectroscopy. They found similar reaction pathways for both carrier materials, which only differed in the reactive basic sites, and concluded that CO2 adsorption on medium basic sites (Ni/Ce0.5Zr0.5O2) results in monodentate carbonates, while CO2 adsorption on strong basic sites (Ni/γ-Al2O3) does not participate in the reaction. It was proposed that medium basic sites enhance the catalytic activity toward CO2 methanation as monodentate formate derived from monodentate carbonate on medium basic sites can be hydrogenated more quickly than bidentate formate derived from hydrogen carbonate. CO2 adsorbed on strong basic sites cannot desorb from the surface until high reaction temperatures and, thus, will not participate in methane formation at lower temperatures [93].
Moreover, Park and McFarland (2009) described the beneficial effect of carbonate formation during CO2 methanation over Pd-based catalysts. They prepared bifunctional Pd–Mg/SiO2 catalysts using the properties of Pd to dissociate molecular hydrogen to hydrogen atoms, which are subsequently transferred and converted with activated surface carbonate species. The carbonate species are formed by the reaction of CO2 with the Mg-containing oxide. It was reported that with carbonate-forming catalyst combinations, meaning MgO-containing catalysts, a high selectivity to methane and a corresponding higher H2 conversion was observed due to stabilization of the intermediate carbonate MeO2–CO and, thus, inhibition of CO desorption [64].
Westermann et al. (2015) studied the catalytic effect of 5–14% Ni on NiUSY and reported that increasing Ni content increases the CO2 conversion (44.9–72.6%) and methane selectivity (60–95%). The mechanism of CO2 hydrogenation on impregnated NiUSY catalysts (5–14%Ni) was also investigated over various temperatures. They stated that the occurrence of CO arises from formate (T ≤ 473 K). Methane formation is found when the system’s energy becomes high enough for CO hydrogenation or dissociation (473 ≤ T ≤ 573 K). The C-O dissociation step is the rate-determining step following HCOO−(ads) → CO(ads) → CH4(g). High CO concentrations are formed on the surface and limit CO hydrogenation and dissociation (573 ≤ T ≤ 623 K). At 573 K, the maximum CO concentration is reached, which limits formate dissociation regarding a Langmuir–Hinshelwood mechanism. As methane concentrations increase along with increasing temperature, it can be assumed that formate can be directly hydrogenated to methane. Due to the vacancy of new active sites, formate is hydrogenated to methane. In addition, CO is obtainable for hydrogenation and dissociation with decreasing CO concentration and increasing methane concentration (T ≥ 623 K). Methane formation is limited by thermodynamics and the rate-determining step (dissociation/hydrogenation of CO). From the studies at different temperatures, Westermann et al. (2015) also proposed a mechanism with NiUSY catalysts, in which initiation occurs via two options: (i) CO2 adsorbs onto exchangeable cations or (ii) monodentate onto EFAL/Ni species. Then, dissociated hydrogen reacts to monodentate formate, which is adsorbed onto Ni (T = 423 K). At 573 K, formate dissociates to carbonyl and methane with a low rate of methane formation. An increase in Ni loading results in an increase in adsorption sites, generating more adsorbed formate species. From the study of zeolites, it was concluded that carbonate is not likely to be directly hydrogenated to methane but mainly via formate formation and dissociation to carbonyls [94].
In addition to Ni-based catalysts on MgO, Kim et al. (2010) suggested a reaction mechanism for CO2 methanation over MgO-supported Pd-based catalysts (Pd-MgO/SiO2) elucidating the decisive role of carbonate formation. The Pd-MgO/SiO2 catalysts were prepared by a reverse microemulsion (ME) method. The reaction mechanism depicts the role of Pd and MgO in CO2 methanation and shows that MgO enables magnesium carbonate formation on the surface. This step adopts a critical role in CO2 methanation. Pd can dissociate hydrogen molecules that are responsible for consecutive magnesium carbonate hydrogenation to methane. This mechanism focuses on the following steps on MgO surfaces: (i) CO2 adsorption occurs on the MgO surface and exposes carbonate on MgO to hydrogen atoms. The oxygen species of carbonate are then hydrogenated with two hydrogen molecules whereby water is formed. (ii) The residual C on the MgO surface is hydrogenated with hydrogen to form methane [41].
The formation of carbonate species during CO2 methanation was also reported by Arellano-Treviño et al. (2019). They investigated the effect of different metal loading on MgO carrier material for three different catalysts including Ni-based catalysts (5% Ru, 0.5% Rh, 10% Ni). They also used Na2O, CaO, and K2O as carrier material. The methanation reaction with 5% Ru_10% MgO/Al2O3 catalysts was performed in a packed-bed reactor at a temperature of 593 K and atmospheric pressure. The feed flow rate was 30 mL min−1 (CO2/N2 = 10:90) plus 30 mL min−1 (H2:N2 = 15:85). With MgO/Al2O as the carrier material, 91% CO2 conversion was achieved showing that MgO/Al2O3 is a suitable carrier material for low-temperature methanation (T < 423 K). This may be dedicated to the fact that bicarbonate and bidentate carbonate (bonds between CO2 and MgO/Al2O3) can be active at low temperatures with both carbonate sites increasing the catalyst activity. However, infrared studies of the Ni catalysts showed that also inactive carbonate species were formed on the Ni surface that resulted in unreacted adsorbed CO2 [95].
Baldauf-Sommerbauer et al. (2016) reported methane formation through hydrogenation of mineral magnesite in a fixed-bed tubular reactor with a flow rate of 500 mL min−1 (H2: N2 = 9:1) in a temperature range of 748 K, 763 K and 778 K, and a pressure of 0.3–1.2 MPa. It was shown that with increasing MgCO3 conversion, the methane yield increased from 4.9 to 36.6% at 748 K, from which it was assumed that the dissociation of carbonates has a positive effect on methane formation [20]. This is in accordance with the findings and conclusion of Arellano-Treviño et al. [95].
Interestingly, carbonate formation was also mentioned for Ni-based catalysts on SiO2 carrier material, whereby it must be noted at this point that SiO2 normally reacts acidically. Xu et al. (2021) studied CO2 methanation over Ni/SiO2 catalysts that were prepared by a combustion-impregnation method and reported 66.9% CO2 conversion and 94.1% selectivity to methane at 593 K. These results were superior than CO2 conversion and methane selectivity with Ni/SiO2 catalysts that were prepared by the conventional impregnation method. They proposed a reaction pathway in which CO2 adsorption occurs on the support sites that are located close to the metal–support interface enabling carbonate, hydroxycarbonate or carbonyl formation. Xu et al. interestingly described basic sites of Ni/SiO2 catalysts that were classified as weak basic sites (bicarbonate) at 323–573 K, medium basic sites (bidentate) at 473–673 K and strong basic sites (monodentate carbonate) at 673–1073 K. They stated that more medium basic sites which were beneficial for the CO2 activation could be created by the combustion-impregnation method. The carbonate species were then hydrogenated to yield methane [96].
