Complement and Fungal Dysbiosis as Prognostic Markers and Potential Targets in PDAC Treatment

 , , , ,
, , , ,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
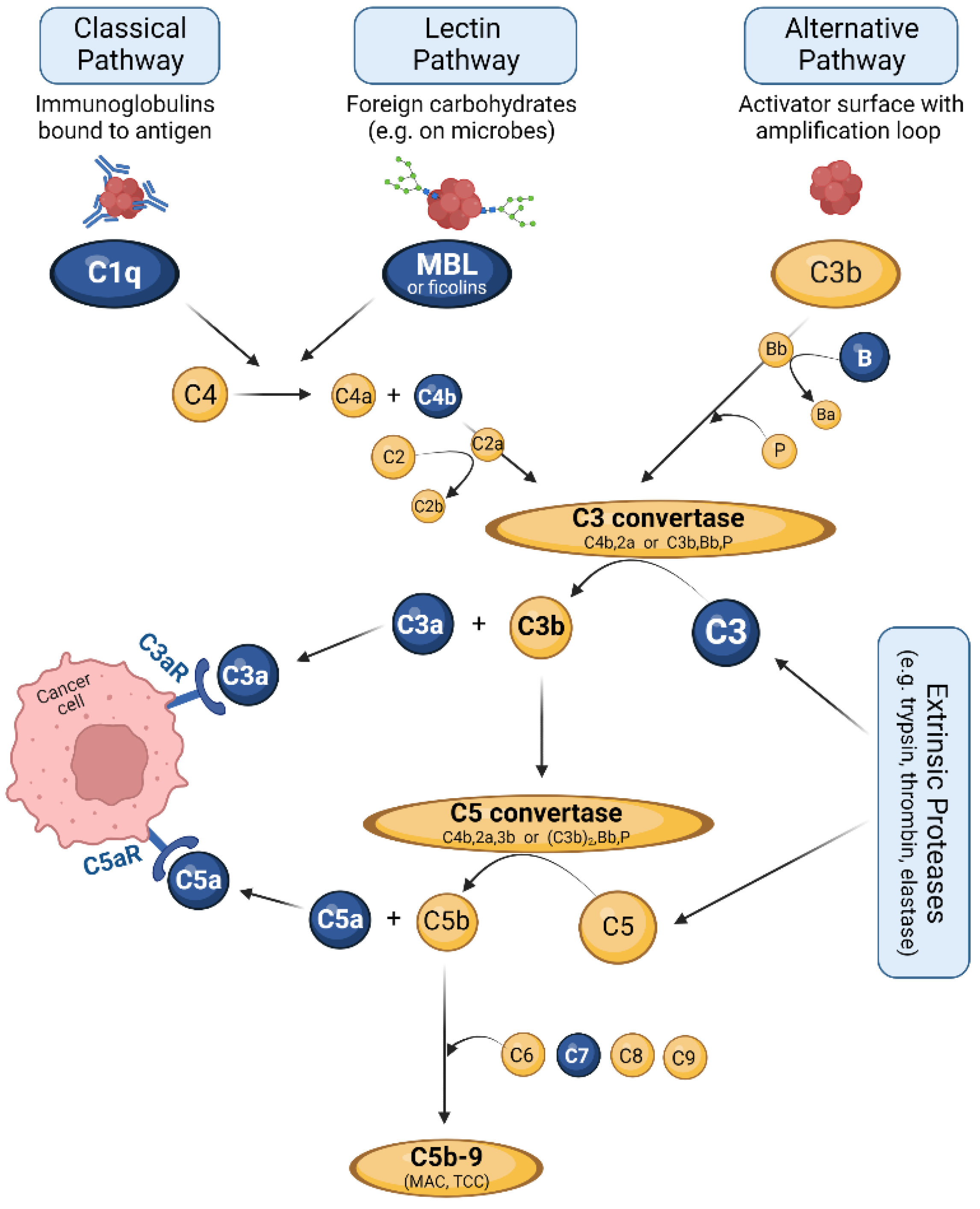
3. The Complement System: Effector and Immune Hub
4. Complement as Part of the TME
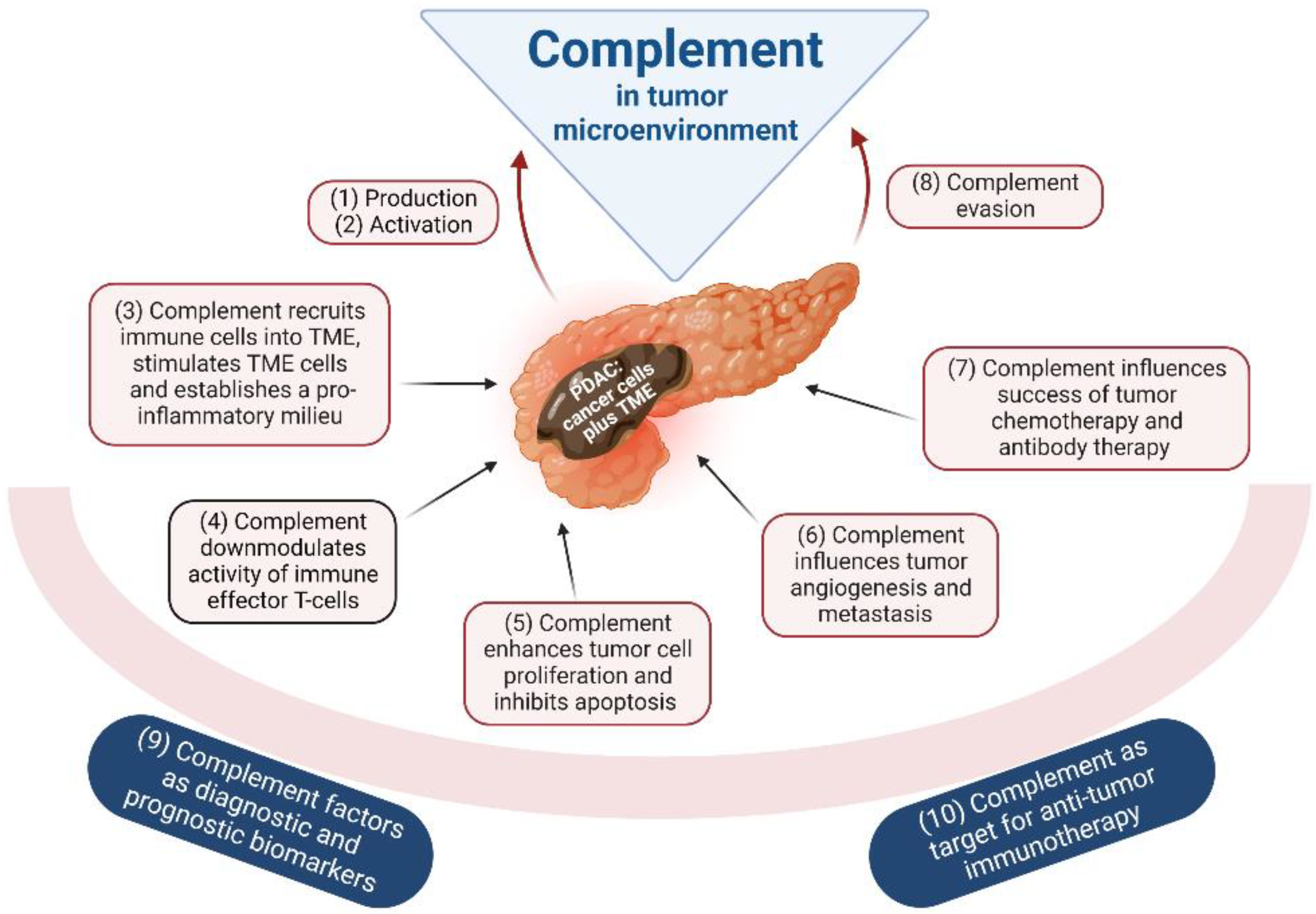
5. The Complement System in Tumorigenic Processes of PDAC
5.1. Complement Production in Tumors
5.2. Complement Activation by the Tumor
5.3. Complement-Mediated Inflammation, TME Cell Recruitment and Activation
5.4. Complement and Local Immunosuppression in the Tumor
5.5. Complement and Tumor Growth
5.6. Complement in Tumor Angiogenesis and Metastasis
5.7. Complement and Efficacy of Anticancer Therapy
5.8. Complement Evasion of Cancer Cells
5.9. Complement as Tumor Biomarker
5.10. Complement as a Therapeutic Target
6. Fungal Dysbiosis and PDAC Development
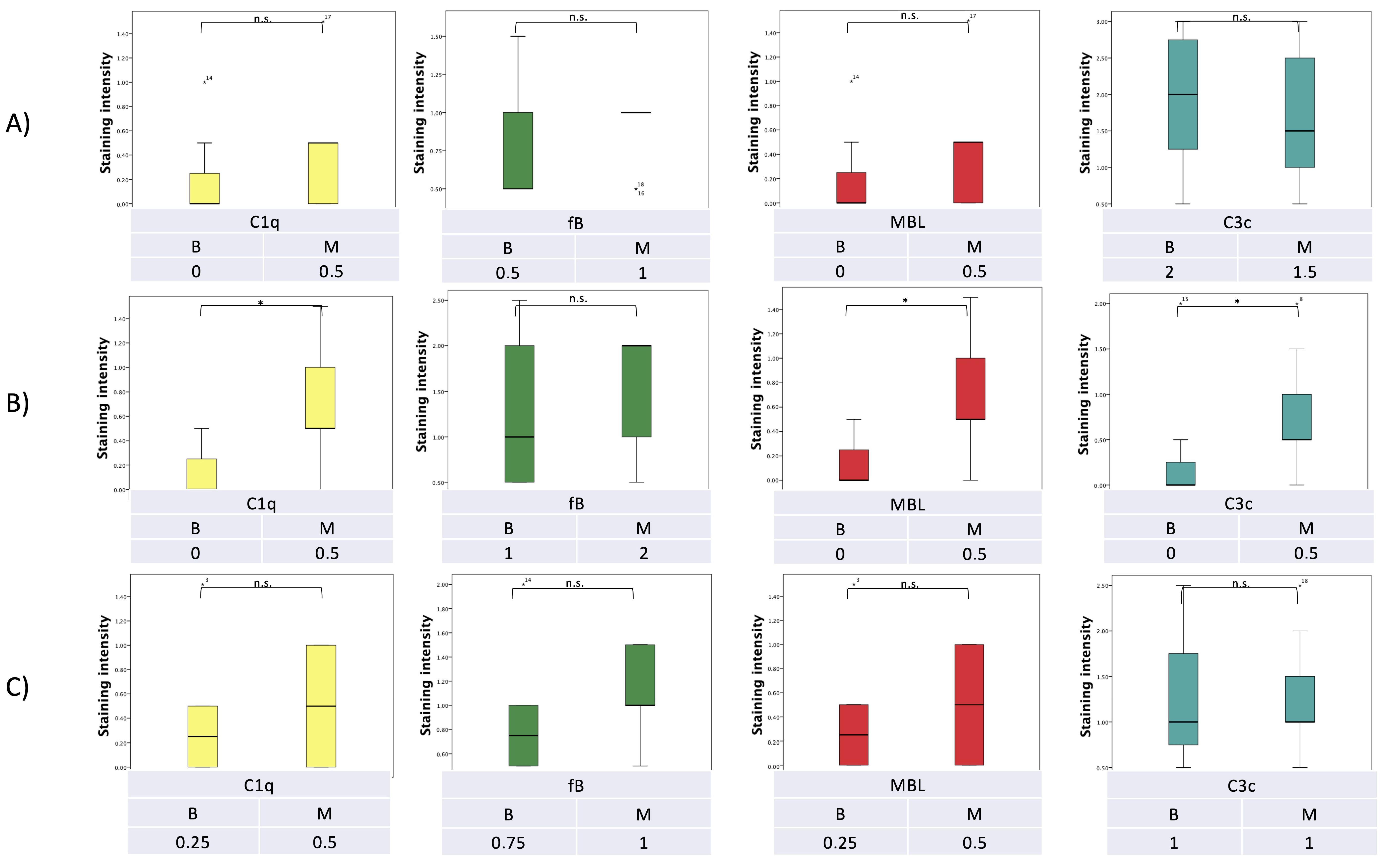
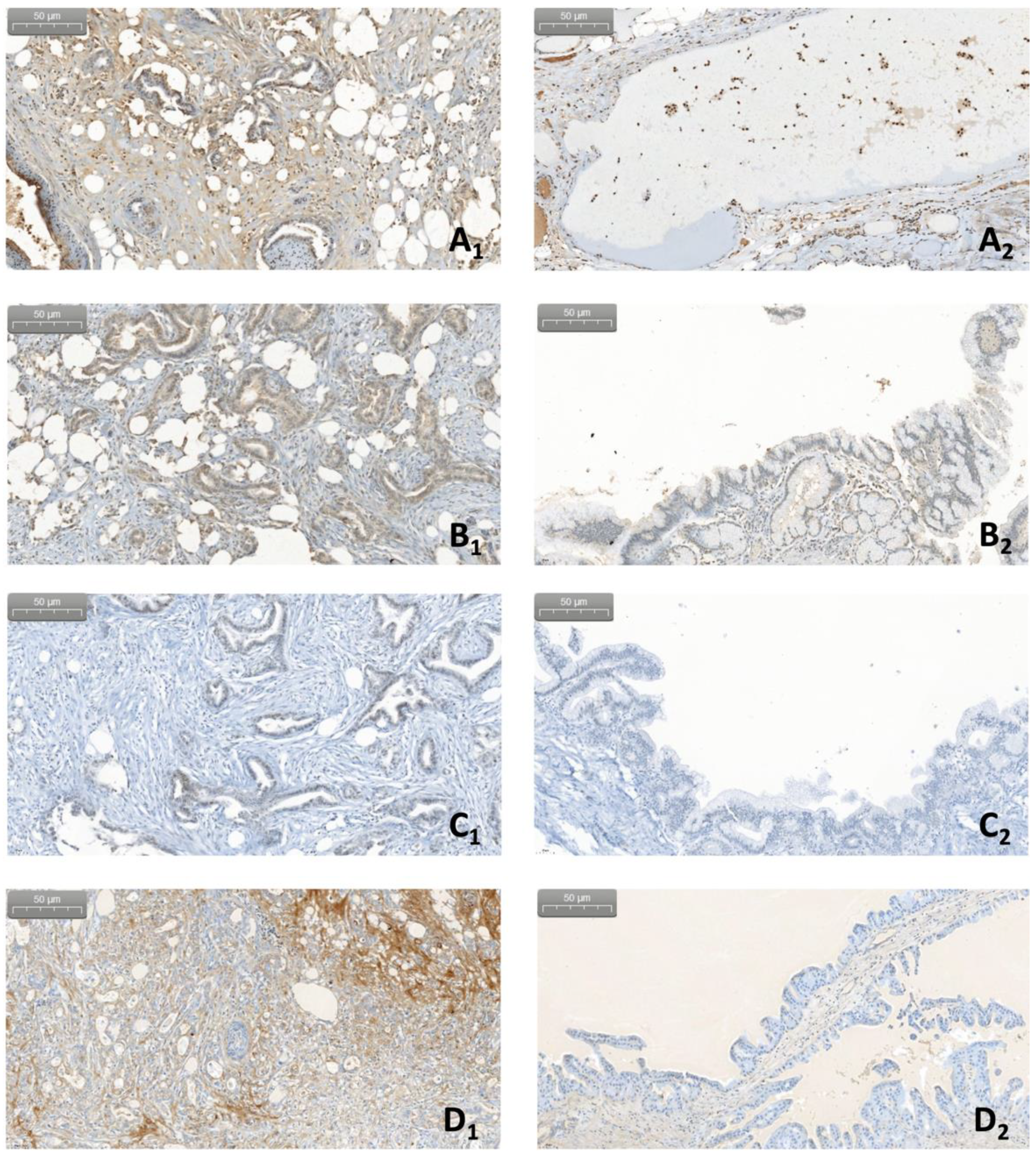
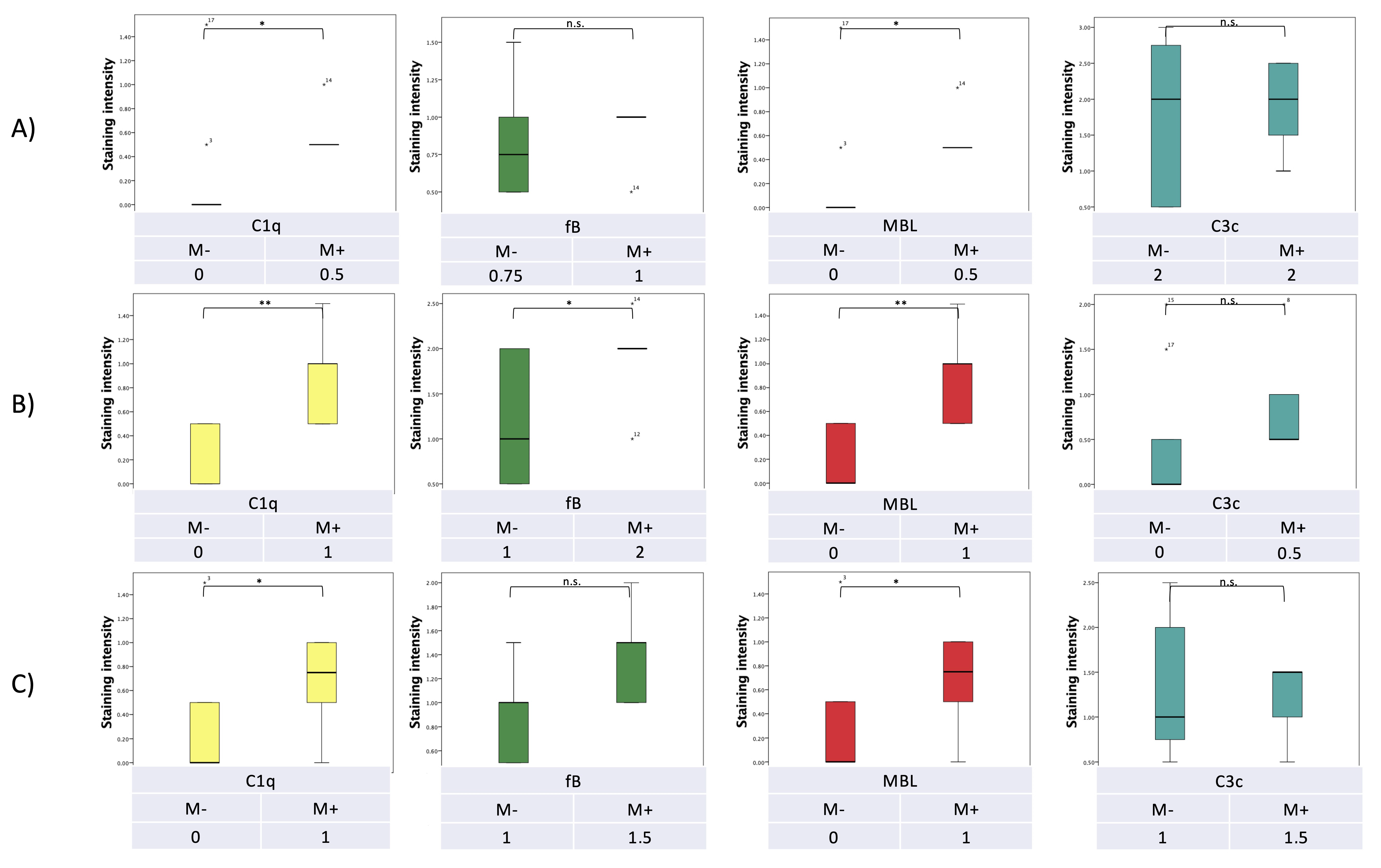
7. Pilot Study: Complement and Malassezia in PDAC
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Materials and Methods
Appendix A.1.1. Patient/Specimen Selection
Appendix A.1.2. DNA-Extraction
Appendix A.1.3. Multiplex PCR
Appendix A.1.4. Immunohistochemistry
Appendix A.1.5. Statistical Analysis
References
- Fong, C.Y.K.; Burke, E.; Cunningham, D.; Starling, N. Up-to-date tailored systemic treatment in pancreatic ductal adenocarcinoma. Gastroenterol. Res. Pract. 2019, 2019, 7135437. [Google Scholar] [CrossRef] [PubMed]
- Versteijne, E.; van Dam, J.L.; Suker, M.; Janssen, Q.P.; Groothuis, K.; Akkermans-Vogelaar, J.M.; Besselink, M.G.; Bonsing, B.A.; Buijsen, J.; Busch, O.R.; et al. Neoadjuvant chemoradiotherapy versus upfront surgery for resectable and borderline resectable pancreatic cancer: Long-term results of the dutch randomized preopanc trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2022, 40, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, W.; Chen, L.; Zhou, H.; Yang, R.; Hu, L.; Zhao, Y. Profiling the potential tumor markers of pancreatic ductal adenocarcinoma using 2D-dige and maldi-tof-ms: Up-regulation of complement c3 and alpha-2-hs-glycoprotein. Pancreatology 2013, 13, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.B.; Steele, N.G.; Carpenter, E.S.; Donahue, K.L.; Bushnell, G.G.; Morris, A.H.; The, S.; Orbach, S.M.; Sirihorachai, V.R.; Nwosu, Z.C.; et al. Pancreatic cancer is marked by complement-high blood monocytes and tumor-associated macrophages. Life Sci. Alliance 2021, 4, e202000935. [Google Scholar] [CrossRef]
- Yang, J.; Lin, P.; Yang, M.; Liu, W.; Fu, X.; Liu, D.; Tao, L.; Huo, Y.; Zhang, J.; Hua, R.; et al. Integrated genomic and transcriptomic analysis reveals unique characteristics of hepatic metastases and pro-metastatic role of complement c1q in pancreatic ductal adenocarcinoma. Genome Biol. 2021, 22, 4. [Google Scholar] [CrossRef]
- Dohlman, A.B.; Klug, J.; Mesko, M.; Gao, I.H.; Lipkin, S.M.; Shen, X.; Iliev, I.D. A pan-cancer mycobiome analysis reveals fungal involvement in gastrointestinal and lung tumors. Cell 2022, 185, 3807–3822.e12. [Google Scholar] [CrossRef]
- Narunsky-Haziza, L.; Sepich-Poore, G.D.; Livyatan, I.; Asraf, O.; Martino, C.; Nejman, D.; Gavert, N.; Stajich, J.E.; Amit, G.; González, A.; et al. Pan-cancer analyses reveal cancer-type-specific fungal ecologies and bacteriome interactions. Cell 2022, 185, 3789–3806.e17. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Pio, R.; Corrales, L.; Lambris, J.D. The role of complement in tumor growth. Adv. Exp. Med. Biol. 2014, 772, 229–262. [Google Scholar] [CrossRef] [Green Version]
- Harpf, V.; Rambach, G.; Würzner, R.; Lass-Flörl, C.; Speth, C. Candida and complement: New aspects in an old battle. Front. Immunol. 2020, 11, 1471. [Google Scholar] [CrossRef]
- Rambach, G.; Wurzner, R.; Speth, C. Complement: An efficient sword of innate immunity. Contrib. Microbiol 2008, 15, 78–100. [Google Scholar]
- Barnum, S.R. Complement: A primer for the coming therapeutic revolution. Pharmacol. Ther. 2017, 172, 63–72. [Google Scholar] [CrossRef]
- Atanes, P.; Ruz-Maldonado, I.; Pingitore, A.; Hawkes, R.; Liu, B.; Zhao, M.; Huang, G.C.; Persaud, S.J.; Amisten, S. C3ar and c5ar1 act as key regulators of human and mouse β-cell function. Cell. Mol. Life Sci. 2018, 75, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Ermert, D.; Blom, A.M. C4b-binding protein: The good, the bad and the deadly. Novel functions of an old friend. Immunol. Lett. 2016, 169, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Kouser, L.; Abdul-Aziz, M.; Nayak, A.; Stover, C.M.; Sim, R.B.; Kishore, U. Properdin and factor h: Opposing players on the alternative complement pathway “see-saw”. Front. Immunol. 2013, 4, 93. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.; Sudhir, P.R.; Zhang, Q. Pancreatic tissue proteomics unveils key proteins, pathways, and networks associated with type 1 diabetes. Proteom. Clin. Appl. 2020, 14, e2000053. [Google Scholar] [CrossRef]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part I—Molecular mechanisms of activation and regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [Green Version]
- Garred, P.; Genster, N.; Pilely, K.; Bayarri-Olmos, R.; Rosbjerg, A.; Ma, Y.J.; Skjoedt, M.O. A journey through the lectin pathway of complement-mbl and beyond. Immunol. Rev. 2016, 274, 74–97. [Google Scholar] [CrossRef]
- Lachmann, P.J.; Lay, E.; Seilly, D.J. Experimental confirmation of the c3 tickover hypothesis by studies with an ab (s77) that inhibits tickover in whole serum. FASEB J. 2018, 32, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Harboe, M.; Mollnes, T.E. The alternative complement pathway revisited. J. Cell. Mol. Med. 2008, 12, 1074–1084. [Google Scholar] [CrossRef] [Green Version]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part II: Role in immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [PubMed]