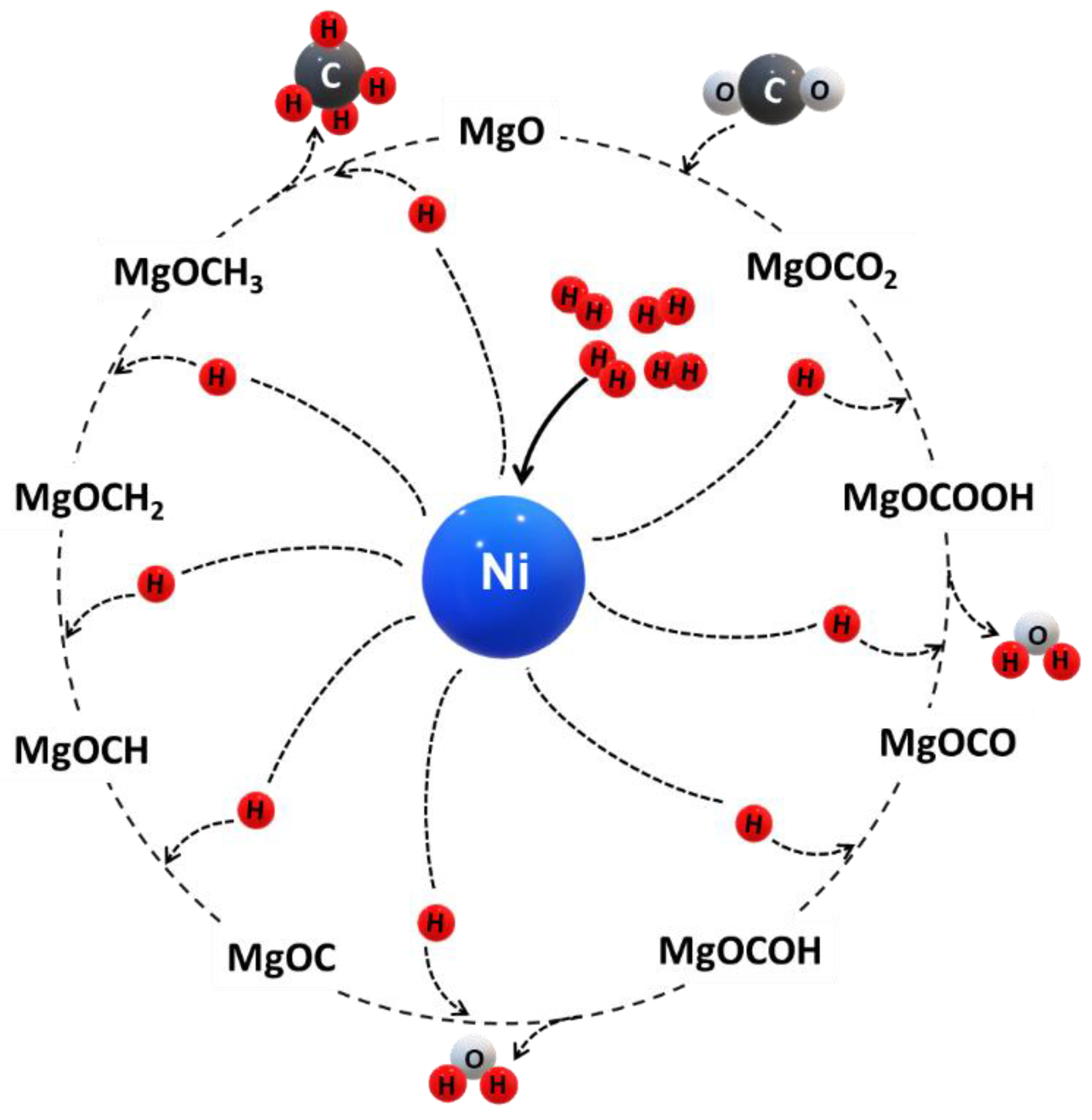
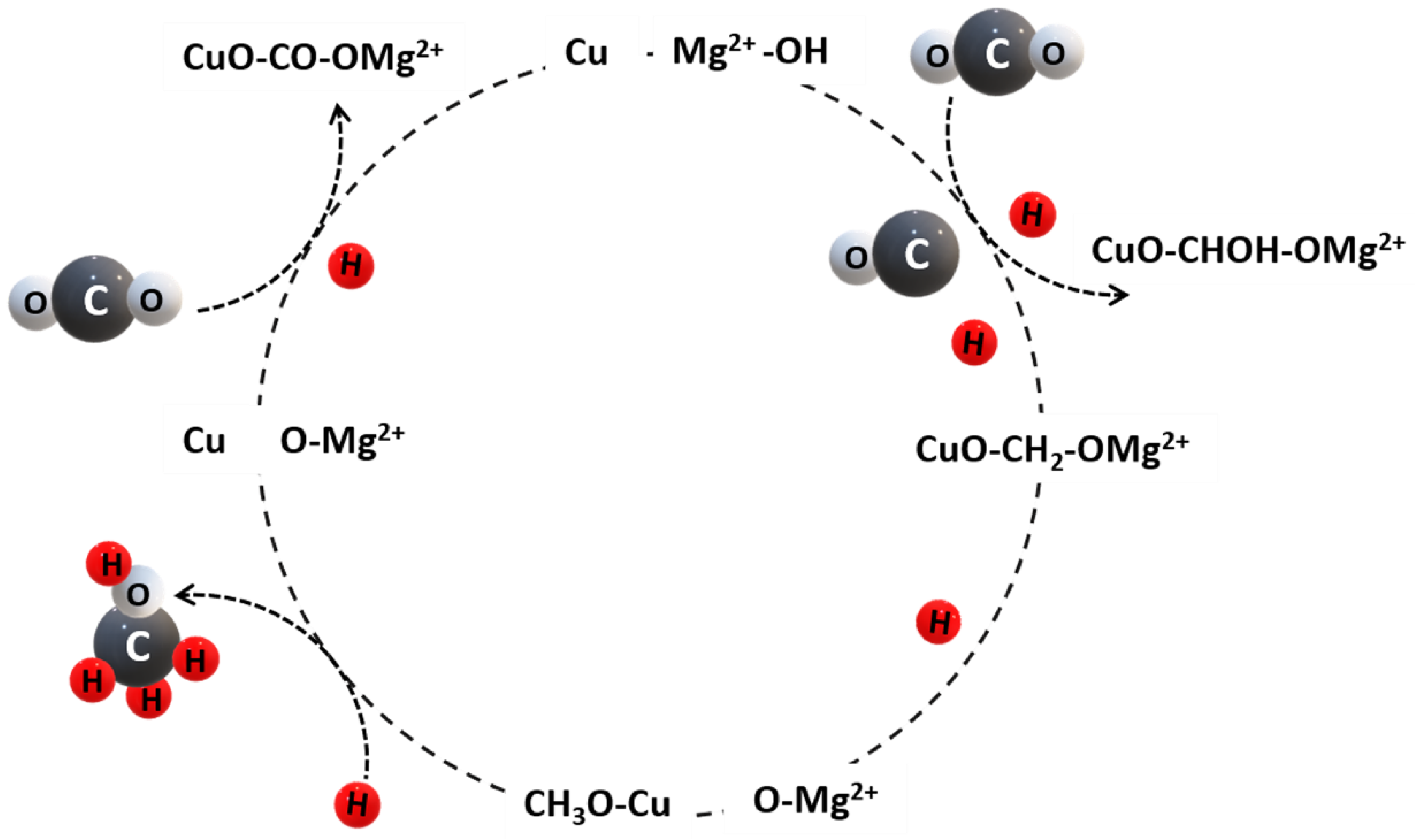
To conclude, the above-mentioned publications elucidate the crucial role of a basic support material enabling carbonate formation through CO2 adsorption during CO2 methanation over Ni-based catalysts. Following the reaction mechanism postulated by Park and McFarland (2009) for a highly dispersed Pd–Mg/SiO2 catalyst, a similar reaction mechanism is conceivable for CO2 methanation over Ni/MgO catalysts (Figure 8). The mechanism is based on the following steps [64]:
- (i)
- CO2 is stabilized by MgO via CO intermediates forming carbonate at the surface. The carbonate formation has a critical role in CO2 methanation. CO dissociation may be rate-determining.
- (ii)
- The carbonate is sequentially hydrogenated producing a carboxy group and water as byproduct.
- (iii)
- The carboxy group reacts with MgO to form MgCOO and is sequentially hydrogenated forming MgCOOH, whereas one molecule of water is generated.
- (iv)
- Hydrogenation with three hydrogen atoms on the MgO surface forming MgOC—MgOCH—MgOCH2—MgOCH3, respectively.
- (v)
- MgOCH3 is hydrogenated with a hydrogen atom to form MgO and methane.
6. Methanol Synthesis over MgO-Supported Catalysts
Various catalysts for methanol synthesis from CO2 have been developed and intensively investigated over the last decades. Analogous to CO2 methanation, the main influencing factors for the catalytic activity, stability and selectivity of the catalysts are the process conditions for the reaction, the preparation method of the catalyst, the choice of the catalytically active material, the catalyst carrier material, and the use of promoters. The target of optimum process conditions, such as temperature, pressure, feed gas composition and flow rate, the amount of catalyst, and continuous or batch operation mode is controlled by the thermodynamics and the kinetics of the reaction. The choice of carrier material, additional promoters, and the preparation method affect catalyst parameters such as particle size, surface area, distribution of the catalytically active metal, acidity and basicity of the carrier material, and temperature and pressure stability of the catalyst. In general, catalysts for methanol synthesis by CO2 hydrogenation can be categorized as follows: Cu-based catalysts, noble metal-based catalysts (Pd, Pt), oxygen-deficient catalysts (In2O3), and bimetallic catalysts (Ni-Ga, Au-Ag) [97,98].
Cu-based catalysts have attracted research interest over the past years, and they are already industrially applied, mainly with the carrier material Al2O3 and the promoter ZnO. However, the bottleneck of Cu/ZnO/Al2O3 catalysts is their deactivation behavior and the relatively low selectivity for methanol because of byproduct formation [99,100,101].
In Cu-catalyzed synthesis of methanol by CO2 hydrogenation, the nature of the carrier material has a pronounced effect on the reaction mechanism [102]. The catalytic activity linearly correlates with the metallic Cu0 surface [103,104], indicating that the reaction takes place at the metallic Cu0 surface [6]. Several studies have shown that the admixture of MgO as an additional promoter increases CuO dispersion, the metallic Cu0 BET surface area and the active basic sites for improved CO2 and also H2 adsorption [6,41,105,106,107,108,109,110].
6.1. Experimental Studies for CO2 Hydrogenation to Methanol over Cu-Based Catalysts with MgO
In the literature, various studies are presented with Cu-based catalysts using MgO either as a carrier material or as promoter (Table 2). However, compared to CO2 methanation, the use of MgO as a sole carrier material is rarely suggested.
To hinder agglomeration and increase the surface area of Cu-based catalysts, alkaline earth metal oxides are mentioned as attractive materials for promoters and catalyst support material [111,112]. Dasireddy et al. (2018) evaluated the effect of alkaline earth metal oxides (MgO, CaO, SrO and BaO) on Cu/Al2O3 catalysts in methanol synthesis by CO2 hydrogenation and compared the results with commercially available Cu/ZnO/Al2O3 catalysts. They reported that according to their electronegativity, the basicity of the catalysts increased in the order of Zn < Mg < Ca < Sr < Ba. The introduction of alkaline earth metal oxides enhanced the interaction between Al2O3 and CuO, which resulted in a weaker reducibility of CuO. The Cu+:Cu0 ratio and Cu0 surface area were higher in all alkaline-earth-metal-containing catalysts, compared to the Cu/ZnO/Al2O3 catalyst, and increased in the order of Ba < Ca < Zn < Sr < Mg. A higher Cu+:Cu0 ratio as well as a higher Cu0 surface area were stated as significant factors for the enhanced CO2 conversion. Moreover, all catalysts showed an enhanced activity in methanol synthesis with increasing GHSV. Best results were obtained with the Cu/MgO/Al2O3 catalyst, which showed an increased number of active sites for CO2 and H2 adsorption. The catalytic performance exceeded even the commercially available HFRI20 and LURGI catalysts [113].