- Huber-Lang, M.; Sarma, J.V.; Zetoune, F.S.; Rittirsch, D.; Neff, T.A.; McGuire, S.R.; Lambris, J.D.; Warner, R.L.; Flierl, M.A.; Hoesel, L.M.; et al. Generation of c5a in the absence of c3: A new complement activation pathway. Nat. Med. 2006, 12, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Kolev, M.; Le Friec, G.; Kemper, C. Complement--tapping into new sites and effector systems. Nat. Rev. Immunol. 2014, 14, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Riedemann, N.C.; Habel, M.; Ziereisen, J.; Hermann, M.; Schneider, C.; Wehling, C.; Kirschfink, M.; Kentouche, K.; Guo, R. Controlling the anaphylatoxin c5a in diseases requires a specifically targeted inhibition. Clin. Immunol. 2017, 180, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Acioli, J.M.; Isobe, M.; Kawasaki, S. Early complement system activation and neutrophil priming in acute pancreatitis: Participation of trypsin. Surgery 1997, 122, 909–917. [Google Scholar] [CrossRef]
- Sarma, J.V.; Ward, P.A. The complement system. Cell Tissue Res. 2011, 343, 227–235. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Novel mechanisms and functions of complement. Nat. Immunol. 2017, 18, 1288–1298. [Google Scholar] [CrossRef]
- Reis, E.S.; Mastellos, D.C.; Hajishengallis, G.; Lambris, J.D. New insights into the immune functions of complement. Nat. Rev. Immunol. 2019, 19, 503–516. [Google Scholar] [CrossRef]
- Zipfel, P.F.; Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (time) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, Q.; Li, T.; Liao, Q.; Zhao, Y. Role of the complement system in the tumor microenvironment. Cancer Cell Int. 2019, 19, 300. [Google Scholar] [CrossRef]
- He, Z.; Zhang, S. Tumor-associated macrophages and their functional transformation in the hypoxic tumor microenvironment. Front. Immunol. 2021, 12, 741305. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Wu, J.; Peng, Y.; Sun, J.; Cheng, P.; Huang, Q. Tumor-associated neutrophils in colorectal cancer development, progression and immunotherapy. Cancers 2022, 14, 4755. [Google Scholar] [CrossRef]
- Petersen, O.H.; Gerasimenko, J.V.; Gerasimenko, O.V.; Gryshchenko, O.; Peng, S. The roles of calcium and atp in the physiology and pathology of the exocrine pancreas. Physiol. Rev. 2021, 101, 1691–1744. [Google Scholar] [CrossRef]
- Xue, R.; Jia, K.; Wang, J.; Yang, L.; Wang, Y.; Gao, L.; Hao, J. A rising star in pancreatic diseases: Pancreatic stellate cells. Front. Physiol. 2018, 9, 754. [Google Scholar] [CrossRef]
- Morishita, K.; Shimizu, K.; Haruta, I.; Kawamura, S.; Kobayashi, M.; Shiratori, K. Engulfment of gram-positive bacteria by pancreatic stellate cells in pancreatic fibrosis. Pancreas 2010, 39, 1002–1007. [Google Scholar] [CrossRef]
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 14. [Google Scholar] [CrossRef]
- Zhang, T.; Ren, Y.; Yang, P.; Wang, J.; Zhou, H. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Cell Death Dis. 2022, 13, 897. [Google Scholar] [CrossRef]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019, 5, e129212. [Google Scholar] [CrossRef] [Green Version]