Ren et al. (2015) investigated the promoting effect of ZnO, ZrO2 and MgO on a Cu/γ-Al2O3 catalyst. The introduction of the metal oxides increased the Cu0 dispersion, the Cu0 surface area and decreased the Cu0 particle size. While the promotional effect of individually added ZnO and ZrO2 on CO2 hydrogenation to methanol was marginal, the simultaneous addition of both oxides increased the CO2 conversion and methanol selectivity significantly. Even better results were achieved with the Cu/ZnO/ZrO2/MgO/γ-Al2O3 catalyst. The optimal ratio of Mg:Zr was observed to be 1:9. A significant increase in STY (space–time yield) was observed for increasing GHSV. The optimal activation temperature of the catalyst was reported to be the hydrogenation temperature. Though lower activation temperatures form smaller Cu0 particles and a higher Cu0 surface area, particles seem to agglomerate if the reaction temperature exceeds the activation temperature [105].
Chatterjee et al. (2019) investigated support interactions in Cu-based catalysts and reported of (strong) metal–support interactions (SMSI). They noted that Cu-supported MgO behaves very much like Cu/Al2O3 and Cu/Ga2O3 [9]. Baseline experiments with pure Cu were in accordance with experimental results from Fichtl et al. (2014) [114], but the results with Cu/MgO were contrary to the ones from Fichtl et al. However, it was speculated that the differences were caused by the preparation methods, which elucidates the critical role of the catalyst preparation method and conditions, and the calcination temperature.
A comprehensive review of Cu-based CO2 hydrogenation to methanol giving insights from experimental work and theoretical analysis was presented by Niu et al. (2022). They investigated structural and surface properties of Cu-based catalysts for methanol synthesis. A stronger inverse kinetic isotope effect of H/D substitution for methanol synthesis compared to CO formation was observed, suggesting that methanol synthesis and CO formation do not share a common intermediate, which indicates that CO2 hydrogenation to methanol is not a combination of the RWGS reaction and CO hydrogenation. CO2 acts as dominant carbon source on Cu0 surfaces, while CO is the dominant carbon source on Cu+. The reaction rate of CO2 hydrogenation was reported to be faster than that of CO hydrogenation. H2 dissociation was described to proceed on the Cu surface, while CO2 adsorption is more favored to occur on the support or the interface [115].
Liu et al. (2016) investigated MgO promotion of Cu/TiO2 catalysts. MgO promotion resulted in a larger specific surface area of TiO2 and increased Cu dispersion. However, an excess of MgO covered the surface of TiO2 and thereby decreased Cu dispersion [108].
Cu-based catalysts are not only the most commonly used catalysts in methanol synthesis from CO2 but are also good candidates for the water–gas shift reaction and the reverse water–gas shift reaction. Thus, there is still a lively debate about the dominant carbon source for methanol synthesis; CO or CO2. It is well known that both CO and CO2 can undergo hydrogenation to methanol, and thus, Robbins et al. (1991) suggested that the dominant carbon source depends on the valence state of the surface Cu. CO2 acts as the dominant carbon source on the Cu0 surface, while CO is the dominant carbon source on the Cu+ surface. In addition, it was found that the reaction rate of CO2 hydrogenation was faster than that of CO [104]. These findings were confirmed by Nielsen et al. (2020) [6], suggesting that methanol is mainly generated via CO2 hydrogenation from the CO/CO2/H2 mixtures.
Cao et al. (2021) gave in his study further insights on CO and CO2 hydrogenation for methanol synthesis and described the key role of adsorbate–adsorbate interactions on Cu and the highly active MgO-Cu interface. They investigated two questions: (i) which of the CO or CO2 hydrogenation routes dominates the methanol synthesis rate, and (ii) what makes irreducible, inert oxides such as MgO efficient promoters for the CO hydrogenation process. The MgO/Cu interface was reported to act as a highly active site proposing a novel lattice oxygen-involved reaction mechanism at the interface for methanol formation. Moreover, formate coverage effects and hydrogen bond formation can significantly lower the activation barrier of the CO2 hydrogenation [116].
Yang et al. (2005) performed a mechanistic study of a novel low-temperature methanol synthesis route on Cu/MgO catalysts from syngas using ethanol as a promoter [110].
The role of the oxide component in the development of Cu-based composite catalysts for methanol synthesis from CO, CO/CO2 mixtures and CO2 was discussed by Zander et al. (2013). It was found that ZnO acts as a geometrical spacer between the Cu nanoparticles and helps to increase and stabilize the Cu dispersion. Thus, ZnO was reported to have two functions in the final catalyst: (i) as nanoparticles, ZnO acts as a physical spacer between the Cu particles, stabilizing the porous microstructure; and (ii) as a thin layer at the surface of the Cu particles, ZnO is an essential ingredient for the active site, and its presence has been shown to affect the adsorption properties. Cu/MgO catalysts showed a slightly smaller BET surface area with smaller crystallites but a 20% higher Cu surface area. It was stated that MgO is an intrinsically better geometrical spacer compared to ZnO as, even at a non-ideal ratio (80:20), Cu particles could be obtained that, with an average size below 10 nm, were similarly small as those found in state-of-the-art Cu/ZnO/Al2O3 catalysts. Thus, it was concluded that the structurally promoting role of ZnO had been successfully replaced by MgO. However, in the hydrogenation of pure CO2, Cu/ZnO showed a much higher activity than Cu/MgO, showing clearly that the methanol synthesis rate was not only a function of the exposed Cu surface area. The low activity of Cu/MgO in CO2 hydrogenation was explained by the absence of the synergetic SMSI effect as MgO is an irreducible oxide that does not show the necessary SMSI in the relevant temperature regime. The addition of 5% ZnO to Cu/MgO increased the BET surface area, the Cu-specific surface area and crystallite size, and also promoted methanol synthesis significantly without promoting the reverse water–gas shift reaction [109].
Kleiber et al. (2022) proposed Cu/MgO catalysts in a novel process concept for direct reduction of siderite ore combined with catalytic CO/CO2 hydrogenation to methanol [14]. In their first study (Kleiber et al. (2021) [117]), preliminary results of the activity of a 38 wt% Cu/MgO catalyst in a semi-continuous tank reactor were presented emphasizing that the catalyst support material MgO was calcined at a low calcining temperature of 823 K. For the chosen calcining conditions, MgO is highly active with respect to CO2 adsorption. Good catalytic activity in CO2 hydrogenation and a high selectivity for methanol were shown for the Cu/MgO catalyst. In repeated cycles with continuous product condensation followed by reactant re-dosing, an overall relative CO2 conversion of 76% and methanol selectivity of 59% were obtained. The maximum methanol selectivity in a single cycle was 88%. However, the long-term stability of the Cu-MgO catalyst was not investigated in this preliminary study.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 2.
Experimental studies using Cu-based catalysts on MgO carrier material for CO2 hydrogenation to methanol.
Table 2.
Experimental studies using Cu-based catalysts on MgO carrier material for CO2 hydrogenation to methanol.
| Catalyst Composition | Preparation Method | Operation Conditions | Performance | Comments | Ref. |
|---|---|---|---|---|---|
| CuO/MgO (wCu = 38 wt%) | impregnation | T = 573 K p = 50 bar v(CO2):v(H2) = 1:3 | XCO2 = 23–29% YCH3OH ~ 28% (after 53 h) SCH3OH ~ 88% (after 53 h) XCO2 = 76% SCH3OH = 59% |
Semi-continuous operation | [117] |
| Cu/MgO | DFT calculations | T = 500–600 K PH2 = 30 bar, PCO2 = PCO = 10 bar | - | [116] | |
| Cu/MgO (wCu = 50 wt%) | precipitation |
T
= 523 K p = 50 bar, H2/CO2/inert = 68/3/29 | YCH3OH = ~20% | [6] | |
| Cu/MgO/Al2O3 Cu:MgO:Al2O3 = 50:30:20 | co-precipitation |
T
= 523 K p = 20 bar n(H2): n(CO2) = 3:1 GHSV = 2000 h−1 and 6000 h−1 | XCO2,2000h−1 = ~3% CH3OH2000h−1 = 0.80 molCH3OH kg−1 h−1 CH3OH6000h−1 = 1.48 molCH3OH kg−1 h−1 TOFCH3OH = 11.9 × 10−4 s−1 | [113] | |
| Cu-ZnO-ZrO2-MgO/Al2O3 | impregnation |
T
= 523 K, p = 20 bar, n(H2): n(CO2) = 3:1, GHSV = 1400 h−1 | XCO2 = 12.1% SCH3OH = 36.0% SCH4 = 2.4% SCO = 61.61% STY (31.0 g kgCat−1 h−1) | mCuO/MgO = 5 g Cu:Zn:Zr:Mg = 2: 1: 0.9: 0.1 | [105] |
6.2. Reaction Mechanism for CO2 Hydrogenation to Methanol over Cu-Based Catalysts
Detailed investigations of the reaction mechanisms of CO2 hydrogenation to methanol are available in the literature (e.g., for Cu/ZnO/Al2O3 [118], Cu/Al2O3 [119], Cu/MgO [116], CuO/CeO2/ZrO2 [120]). The so-called formate pathway is a potential pathway for methanol synthesis from CO2:
CO2* → HCOO* → HCOOH* → CH3O2* → CH2O* → CH3O* → CH3OH*
Tabatabaei et al. (2006) reported that CO2 adsorption on Cu/ZnO catalysts forms a formate species (HCO2−) that is displaced by monodentate at an anion vacancy. Moreover, bidentate formate was found on Zn-terminated ZnO surfaces at 530 K [121]. Similarly, Shido and Iwasawa (1993) also reported bidentate formate formation on ZnO [122]. Furthermore, Nielsen et al. (2020), who studied the mechanism of CO and CO2 hydrogenation to methanol on bifunctional Cu-based catalysts, described that the bifunctional catalysts combine the effect of copper and basic oxide with (i) basic oxide sites that activate CO as formate at the metal/oxide interface, and (ii) metal-assisted hydrogenation of the interfacial formate species. The results for the CO/H2 feed showed formate and methoxide formation at 523 K and methanol formation at 423 K over Cu/MgO catalyst. The majority of formate species on MgO were less reactive and more stable, which is pronounced for methanol production, and less displaced by carbonate [6].
Regarding the formate pathway, carbonate formation has also been considered and critically discussed for Cu-based catalysts. With Cu/MgO catalysts for CO2/CO/H2 feed streams, the presence of CO2 was reported to create carbonate and bicarbonate species (the formate species react at the metal/oxide interface and are displaced by carbonate) and, thus, deteriorates methanol production.
Nielsen et al. (2020) concluded that the presence of CO2 in mixed CO/CO2 feed streams has a negative effect for basic oxide sites and, thus, the bifunctional interaction results in a deactivation effect of CO2 on Cu/MgO catalysts due to carbonate blocking [6]. This agrees well with the findings of Cao et al. (2021) who discussed the adsorbate–adsorbent interaction through microkinetic modeling. They described formate formation at the surface that was poisoned and reduced the accessible sites for other intermediates, leading to low methanol synthesis rates [116].
To conclude, the above-mentioned, sometimes contradictory, findings discussed above point out that carbonate formation has a decisive role when MgO is used as support material for metal-based catalysts. However, carbonate formation does not always have a positive effect. It may also result in blocking and, for CO2 hydrogenation, limits methanol formation. Following the reaction mechanism postulated by Nielsen et al. (2020) [6] for supported Cu catalysts, the reaction mechanism depicted in Figure 9 is conceivable for CO2 and CO hydrogenation to methanol over Cu/MgO catalysts, indicating the deactivating effect of CO2. It is suggested that with CO2, the basic magnesium sites are blocked as inactive carbonate species.
7. Conclusions
In this review, the role of MgO as support material for Ni-based catalysts for methane synthesis and Cu-based catalysts for methanol synthesis was discussed. Due to its basicity, the alkaline earth metal oxide MgO shows pronounced CO2 adsorption capacity giving access to bifunctional catalysts via carbonate formation and, thus, CO2 activation. This has been intensively described for CO2 methanation. A similar advantageous role would also be conceivable for methanol synthesis from CO2. However, a disadvantageous effect is described in the literature. It can be assumed that carbonate formation and, thus, strongly bound CO2 is somehow deactivating the catalyst for methanol synthesis. Be that as it may, the results in the literature are not totally consistent. It can be assumed that the type of catalyst preparation and the preparation conditions are also decisive for the role of carbonate formation in methanol synthesis and therefore greater attention should be paid to the calcination temperature in future studies.
Author Contributions
Conceptualization, S.L.; methodology, S.L.; data curation, K.S., S.K. and S.L.; writing—original draft preparation, K.S., S.K. and S.L.; writing—review and editing, S.L.; visualization, K.S.; supervision, S.L.; project administration, S.L. All authors have read and agreed to the published version of the manuscript.
Funding
Supported by TU Graz Open Access Publishing Fund.
Data Availability Statement
The data presented in this study are available in the cited references.
Acknowledgments
The authors gratefully acknowledge the support from the NAWI Graz program.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- International Energy Agency. Global Energy Review: CO2 Emissions in 2021. 2022. Available online: https://www.iea.org/reports/global-energy-review-co2-emissions-in-2021-2 (accessed on 21 March 2023).
- Topham, S.; Bazzanella, A.; Schiebahn, S.; Luhr, S.; Zhao, L.; Otto, A.; Stolten, D. Carbon Dioxide. Ullmann’s Encycl. Ind. Chem. 2014, 1–43. [Google Scholar] [CrossRef]
- Wei, W.; Jinlong, G. Methanation of carbon dioxide: An overview. Front. Chem. Sci. Eng. 2011, 5, 2–10. [Google Scholar] [CrossRef]
- Guil-López, R.; Mota, N.; Llorente, J.; Millán, E.; Pawelec, B.; Fierro, J.L.G.; Navarro, R.M. Methanol synthesis from CO2: A Review of the latest developments in heterogeneous catalysis. Materials 2019, 12, 3902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Nie, X.; Guo, X.; Song, C.; Chen, J.G. Recent advances in carbon dioxide hydrogenation to methanol via heterogeneous catalysis. Chem. Rev. 2020, 120, 7984–8034. [Google Scholar] [CrossRef]
- Nielsen, N.D.; Thrane, J.; Jensen, A.D.; Christensen, J.M. Bifunctional synergy in CO hydrogenation to methanol with supported Cu. Catal. Lett. 2020, 150, 1427–1433. [Google Scholar] [CrossRef]
- Joo, O.-S.; Jung, K.-D.; Moon, I.; Rozovskii, A.Y.; Lin, G.I.; Han, S.-H.; Uhm, S.-J. Carbon dioxide hydrogenation to form methanol via a reverse-water-gas-shift reaction (the CAMERE process). Ind. Eng. Chem. Res. 1999, 38, 1808–1812. [Google Scholar] [CrossRef]
- Goehna, H.; Koenig, P. Producing methanol from CO2. ChemTech 1994, 24. Available online: https://www.osti.gov/biblio/7157792 (accessed on 21 March 2023).
- Chatterjee, R.; Kuld, S.; van den Berg, R.; Chen, A.; Shen, W.; Christensen, J.M.; Jensen, A.D.; Sehested, J. Mapping support interactions in copper catalysts. Top. Catal. 2019, 62, 649–659. [Google Scholar] [CrossRef]
- Shen, L.; Xu, J.; Zhu, M.; Han, Y.-F. Essential Role of the Support for Nickel-Based CO2 Methanation Catalysts. ACS Catal. 2020, 10, 14581–14591. [Google Scholar] [CrossRef]
- Julkapli, N.M.; Bagheri, S. Magnesium oxide as a heterogeneous catalyst support. Rev. Inorg. Chem. 2016, 36, 1–41. [Google Scholar] [CrossRef]
- Jensen, M.B.; Pettersson, L.G.M.; Swang, O.; Olsbye, U. CO2 sorption on MgO and CaO surfaces: A comparative quantum chemical cluster study. J. Phys. Chem. B 2005, 109, 16774–16781. [Google Scholar] [CrossRef] [PubMed]
- Loder, A.; Siebenhofer, M.; Lux, S. The reaction kinetics of CO2 methanation on a bifunctional Ni/MgO catalyst. J. Ind. Eng. Chem. 2020, 85, 196–207. [Google Scholar] [CrossRef]
- Kleiber, S.; Loder, A.; Siebenhofer, M.; Böhm, A.; Lux, S. Direct reduction of siderite ore combined with catalytic CO/CO2 hydrogenation to methane and methanol: A technology concept. Chem. Ing. Tech. 2022, 94, 701–711. [Google Scholar] [CrossRef]
- Baldauf-Sommerbauer, G.; Lux, S.; Aniser, W.; Bitschnau, B.; Letofsky-Papst, I.; Siebenhofer, M. Steady-state and controlled heating rate methanation of CO2 on Ni/MgO in a bench-scale fixed bed tubular reactor. J. CO2 Util. 2018, 23, 1–9. [Google Scholar] [CrossRef]
- Bowles, J.F.W. (Ed.) Encyclopedia of Geology, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2021; ISBN 9780081029091. [Google Scholar]
- Seeger, M.; Otto, W.; Flick, W.; Bickelhaupt, F.; Akkerman, O.S. Magnesium compounds. Ullmann’s Encycl. Ind. Chem. 2011. [Google Scholar] [CrossRef]
- Amodeo, J.; Merkel, S.; Tromas, C.; Carrez, P.; Korte-Kerzel, S.; Cordier, P.; Chevalier, J. Dislocations and plastic deformation in MgO crystals: A review. Crystals 2018, 8, 240. [Google Scholar] [CrossRef] [Green Version]
- European Commission. Best Available Techniques (BAT) Reference Document for the Production of Cement, Lime and Magnesium Oxide, Industrial Emissions Directive 2010/75/EU. Available online: https://eippcb.jrc.ec.europa.eu/sites/default/files/2019-11/CLM_Published_def_0.pdf (accessed on 21 March 2023).