- Griffin, J.L.; Kauppinen, R.A. Tumour metabolomics in animal models of human cancer. J. Proteome Res. 2007, 6, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, T.; Takahashi, K.; Moriya, S.; Hata, K.; Yamamoto, K.; Wada, T.; Yamaguchi, K.; Shiozaki, K. Altered expression of sialidases in human cancer. Adv. Exp. Med. Biol. 2012, 749, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Ain, D.; Shaikh, T.; Manimala, S.; Ghebrehiwet, B. The role of complement in the tumor microenvironment. Fac. Rev. 2021, 10, 80. [Google Scholar] [CrossRef]
- Zheng, Z.; Li, Y.N.; Jia, S.; Zhu, M.; Cao, L.; Tao, M.; Jiang, J.; Zhan, S.; Chen, Y.; Gao, P.J.; et al. Lung mesenchymal stromal cells influenced by th2 cytokines mobilize neutrophils and facilitate metastasis by producing complement c3. Nat. Commun. 2021, 12, 6202. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Ma, Y.; Wei, S.; Liang, X. The dual role of complement in cancers, from destroying tumors to promoting tumor development. Cytokine 2021, 143, 155522. [Google Scholar] [CrossRef]
- D’Angelo, A.; Sobhani, N.; Roviello, G.; Bagby, S.; Bonazza, D.; Bottin, C.; Giudici, F.; Zanconati, F.; De Manzini, N.; Guglielmi, A.; et al. Tumour infiltrating lymphocytes and immune-related genes as predictors of outcome in pancreatic adenocarcinoma. PLoS ONE 2019, 14, e0219566. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Na, K.; Jeong, S.K.; Lim, J.S.; Kim, S.A.; Lee, M.J.; Song, S.Y.; Kim, H.; Hancock, W.S.; Paik, Y.K. Identification of human complement factor b as a novel biomarker candidate for pancreatic ductal adenocarcinoma. J. Proteome Res. 2014, 13, 4878–4888. [Google Scholar] [CrossRef]
- Chen, K.; Wang, Q.; Li, M.; Guo, H.; Liu, W.; Wang, F.; Tian, X.; Yang, Y. Single-cell rna-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression. EBioMedicine 2021, 66, 103315. [Google Scholar] [CrossRef]
- Chen, J.; Wu, W.; Zhen, C.; Zhou, H.; Yang, R.; Chen, L.; Hu, L. Expression and clinical significance of complement c3, complement c4b1 and apolipoprotein e in pancreatic cancer. Oncol. Lett. 2013, 6, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Ajona, D.; Pajares, M.J.; Corrales, L.; Perez-Gracia, J.L.; Agorreta, J.; Lozano, M.D.; Torre, W.; Massion, P.P.; de-Torres, J.P.; Jantus-Lewintre, E.; et al. Investigation of complement activation product c4d as a diagnostic and prognostic biomarker for lung cancer. J. Natl. Cancer Inst. 2013, 105, 1385–1393. [Google Scholar] [CrossRef] [Green Version]
- Mäkelä, K.; Helén, P.; Haapasalo, H.; Paavonen, T. Complement activation in astrocytomas: Deposition of c4d and patient outcome. BMC Cancer 2012, 12, 565. [Google Scholar] [CrossRef]
- Berraondo, P.; Minute, L.; Ajona, D.; Corrales, L.; Melero, I.; Pio, R. Innate immune mediators in cancer: Between defense and resistance. Immunol. Rev. 2016, 274, 290–306. [Google Scholar] [CrossRef]
- Geng, X.; Chen, H.; Zhao, L.; Hu, J.; Yang, W.; Li, G.; Cheng, C.; Zhao, Z.; Zhang, T.; Li, L.; et al. Cancer-associated fibroblast (caf) heterogeneity and targeting therapy of cafs in pancreatic cancer. Front. Cell Dev. Biol. 2021, 9, 655152. [Google Scholar] [CrossRef]