- Baldauf-Sommerbauer, G.; Lux, S.; Aniser, W.; Siebenhofer, M. Reductive Calcination of Mineral Magnesite: Hydrogenation of Carbon Dioxide without Catalysts. Chem. Eng. Technol. 2016, 39, 2035–2041. [Google Scholar] [CrossRef]
- Védrine, J.C. (Ed.) Metal Oxides in Heterogeneous Catalysis; Elsevier: Paris, France, 2018. [Google Scholar]
- Di Cosimo, J.I.; Díez, V.K.; Ferretti, C.; Apesteguía, C.R. Basic Catalysis on MgO: Generation, Characterization and Catalytic Properties of Active Sites; Royal Society of Chemistry: London, UK, 2014. [Google Scholar]
- Wu, M.-C.; Goodman, D.W. Acid/base properties of MgO studied by high resolution electron energy loss spectroscopy. Catal. Lett. 1992, 15, 1–11. [Google Scholar] [CrossRef]
- Kumar, S.; Saxena, S.K. A comparative study of CO2 sorption properties for different oxides. Mater. Renew. Sustain. Energy 2014, 3, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Abedin, A.H.; Rosen, M.A. A critical review of thermochemical energy storage systems. Open Renew. Energy J. 2011, 4, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Yu, C.; Lu, J.; Wei, X.; Wang, W.; Pan, G. Enhanced CO2 adsorption of MgO with alkali metal nitrates and carbonates. Appl. Energy 2020, 263, 114681. [Google Scholar] [CrossRef]
- Han, X.; Wang, L.; Ling, H.; Ge, Z.; Lin, X.; Dai, X.; Chen, H. Critical review of thermochemical energy storage systems based on cobalt, manganese, and copper oxides. Renew. Sustain. Energy Rev. 2022, 158, 112076. [Google Scholar] [CrossRef]
- Kyaw, K.; Matsuda, H.; Hasatani, M. Applicability of carbonation/decarbonation reactions to high-temperature thermal energy storage and temperature upgrading. J. Chem. Eng. Japan 1996, 29, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Pardo, P.; Deydier, A.; Anxionnaz-Minvielle, Z.; Rougé, S.; Cabassud, M.; Cognet, P. A review on high temperature thermochemical heat energy storage. Renew. Sustain. Energy Rev. 2014, 32, 591–610. [Google Scholar] [CrossRef] [Green Version]
- Müller, D.; Knoll, C.; Gravogl, G.; Artner, W.; Werner, A.; Welch, J.M.; Harasek, M.; Miletich, R.; Weinberger, P. Low-temperature carbonatization of metal oxides. Energy Procedia 2019, 158, 4870–4881. [Google Scholar] [CrossRef]
- Shkatulov, A.; Miura, H.; Kim, S.T.; Zamengo, M.; Harada, T.; Takasu, H.; Kato, Y.; Aristov, Y. Thermochemical storage of medium-temperature heat using MgO promoted with eutectic ternary mixture LiNO3-NaNO3-KNO3. J. Energy Storage 2022, 51, 104409. [Google Scholar] [CrossRef]
- Zhang, L.; Zheng, Y.; Guo, Y.; Bai, S.; Song, M.; Huang, P.; Wei, X.; Sun, J.; Li, C.; Zhang, J.; et al. Alkali metal nitrates promoted MgO composites with high CO2 uptake for thermochemical energy storage. ACS Appl. Energy Mater. 2021, 4, 9513–9524. [Google Scholar] [CrossRef]
- Flegkas, S.; Birkelbach, F.; Winter, F.; Groenewold, H.; Werner, A. Profitability analysis and capital cost estimation of a thermochemical energy storage system utilizing fluidized bed reactors and the reaction system MgO/Mg(OH)2. Energies 2019, 12, 4788. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Zhou, T.; Gao, Y.; Louis, B.; O’Hare, D.; Wang, Q. Molten salts-modified MgO-based adsorbents for intermediate-temperature CO2 capture: A review. J. Energy Chem. 2017, 26, 830–838. [Google Scholar] [CrossRef] [Green Version]
- Hiremath, V.; Shavi, R.; Seo, J.G. Controlled oxidation state of Ti in MgO-TiO2 composite for CO2 capture. Chem. Eng. J. 2017, 308, 177–183. [Google Scholar] [CrossRef]
- Ruhaimi, A.H.; Aziz, M.; Jalil, A.A. Magnesium oxide-based adsorbents for carbon dioxide capture: Current progress and future opportunities. J. CO2 Util. 2021, 43, 101357. [Google Scholar] [CrossRef]
- Wang, Q.; Luo, J.; Zhong, Z.; Borgna, A. CO2 capture by solid adsorbents and their applications: Current status and new trends. Energy Environ. Sci. 2011, 4, 42–55. [Google Scholar] [CrossRef]
- Meis, N.N.A.H.; Bitter, J.H.; de Jong, K.P. Support and size effects of activated hydrotalcites for precombustion CO2 capture. Ind. Eng. Chem. Res. 2010, 49, 1229–1235. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, Y.; Takaoka, K.; Yamabe, S.; Ito, T. Interaction of CO2 with magnesium oxide surfaces: A TPD, FTIR, and cluster-model calculation study. J. Phys. Chem. 1995, 99, 3704–3710. [Google Scholar] [CrossRef]
- Huang, J.; Li, X.; Wang, X.; Fang, X.; Wang, H.; Xu, X. New insights into CO2 methanation mechanisms on Ni/MgO catalysts by DFT calculations: Elucidating Ni and MgO roles and support effects. J. CO2 Util. 2019, 33, 55–63. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, H.M.; Park, J.-N. Bifunctional mechanism of CO2 methanation on Pd-MgO/SiO2 catalyst: Independent roles of MgO and Pd on CO2 methanation. J. Phys. Chem. C 2010, 114, 7128–7131. [Google Scholar] [CrossRef]
- Falsig, H.; Hvolbæk, B.; Kristensen, I.S.; Jiang, T.; Bligaard, T.; Christensen, C.H.; Nørskov, J.K. Trends in the catalytic CO oxidation activity of nanoparticles. Angew. Chem. 2008, 120, 4913–4917. [Google Scholar] [CrossRef]
- Moses, P.G.; Hinnemann, B.; Topsøe, H.; Nørskov, J.K. The effect of Co-promotion on MoS2 catalysts for hydrodesulfurization of thiophene: A density functional study. J. Catal. 2009, 268, 201–208. [Google Scholar] [CrossRef]
- Lauritsen, J.V.; Kibsgaard, J.; Helveg, S.; Topsøe, H.; Clausen, B.S.; Laegsgaard, E.; Besenbacher, F. Size-dependent structure of MoS2 nanocrystals. Nat. Nanotechnol. 2007, 2, 53–58. [Google Scholar] [CrossRef]
- Gao, L.Z.; Au, C.T. CO2 hydrogenation to methanol on a YBa2Cu3O7 catalyst. J. Catal. 2000, 189, 1–15. [Google Scholar] [CrossRef]
- Causa, M.; Dovesi, R.; Kotomin, E.; Pisani, C. The MgO(110) surface and CO adsorption thereon. I. Clean (110) surface. J. Phys. C Solid State Phys. 1987, 20, 4983–4990. [Google Scholar] [CrossRef]
- Usseinov, A.B.; Akilbekov, A.T.; Kotomin, E.A.; Popov, A.I.; Seitov, D.D.; Nekrasov, K.A.; Giniyatova, S.G.; Karipbayev, Z.T. The first principles calculations of CO2 adsorption on (1010) ZnO surface. In Proceedings of the Physics, Technologies and Innovation (Pti-2019): Proceedings of the Vi International Young Researchers’ Conference, Ekaterinburg, Russia, 20–23 May 2019; p. 20181. [Google Scholar]
- Kantorovich, L.N.; Shluger, A.L.; Gillan, M.J. What Can We Learn About Perfect and Defective MgO (001) Surface Using Density Functional Theory? In Defects and Surface-Induced Effects in Advanced Perovskites; Borstel, G., Krumins, A., Millers, D., Eds.; Springer: Dordrecht, The Netherlands, 2000; pp. 49–60. ISBN 978-94-011-4030-0. [Google Scholar]
- Yu, H.; Wang, X.; Shu, Z.; Fujii, M.; Song, C. Al2O3 and CeO2-promoted MgO sorbents for CO2 capture at moderate temperatures. Front. Chem. Sci. Eng. 2018, 12, 83–93. [Google Scholar] [CrossRef]
- Jin, S.; Bang, G.; Liu, L.; Lee, C.-H. Synthesis of mesoporous MgO–CeO2 composites with enhanced CO2 capture rate via controlled combustion. Microporous Mesoporous Mater. 2019, 288, 109587. [Google Scholar] [CrossRef]
- Jin, S.; Ko, K.-J.; Song, Y.-G.; Lee, K.; Lee, C.-H. Fabrication and kinetic study of spherical MgO agglomerates via water-in-oil method for pre-combustion CO2 capture. Chem. Eng. J. 2019, 359, 285–297. [Google Scholar] [CrossRef]
- Gao, W.; Zhou, T.; Gao, Y.; Wang, Q.; Lin, W. Study on MNO3/NO2 (M = Li, Na, and K)/MgO composites for intermediate-temperature CO2 capture. Energy Fuels 2019, 33, 1704–1712. [Google Scholar] [CrossRef]
- Chen, J.L.; Dong, X.Y.M.; Shi, C.L.; Li, S.H.; Wang, Y.; Zhu, J.H. Fabrication of strong solid base FeO–MgO for warm CO2 capture. Clean—Soil Air Water 2019, 47, 1800447. [Google Scholar] [CrossRef]
- Li, P.; Chen, R.; Lin, Y.; Li, W. General approach to facile synthesis of MgO-based porous ultrathin nanosheets enabling high-efficiency CO2 capture. Chem. Eng. J. 2021, 404, 126459. [Google Scholar] [CrossRef]
- Wang, J.; Huang, L.; Yang, R.; Zhang, Z.; Wu, J.; Gao, Y.; Wang, Q.; O’Hare, D.; Zhong, Z. Recent advances in solid sorbents for CO2 capture and new development trends. Energy Environ. Sci. 2014, 7, 3478–3518. [Google Scholar] [CrossRef]
- Xiao, G.; Singh, R.; Chaffee, A.; Webley, P. Advanced adsorbents based on MgO and K2CO3 for capture of CO2 at elevated temperatures. Int. J. Greenh. Gas Control 2011, 5, 634–639. [Google Scholar] [CrossRef]
- Ito, T. Initial sintering of magnesium oxide in carbon dioxide. J. Chem. Soc. Faraday Trans. 1982, 1, 1603–1613. [Google Scholar] [CrossRef]
- Ho, K.; Jin, S.; Zhong, M.; Vu, A.-T.; Lee, C.-H. Sorption capacity and stability of mesoporous magnesium oxide in post-combustion CO2 capture. Mater. Chem. Phys. 2017, 198, 154–161. [Google Scholar] [CrossRef]
- Vu, A.-T.; Ho, K.; Jin, S.; Lee, C.-H. Double sodium salt-promoted mesoporous MgO sorbent with high CO2 sorption capacity at intermediate temperatures under dry and wet conditions. Chem. Eng. J. 2016, 291, 161–173. [Google Scholar] [CrossRef]
- Hiremath, V.; Shavi, R.; Gil Seo, J. Mesoporous magnesium oxide nanoparticles derived via complexation-combustion for enhanced performance in carbon dioxide capture. J. Colloid Interface Sci. 2017, 498, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Tuan, V.A.; Lee, C.H. Preparation of rod-like MgO by simple precipitation method for CO2 capture at ambient temperature. Vietnam. J. Chem. 2018, 56, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Zhou, T.; Wang, Q. Controlled synthesis of MgO with diverse basic sites and its CO2 capture mechanism under different adsorption conditions. Chem. Eng. J. 2018, 336, 710–720. [Google Scholar] [CrossRef]
- Chen, A.; Yu, Y.; Li, Y.; Li, Y.; Jia, M. Solid-state grinding synthesis of ordered mesoporous MgO/carbon spheres composites for CO2 capture. Mater. Lett. 2016, 164, 520–523. [Google Scholar] [CrossRef]
- Park, J.-N.; McFarland, E.W. A highly dispersed Pd–Mg/SiO2 catalyst active for methanation of CO2. J. Catal. 2009, 266, 92–97. [Google Scholar] [CrossRef]
- Fukuda, Y.; Tanabe, K. Infrared study of carbon dioxide adsorbed on magnesium and calcium oxides. BCSJ 1973, 46, 1616–1619. [Google Scholar] [CrossRef] [Green Version]
- Ahn, C.-I.; Jeong, D.-W.; Cho, J.M.; Na, H.-S.; Jang, W.-J.; Roh, H.-S.; Choi, J.-H.; Um, S.H.; Bae, J.W. Water gas shift reaction on the Mn-modified ordered mesoporous Co3O4. Microporous Mesoporous Mater. 2016, 221, 204–211. [Google Scholar] [CrossRef]
- Highfield, J.; Bu, J.; Fagerlund, J.; Zevenhoven, R. The promoter effect of steam in gas-solid CO2 mineralisation. In Proceedings of the Conference: 11th International Conference on Carbon Dioxide Utilization (ICCDU XI), Dijon, France, 27–30 June 2011. [Google Scholar]
- Ding, Y.-D.; Song, G.; Liao, Q.; Zhu, X.; Chen, R. Bench scale study of CO2 adsorption performance of MgO in the presence of water vapor. Energy 2016, 112, 101–110. [Google Scholar] [CrossRef]
- Ram Reddy, M.K.; Xu, Z.P.; Lu, G.Q.; Da Diniz Costa, J.C. Influence of water on high-temperature CO2 capture using layered double hydroxide derivatives. Ind. Eng. Chem. Res. 2008, 47, 2630–2635. [Google Scholar] [CrossRef]
- Duan, Y.; Sorescu, D.C. CO2 capture properties of alkaline earth metal oxides and hydroxides: A combined density functional theory and lattice phonon dynamics study. J. Chem. Phys. 2010, 133, 74508. [Google Scholar] [CrossRef]
- Fagerlund, J.; Highfield, J.; Zevenhoven, R. Kinetics studies on wet and dry gas–solid carbonation of MgO and Mg(OH)2 for CO2 sequestration. RSC Adv. 2012, 2, 10380. [Google Scholar] [CrossRef]
- Baldauf-Sommerbauer, G.; Lux, S.; Aniser, W.; Siebenhofer, M. Synthesis of carbon monoxide from hydrogen and magnesite/dolomite. Chem. Ing. Tech. 2017, 89, 172–179. [Google Scholar] [CrossRef]
- Smith, H.J.; Fahrenkamp-Uppenbrink, J.; Coontz, R. Carbon capture and sequestration. Clearing the air. Introduction. Science 2009, 325, 1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, S. Carbon capture and sequestration. Science 2009, 325, 1599. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-E.; Yoo, J.S. New CO2 chemistry—Recent advances in utilizing CO2 as an oxidant and current understanding on its role. In Carbon Dioxide Utilization for Global Sustainability, Proceedings of 7th the International Conference on Carbon Dioxide Utilization; Elsevier: Amsterdam, The Netherlands, 2004; pp. 303–314. ISBN 9780444516008. [Google Scholar]
- Steeneveldt, R.; Berger, B.; Torp, T.A. CO2 capture and storage. Chem. Eng. Res. Des. 2006, 84, 739–763. [Google Scholar] [CrossRef]
- Li, L.; King, D.L.; Nie, Z.; Howard, C. Magnesia-stabilized calcium oxide absorbents with improved durability for high temperature CO2 capture. Ind. Eng. Chem. Res. 2009, 48, 10604–10613. [Google Scholar] [CrossRef]
- Manovic, V.; Anthony, E.J. CaO-based pellets supported by calcium aluminate cements for high-temperature CO2 capture. Environ. Sci. Technol. 2009, 43, 7117–7122. [Google Scholar] [CrossRef]
- Sandru, M.; Haukebø, S.H.; Hägg, M.-B. Composite hollow fiber membranes for CO2 capture. J. Membr. Sci. 2010, 346, 172–186. [Google Scholar] [CrossRef]
- Bachu, S. CO2 storage in geological media: Role, means, status and barriers to deployment. Prog. Energy Combust. Sci. 2008, 34, 254–273. [Google Scholar] [CrossRef]
- Haszeldine, R.S. Carbon capture and storage: How green can black be? Science 2009, 325, 1647–1652. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Xu, J.; Liang, B.; Duan, H.; Hou, B.; Huang, Y. Catalytic carbon dioxide hydrogenation to methane: A review of recent studies. J. Energy Chem. 2016, 25, 553–565. [Google Scholar] [CrossRef]
- Guo, M.; Lu, G. The effect of impregnation strategy on structural characters and CO2 methanation properties over MgO modified Ni/SiO2 catalysts. Catal. Commun. 2014, 54, 55–60. [Google Scholar] [CrossRef]
- Duyar, M.S.; Wang, S.; Arellano-Treviño, M.A.; Farrauto, R.J. CO2 utilization with a novel dual function material (DFM) for capture and catalytic conversion to synthetic natural gas: An update. J. CO2 Util. 2016, 15, 65–71. [Google Scholar] [CrossRef]
- Varun, Y.; Sreedhar, I.; Singh, S.A. Highly stable M/NiO–MgO (M = Co, Cu and Fe) catalysts towards CO2 methanation. Int. J. Hydrog. Energy 2020, 45, 28716–28731. [Google Scholar] [CrossRef]
- Fan, M.T.; Lin, J.D.; Zhang, H.B.; Liao, D.W. In situ growth of carbon nanotubes on Ni/MgO: A facile preparation of efficient catalysts for the production of synthetic natural gas from syngas. Chem. Commun. 2015, 51, 15720–15723. [Google Scholar] [CrossRef]
- Nakayama, T.; Ichikuni, N.; Sato, S.; Nozaki, F. Ni/MgO catalyst prepared using citric acid for hydrogenation of carbon dioxide. Appl. Catal. A Gen. 1997, 158, 185–199. [Google Scholar] [CrossRef]
- Takezawa, N.; Terunuma, H.; Shimokawabe, M.; Kobayashib, H. Methanation of carbon dioxide: Preparation of Ni/MgO catalysts and their performance. Appl. Catal. 1986, 23, 291–298. [Google Scholar] [CrossRef]