- Markiewski, M.M.; DeAngelis, R.A.; Benencia, F.; Ricklin-Lichtsteiner, S.K.; Koutoulaki, A.; Gerard, C.; Coukos, G.; Lambris, J.D. Modulation of the antitumor immune response by complement. Nat. Immunol. 2008, 9, 1225–1235. [Google Scholar] [CrossRef] [Green Version]
- Ajona, D.; Ortiz-Espinosa, S.; Pio, R. Complement anaphylatoxins c3a and c5a: Emerging roles in cancer progression and treatment. Semin. Cell Dev. Biol. 2019, 85, 153–163. [Google Scholar] [CrossRef]
- Metzemaekers, M.; Gouwy, M.; Proost, P. Neutrophil chemoattractant receptors in health and disease: Double-edged swords. Cell. Mol. Immunol. 2020, 17, 433–450. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Hussain, N.; Das, D.; Pramanik, A.; Pandey, M.K.; Joshi, V.; Pramanik, K.C. Targeting the complement system in pancreatic cancer drug resistance: A novel therapeutic approach. Cancer Drug Resist. 2022, 5, 317–327. [Google Scholar] [CrossRef]
- Shimazaki, R.; Takano, S.; Satoh, M.; Takada, M.; Miyahara, Y.; Sasaki, K.; Yoshitomi, H.; Kagawa, S.; Furukawa, K.; Takayashiki, T.; et al. Complement factor b regulates cellular senescence and is associated with poor prognosis in pancreatic cancer. Cell. Oncol. 2021, 44, 937–950. [Google Scholar] [CrossRef]
- Sendler, M.; Beyer, G.; Mahajan, U.M.; Kauschke, V.; Maertin, S.; Schurmann, C.; Homuth, G.; Völker, U.; Völzke, H.; Halangk, W.; et al. Complement component 5 mediates development of fibrosis, via activation of stellate cells, in 2 mouse models of chronic pancreatitis. Gastroenterology 2015, 149, 765–776.e10. [Google Scholar] [CrossRef] [Green Version]
- Riedl, M.; Noone, D.G.; Khan, M.A.; Pluthero, F.G.; Kahr, W.H.A.; Palaniyar, N.; Licht, C. Complement activation induces neutrophil adhesion and neutrophil-platelet aggregate formation on vascular endothelial cells. Kidney Int. Rep. 2017, 2, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Takano, S.; Tomizawa, S.; Miyahara, Y.; Furukawa, K.; Takayashiki, T.; Kuboki, S.; Takada, M.; Ohtsuka, M. C4b-binding protein α-chain enhances antitumor immunity by facilitating the accumulation of tumor-infiltrating lymphocytes in the tumor microenvironment in pancreatic cancer. J. Exp. Clin. Cancer Res.: CR 2021, 40, 212. [Google Scholar] [CrossRef] [PubMed]
- Tegla, C.A.; Cudrici, C.; Patel, S.; Trippe, R., 3rd; Rus, V.; Niculescu, F.; Rus, H. Membrane attack by complement: The assembly and biology of terminal complement complexes. Immunol. Res. 2011, 51, 45–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, R.M.; Cannon, A.; Reynolds, J.V.; Lysaght, J.; Lynam-Lennon, N. Complement in tumourigenesis and the response to cancer therapy. Cancers 2021, 13, 1209. [Google Scholar] [CrossRef]
- Ahmad, R.S.; Eubank, T.D.; Lukomski, S.; Boone, B.A. Immune cell modulation of the extracellular matrix contributes to the pathogenesis of pancreatic cancer. Biomolecules 2021, 11, 901. [Google Scholar] [CrossRef]
- Jin, L.; Kim, H.S.; Shi, J. Neutrophil in the pancreatic tumor microenvironment. Biomolecules 2021, 11, 1170. [Google Scholar] [CrossRef]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of mbl. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef]
- Wang, H.; Capula, M.; Krom, B.P.; Yee, D.; Giovannetti, E.; Deng, D. Of fungi and men: Role of fungi in pancreatic cancer carcinogenesis. Ann. Transl. Med. 2020, 8, 1257. [Google Scholar] [CrossRef]
- Bulla, R.; Tripodo, C.; Rami, D.; Ling, G.S.; Agostinis, C.; Guarnotta, C.; Zorzet, S.; Durigutto, P.; Botto, M.; Tedesco, F. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat. Commun. 2016, 7, 10346. [Google Scholar] [CrossRef] [Green Version]
- Corrales, L.; Ajona, D.; Rafail, S.; Lasarte, J.J.; Riezu-Boj, J.I.; Lambris, J.D.; Rouzaut, A.; Pajares, M.J.; Montuenga, L.M.; Pio, R. Anaphylatoxin c5a creates a favorable microenvironment for lung cancer progression. J. Immunol. 2012, 189, 4674–4683. [Google Scholar] [CrossRef] [Green Version]
- Gumberger, P.; Bjornsson, B.; Sandström, P.; Bojmar, L.; Zambirinis, C.P. The liver pre-metastatic niche in pancreatic cancer: A potential opportunity for intervention. Cancers 2022, 14, 3028. [Google Scholar] [CrossRef]
- Xie, Z.; Gao, Y.; Ho, C.; Li, L.; Jin, C.; Wang, X.; Zou, C.; Mao, Y.; Wang, X.; Li, Q.; et al. Exosome-delivered cd44v6/c1qbp complex drives pancreatic cancer liver metastasis by promoting fibrotic liver microenvironment. Gut 2022, 71, 568–579. [Google Scholar] [CrossRef]
- Derynck, R.; Weinberg, R.A. Emt and cancer: More than meets the eye. Dev. Cell 2019, 49, 313–316. [Google Scholar] [CrossRef]
- Suzuki, R.; Okubo, Y.; Takagi, T.; Sugimoto, M.; Sato, Y.; Irie, H.; Nakamura, J.; Takasumi, M.; Kato, T.; Hashimoto, M.; et al. The complement c3a-c3a receptor axis regulates epithelial-to-mesenchymal transition by activating the erk pathway in pancreatic ductal adenocarcinoma. Anticancer. Res. 2022, 42, 1207–1215. [Google Scholar] [CrossRef]
- Saito, K.; Iioka, H.; Maruyama, S.; Sumardika, I.W.; Sakaguchi, M.; Kondo, E. Podxl1 promotes metastasis of the pancreatic ductal adenocarcinoma by activating the c5ar/c5a axis from the tumor microenvironment. Neoplasia 2019, 21, 1121–1132. [Google Scholar] [CrossRef]
- Taylor, R.P.; Lindorfer, M.A. The role of complement in mab-based therapies of cancer. Methods 2014, 65, 18–27. [Google Scholar] [CrossRef]
- Kolev, M.; Towner, L.; Donev, R. Complement in cancer and cancer immunotherapy. Arch. Immunol. Et Ther. Exp. 2011, 59, 407–419. [Google Scholar] [CrossRef]
- Ollert, M.W.; Frade, R.; Fiandino, A.; Panneerselvam, M.; Petrella, E.C.; Barel, M.; Pangburn, M.K.; Bredehorst, R.; Vogel, C.W. C3-cleaving membrane proteinase. A new complement regulatory protein of human melanoma cells. J. Immunol. 1990, 144, 3862–3867. [Google Scholar]
- Moskovich, O.; Fishelson, Z. Live cell imaging of outward and inward vesiculation induced by the complement c5b-9 complex. J. Biol. Chem. 2007, 282, 29977–29986. [Google Scholar] [CrossRef] [Green Version]