- Bette, N.; Thielemann, J.; Schreiner, M.; Mertens, F. Methanation of CO2 over a (Mg,Al)Ox Supported Nickel Catalyst Derived from a (Ni,Mg,Al)-Hydrotalcite-like Precursor. ChemCatChem 2016, 8, 2903–2906. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects. Green Chem. 2015, 17, 2647–2663. [Google Scholar] [CrossRef]
- Kattel, S.; Liu, P.; Chen, J.G. Tuning selectivity of CO2 hydrogenation reactions at the metal/oxide interface. J. Am. Chem. Soc. 2017, 139, 9739–9754. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Zhang, X.; Rui, N.; Hu, X.; Liu, C. Structural effect of Ni/ZrO2 catalyst on CO2 methanation with enhanced activity. Appl. Catal. B Environ. 2019, 244, 159–169. [Google Scholar] [CrossRef]
- Pan, Q.; Peng, J.; Sun, T.; Wang, S.; Wang, S. Insight into the reaction route of CO2 methanation: Promotion effect of medium basic sites. Catal. Commun. 2014, 45, 74–78. [Google Scholar] [CrossRef]
- Westermann, A.; Azambre, B.; Bacariza, M.C.; Graça, I.; Ribeiro, M.F.; Lopes, J.M.; Henriques, C. Insight into CO2 methanation mechanism over NiUSY zeolites: An operando IR study. Appl. Catal. B Environ. 2015, 174–175, 120–125. [Google Scholar] [CrossRef]
- Arellano-Treviño, M.A.; He, Z.; Libby, M.C.; Farrauto, R.J. Catalysts and adsorbents for CO2 capture and conversion with dual function materials: Limitations of Ni-containing DFMs for flue gas applications. J. CO2 Util. 2019, 31, 143–151. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, Y.; Li, J.; Wei, S.; Gao, X.; Wang, P. Combustion-impregnation preparation of Ni/SiO2 catalyst with improved low-temperature activity for CO2 methanation. Int. J. Hydrog. Energy 2021, 46, 20919–20929. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, C.; Gao, P.; Wang, H.; Li, X.; Zhong, L.; Wei, W.; Sun, Y. A review of the catalytic hydrogenation of carbon dioxide into value-added hydrocarbons. Catal. Sci. Technol. 2017, 7, 4580–4598. [Google Scholar] [CrossRef]
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef]
- Ban, H.; Li, C.; Asami, K.; Fujimoto, K. Influence of rare-earth elements (La, Ce, Nd and Pr) on the performance of Cu/Zn/Zr catalyst for CH3OH synthesis from CO2. Catal. Commun. 2014, 54, 50–54. [Google Scholar] [CrossRef]
- Guo, X.; Mao, D.; Lu, G.; Wang, S.; Wu, G. Glycine–nitrate combustion synthesis of CuO–ZnO–ZrO2 catalysts for methanol synthesis from CO2 hydrogenation. J. Catal. 2010, 271, 178–185. [Google Scholar] [CrossRef]
- Hu, B.; Yin, Y.; Liu, G.; Chen, S.; Hong, X.; Tsang, S.C.E. Hydrogen spillover enabled active Cu sites for methanol synthesis from CO2 hydrogenation over Pd doped CuZn catalysts. J. Catal. 2018, 359, 17–26. [Google Scholar] [CrossRef]
- Fujitani, T.; Saito, M.; Kanai, Y.; Kakumoto, T.; Watanabe, T.; Nakamura, J.; Uchijima, T. The role of metal oxides in promoting a copper catalyst for methanol synthesis. Catal. Lett. 1994, 25, 271–276. [Google Scholar] [CrossRef]
- Baltes, C.; Vukojevic, S.; Schuth, F. Correlations between synthesis, precursor, and catalyst structure and activity of a large set of CuO/ZnO/Al2O3 catalysts for methanol synthesis. J. Catal. 2008, 258, 334–344. [Google Scholar] [CrossRef]
- Robbins, J.L.; Iglesia, E.; Kelkar, C.P.; DeRites, B. Methanol synthesis over Cu/SiO2 catalysts. Catal. Lett. 1991, 10, 1–10. [Google Scholar] [CrossRef]
- Ren, H.; Xu, C.-H.; Zhao, H.-Y.; Wang, Y.-X.; Liu, J.; Liu, J.-Y. Methanol synthesis from CO2 hydrogenation over Cu/γ-Al2O3 catalysts modified by ZnO, ZrO2 and MgO. J. Ind. Eng. Chem. 2015, 28, 261–267. [Google Scholar] [CrossRef]
- Dasireddy, V.D.; Neja, S.Š.; Blaž, L. Correlation between synthesis pH, structure and Cu/MgO/Al2O3 heterogeneous catalyst activity and selectivity in CO2 hydrogenation to methanol. J. CO2 Util. 2018, 28, 189–199. [Google Scholar] [CrossRef]
- Zhan, H.; Li, F.; Gao, P.; Zhao, N.; Xiao, F.; Wei, W.; Zhong, L.; Sun, Y. Methanol synthesis from CO2 hydrogenation over La–M–Cu–Zn–O (M = Y, Ce, Mg, Zr) catalysts derived from perovskite-type precursors. J. Power Sources 2014, 251, 113–121. [Google Scholar] [CrossRef]
- Liu, C.; Guo, X.; Guo, Q.; Mao, D.; Yu, J.; Lu, G. Methanol synthesis from CO2 hydrogenation over copper catalysts supported on MgO-modified TiO2. J. Mol. Catal. A Chem. 2016, 425, 86–93. [Google Scholar] [CrossRef]
- Zander, S.; Kunkes, E.L.; Schuster, M.E.; Schumann, J.; Weinberg, G.; Teschner, D.; Jacobsen, N.; Schlögl, R.; Behrens, M. The role of the oxide component in the development of copper composite catalysts for methanol synthesis. Angew. Chem. Int. Ed. Engl. 2013, 52, 6536–6540. [Google Scholar] [CrossRef]
- Yang, R.; Zhang, Y.; Iwama, Y.; Tsubaki, N. Mechanistic study of a new low-temperature methanol synthesis on Cu/MgO catalysts. Appl. Catal. A Gen. 2005, 288, 126–133. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, H.; Wei, W.; Sun, Y. Solid base and their performance in synthesis of propylene glycol methyl ether. J. Mol. Catal. A Chem. 2005, 231, 83–88. [Google Scholar] [CrossRef]
- Madeira, L.M.; Martín-Aranda, R.M.; Maldonado-Hódar, F.J.; Fierro, J.; Portela, M.F. Oxidative dehydrogenation of n-butane over alkali and alkaline earth-promoted α-NiMoO4 catalysts. J. Catal. 1997, 169, 469–479. [Google Scholar] [CrossRef]
- Dasireddy, V.D.; Štefančič, N.S.; Huš, M.; Likozar, B. Effect of alkaline earth metal oxide (MO) Cu/MO/Al2O3 catalysts on methanol synthesis activity and selectivity via CO2 reduction. Fuel 2018, 233, 103–112. [Google Scholar] [CrossRef]
- Fichtl, M.B.; Schumann, J.; Kasatkin, I.; Jacobsen, N.; Behrens, M.; Schlögl, R.; Muhler, M.; Hinrichsen, O. Counting of oxygen defects versus metal surface sites in methanol synthesis catalysts by different probe molecules. Angew. Chem. Int. Ed. Engl. 2014, 53, 7043–7047. [Google Scholar] [CrossRef]
- Niu, J.; Liu, H.; Jin, Y.; Fan, B.; Qi, W.; Ran, J. Comprehensive review of Cu-based CO2 hydrogenation to CH3OH: Insights from experimental work and theoretical analysis. Int. J. Hydrog. Energy 2022, 47, 9183–9200. [Google Scholar] [CrossRef]
- Cao, A.; Wang, Z.; Li, H.; Elnabawy, A.O.; Nørskov, J.K. New insights on CO and CO2 hydrogenation for methanol synthesis: The key role of adsorbate-adsorbate interactions on Cu and the highly active MgO-Cu interface. J. Catal. 2021, 400, 325–331. [Google Scholar] [CrossRef]
- Kleiber, S.; Pallua, M.; Siebenhofer, M.; Lux, S. Catalytic hydrogenation of CO2 to methanol over Cu/MgO catalysts in a semi-continuous reactor. Energies 2021, 14, 4319. [Google Scholar] [CrossRef]
- Grabow, L.C.; Mavrikakis, M. Mechanism of methanol synthesis on Cu through CO2 and CO hydrogenation. ACS Catal. 2011, 1, 365–384. [Google Scholar] [CrossRef]
- Lam, E.; Corral-Pérez, J.J.; Larmier, K.; Noh, G.; Wolf, P.; Comas-Vives, A.; Urakawa, A.; Copéret, C. CO2 hydrogenation on Cu/Al2O3: Role of the metal/support Interface in driving activity and selectivity of a bifunctional catalyst. Angew. Chem. Int. Ed. Engl. 2019, 58, 13989–13996. [Google Scholar] [CrossRef]
- Poto, S.; van Vico Berkel, D.; Gallucci, F.; Fernanda Neira d’Angelo, M. Kinetic modelling of the methanol synthesis from CO2 and H2 over a CuO/CeO2/ZrO2 catalyst: The role of CO2 and CO hydrogenation. Chem. Eng. J. 2022, 435, 134946. [Google Scholar] [CrossRef]
- Tabatabaei, J.; Sakakini, B.H.; Waugh, K.C. On the mechanism of methanol synthesis and the water-gas shift reaction on ZnO. Catal. Lett. 2006, 110, 77–84. [Google Scholar] [CrossRef]
- Shido, T.; Iwasawa, Y. The effect of coadsorbates in reverse water-gas shift reaction on ZnO, in relation to reactant-promoted reaction mechanism. J. Catal. 1993, 140, 575–584. [Google Scholar] [CrossRef]
Figure 1.
Standard free enthalpies of reaction ΔRG0 for carbonate formation from metal oxides between 300 and 1200 K at ambient pressure: calculated with HSC Chemistry 8; MeO + CO2 ⇌ MeCO3; (a) alkaline-earth-metal oxides and (b) transition-metal oxides.
Figure 1.
Standard free enthalpies of reaction ΔRG0 for carbonate formation from metal oxides between 300 and 1200 K at ambient pressure: calculated with HSC Chemistry 8; MeO + CO2 ⇌ MeCO3; (a) alkaline-earth-metal oxides and (b) transition-metal oxides.

Figure 2.
Equilibrium solid phase diagram of the system MgO–CO2: solid phase fields were predicted as a function of the temperature and the feed hydrogen mole fraction; gaseous products are not shown; data calculated with FactSage 8.0™ (Thermfact and GTT-Technologies).
Figure 2.
Equilibrium solid phase diagram of the system MgO–CO2: solid phase fields were predicted as a function of the temperature and the feed hydrogen mole fraction; gaseous products are not shown; data calculated with FactSage 8.0™ (Thermfact and GTT-Technologies).

Figure 3.
Formation of the carbonate species monodentate carbonate, bidentate carbonate and bicarbonate during adsorption of CO2 on MgO-based adsorbents (adapted from [36]).
Figure 3.
Formation of the carbonate species monodentate carbonate, bidentate carbonate and bicarbonate during adsorption of CO2 on MgO-based adsorbents (adapted from [36]).

Figure 4.
Standard free enthalpies of reaction ΔRG0 for the reaction of CO2 with MgO (MgO + CO2 ⇋ MgCO3) and Mg(OH)2 (Mg(OH)2 + CO2 ⇋ MgCO3 + H2O), and the reaction of H2O with MgO (MgO + H2O ⇋ Mg(OH)2) in a temperature range of 300 to 1200 K at ambient pressure; calculated with HSC Chemistry 8.
Figure 4.
Standard free enthalpies of reaction ΔRG0 for the reaction of CO2 with MgO (MgO + CO2 ⇋ MgCO3) and Mg(OH)2 (Mg(OH)2 + CO2 ⇋ MgCO3 + H2O), and the reaction of H2O with MgO (MgO + H2O ⇋ Mg(OH)2) in a temperature range of 300 to 1200 K at ambient pressure; calculated with HSC Chemistry 8.

Figure 5.
Equilibrium solid phase diagram of the system MgO–H2O: solid phase fields were predicted as a function of the temperature and the feed hydrogen mole fraction; gaseous products are not shown; data calculated with FactSage 8.0™ (Thermfact and GTT-Technologies). The solid phases at equilibrium were predicted as a function of the temperature and the feed H2O (/( + )), where n0 denotes the number of moles of the feed component.
Figure 5.
Equilibrium solid phase diagram of the system MgO–H2O: solid phase fields were predicted as a function of the temperature and the feed hydrogen mole fraction; gaseous products are not shown; data calculated with FactSage 8.0™ (Thermfact and GTT-Technologies). The solid phases at equilibrium were predicted as a function of the temperature and the feed H2O (/( + )), where n0 denotes the number of moles of the feed component.

Figure 6.
Equilibrium solid phase diagram of the system MgCO3–H2O: solid phase fields were predicted as a function of the temperature and the feed hydrogen mole fraction; gaseous products are not shown; data calculated with FactSage 8.0™ (Thermfact and GTT-Technologies). The solid phases at equilibrium were predicted as a function of the temperature and the feed H2O (/( + )), where n0 denotes the number of moles of the feed component.
Figure 6.
Equilibrium solid phase diagram of the system MgCO3–H2O: solid phase fields were predicted as a function of the temperature and the feed hydrogen mole fraction; gaseous products are not shown; data calculated with FactSage 8.0™ (Thermfact and GTT-Technologies). The solid phases at equilibrium were predicted as a function of the temperature and the feed H2O (/( + )), where n0 denotes the number of moles of the feed component.

Figure 7.
Equilibrium solid phase diagram of the system MgCO3–H2: solid phase fields were predicted as a function of the temperature and the feed hydrogen mole fraction; gaseous products are not shown; data calculated with FactSage 8.0™ (Thermfact and GTT-Technologies). The solid phases at equilibrium were predicted as a function of the temperature and the feed H2 (/( + )), where n0 denotes the number of moles of the feed component.
Figure 7.
Equilibrium solid phase diagram of the system MgCO3–H2: solid phase fields were predicted as a function of the temperature and the feed hydrogen mole fraction; gaseous products are not shown; data calculated with FactSage 8.0™ (Thermfact and GTT-Technologies). The solid phases at equilibrium were predicted as a function of the temperature and the feed H2 (/( + )), where n0 denotes the number of moles of the feed component.

Figure 8.
A potential bifunctional mechanism for Ni–MgO whereby spillover of atomic hydrogen from Ni in contact with MgCO3 sequentially hydrogenates carbon until the product methane desorbs (adapted from Pd-Mg/SiO2 catalysts from [64]).
Figure 8.
A potential bifunctional mechanism for Ni–MgO whereby spillover of atomic hydrogen from Ni in contact with MgCO3 sequentially hydrogenates carbon until the product methane desorbs (adapted from Pd-Mg/SiO2 catalysts from [64]).

Figure 9.
Potential mechanism of methanol synthesis from CO2 and CO via formate and methoxide over a Cu/MgO catalyst (adapted from [6]).
Figure 9.
Potential mechanism of methanol synthesis from CO2 and CO via formate and methoxide over a Cu/MgO catalyst (adapted from [6]).

Table 1.
Experimental studies using Ni-based catalysts on MgO carrier material for CO2 methanation.
| Catalyst Composition | Preparation Method | Operation Conditions | Performance | Comments | Ref. |
|---|---|---|---|---|---|
| Ni/MgO (wNi = 0–27 wt%) | wet impregnation | T = 533–648 K GHSV = 3.7 m3 kg−1 h−1 v(H2):v(CO2):v(N2) = 4:1:5 | XCO2 = 87% YCH4 = 99% | [13] | |
| Ni/MgO (wNi = 11 and 17 wt%) | wetimpregnation | T = 603 K GHSV = 1.24–5 m3 kg−1 h−1 | XCO2 = 70% YCH4 = 99 % | [15] | |
| NiO/MgO (with and without impregnation with 2% Co, Cu, Fe) | sonochemicalmethod | T = 673 K, GHSV = 47.76 h−1 | XCO2 = 85% YCH4 = 98% (XCO2 = 90% (Co), 86% (Cu), 89% (Fe) YCH4 = 99% (Co), 94% (Cu), 96% (Fe)) | [85] | |
| Ni/MgO-CNTs 1 | precipitation | T = 473–713 K GHSV = 40 m3 kg−1 h−1 v(H2):v(CO):v(N2):v(CO2) = 75:15:5:5 | X(CO+CO2) = ~100% YCH4 = ~100% | [86] | |
| Ni/MgO (wNi = 30–90 wt%) | citric acid complex method | T = 553 K v(CO2):v(H2) = 1:8 | XCO2 = 62–85% SCH4 = 99–100% | space–time yield of CH4: 8.7–28.2 min−1 | [87] |
| Ni/MgO (wNi = 70 w%t) | coprecipitation | T = 553 K v(CO2):v(H2) = 1:8 | XCO2 = 32% SCH4 = 68% | space–time yield of CH4: 2.7 min−1 | [87] |
| Mg-Al-CO3 LDH 2 catalyst | coprecipitation | T = 473–573 K v(CO2):v(O2):v(N2) = 14:4:82 | CO2 sorption: 2.72% (dry sorption), 3.14% (wet condition, 12% water) | [69] | |
| Ni/MgO Ni(111), MgO (110) | DFT calculations | [40] |
1 carbon nanotubes, 2 layered double hydroxide.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Suksumrit, K.; Kleiber, S.; Lux, S. The Role of Carbonate Formation during CO2 Hydrogenation over MgO-Supported Catalysts: A Review on Methane and Methanol Synthesis. Energies 2023, 16, 2973. https://doi.org/10.3390/en16072973
AMA Style
Suksumrit K, Kleiber S, Lux S. The Role of Carbonate Formation during CO2 Hydrogenation over MgO-Supported Catalysts: A Review on Methane and Methanol Synthesis. Energies. 2023; 16(7):2973. https://doi.org/10.3390/en16072973
Chicago/Turabian StyleSuksumrit, Kamonrat, Sascha Kleiber, and Susanne Lux. 2023. "The Role of Carbonate Formation during CO2 Hydrogenation over MgO-Supported Catalysts: A Review on Methane and Methanol Synthesis" Energies 16, no. 7: 2973. https://doi.org/10.3390/en16072973
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.