- Juhl, H.; Helmig, F.; Baltzer, K.; Kalthoff, H.; Henne-Bruns, D.; Kremer, B. Frequent expression of complement resistance factors cd46, cd55, and cd59 on gastrointestinal cancer cells limits the therapeutic potential of monoclonal antibody 17-1a. J. Surg. Oncol. 1997, 64, 222–230. [Google Scholar] [CrossRef]
- Schiarea, S.; Solinas, G.; Allavena, P.; Scigliuolo, G.M.; Bagnati, R.; Fanelli, R.; Chiabrando, C. Secretome analysis of multiple pancreatic cancer cell lines reveals perturbations of key functional networks. J. Proteome Res. 2010, 9, 4376–4392. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.A.; Schwaeble, W.; Wittig, B.M.; Meyer zum Büschenfelde, K.H.; Dippold, W.G. Expression and regulation by interferon-gamma of the membrane-bound complement regulators cd46 (mcp), cd55 (daf) and cd59 in gastrointestinal tumours. Eur. J. Cancer 1999, 35, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Roeise, O.; Sivertsen, S.; Ruud, T.E.; Bouma, B.N.; Stadaas, J.O.; Aasen, A.O. Studies on components of the contact phase system in patients with advanced gastrointestinal cancer. Cancer 1990, 65, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Osther, K.; Förnvik, K.; Liljedahl, E.; Salford, L.G.; Redebrandt, H.N. Upregulation of c1-inhibitor in pancreatic cancer. Oncotarget 2019, 10, 5703–5712. [Google Scholar] [CrossRef] [Green Version]
- Bettac, L.; Denk, S.; Seufferlein, T.; Huber-Lang, M. Complement in pancreatic disease-perpetrator or savior? Front. Immunol. 2017, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Lee, M.J.; Hwang, H.K.; Lee, S.H.; Kim, H.; Paik, Y.K.; Kang, C.M. Prognostic potential of the preoperative plasma complement factor b in resected pancreatic cancer: A pilot study. Cancer Biomark. Sect. A Dis. Markers 2019, 24, 335–342. [Google Scholar] [CrossRef]
- Narasimhan, A.; Shahda, S.; Kays, J.K.; Perkins, S.M.; Cheng, L.; Schloss, K.N.H.; Schloss, D.E.I.; Koniaris, L.G.; Zimmers, T.A. Identification of potential serum protein biomarkers and pathways for pancreatic cancer cachexia using an aptamer-based discovery platform. Cancers 2020, 12, 3787. [Google Scholar] [CrossRef]
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 2019, 68, 654–662. [Google Scholar] [CrossRef]
- Zhu, F.; Willette-Brown, J.; Song, N.Y.; Lomada, D.; Song, Y.; Xue, L.; Gray, Z.; Zhao, Z.; Davis, S.R.; Sun, Z.; et al. Autoreactive t cells and chronic fungal infection drive esophageal carcinogenesis. Cell Host Microbe 2017, 21, 478–493.e77. [Google Scholar] [CrossRef] [Green Version]
- Del Castillo, E.; Meier, R.; Chung, M.; Koestler, D.C.; Chen, T.; Paster, B.J.; Charpentier, K.P.; Kelsey, K.T.; Izard, J.; Michaud, D.S. The microbiomes of pancreatic and duodenum tissue overlap and are highly subject specific but differ between pancreatic cancer and noncancer subjects. Cancer Epidemiol. Biomark. Prev. 2019, 28, 370–383. [Google Scholar] [CrossRef] [Green Version]
- Bellotti, R.; Speth, C.; Adolph, T.E.; Lass-Flörl, C.; Effenberger, M.; Öfner, D.; Maglione, M. Micro- and mycobiota dysbiosis in pancreatic ductal adenocarcinoma development. Cancers 2021, 13, 3431. [Google Scholar] [CrossRef]
- Alam, A.; Levanduski, E.; Denz, P.; Villavicencio, H.S.; Bhatta, M.; Alhorebi, L.; Zhang, Y.; Gomez, E.C.; Morreale, B.; Senchanthisai, S.; et al. Fungal mycobiome drives il-33 secretion and type 2 immunity in pancreatic cancer. Cancer Cell 2022, 40, 153–167.e11. [Google Scholar] [CrossRef]
- Suhr, M.J.; Banjara, N.; Hallen-Adams, H.E. Sequence-based methods for detecting and evaluating the human gut mycobiome. Lett. Appl. Microbiol. 2016, 62, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Alnuaimi, A.D.; Ramdzan, A.N.; Wiesenfeld, D.; O’Brien-Simpson, N.M.; Kolev, S.D.; Reynolds, E.C.; McCullough, M.J. Candida virulence and ethanol-derived acetaldehyde production in oral cancer and non-cancer subjects. Oral Dis. 2016, 22, 805–814. [Google Scholar] [CrossRef]
- Gaitanis, G.; Magiatis, P.; Hantschke, M.; Bassukas, I.D.; Velegraki, A. The malassezia genus in skin and systemic diseases. Clin. Microbiol. Rev. 2012, 25, 106–141. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, S.; Elhance, A.; Van Duzer, A.; Kumar, S.; Leitenberger, J.J.; Oshimori, N. Tumor-initiating cells establish an il-33-tgf-β niche signaling loop to promote cancer progression. Science 2020, 369, eaay1813. [Google Scholar] [CrossRef]
- Uchida, M.; Anderson, E.L.; Squillace, D.L.; Patil, N.; Maniak, P.J.; Iijima, K.; Kita, H.; O’Grady, S.M. Oxidative stress serves as a key checkpoint for il-33 release by airway epithelium. Allergy 2017, 72, 1521–1531. [Google Scholar] [CrossRef]
- Collard, C.D.; Väkevä, A.; Morrissey, M.A.; Agah, A.; Rollins, S.A.; Reenstra, W.R.; Buras, J.A.; Meri, S.; Stahl, G.L. Complement activation after oxidative stress: Role of the lectin complement pathway. Am. J. Pathol. 2000, 156, 1549–1556. [Google Scholar] [CrossRef]
- West, P.W.; Bahri, R.; Garcia-Rodriguez, K.M.; Sweetland, G.; Wileman, G.; Shah, R.; Montero, A.; Rapley, L.; Bulfone-Paus, S. Interleukin-33 amplifies human mast cell activities induced by complement anaphylatoxins. Front. Immunol. 2020, 11, 615236. [Google Scholar] [CrossRef]
- Arbore, G.; West, E.E.; Spolski, R.; Robertson, A.A.B.; Klos, A.; Rheinheimer, C.; Dutow, P.; Woodruff, T.M.; Yu, Z.X.; O’Neill, L.A.; et al. T helper 1 immunity requires complement-driven nlrp3 inflammasome activity in cd4⁺ t cells. Science 2016, 352, aad1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, P.; Xu, Y.; Li, L.; Lv, X.; Li, L.; Chen, J.; Zhou, D.; Wang, X.; Wang, Q.; Zhang, W.; et al. Intracellular complement c5a/c5ar1 stabilizes β-catenin to promote colorectal tumorigenesis. Cell Rep. 2022, 39, 110851. [Google Scholar] [CrossRef] [PubMed]
- Vuran, E.; Karaarslan, A.; Karasartova, D.; Turegun, B.; Sahin, F. Identification of malassezia species from pityriasis versicolor lesions with a new multiplex pcr method. Mycopathologia 2014, 177, 41–49. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | n (%) |
|---|---|
| Sex ratio (M:F) | 11:8 |
| BMI * | 25.6 (16–43) |
| Diabetes mellitus | |
| Type I | 1 (5.3) |
| Type II | 3 (15.8) |
| Tobacco | 7 (36.8) |
| Alcohol | 7 (36.8) |
| Serum Bilirubin (mg/dL) * | 0.79 (0.0–13.0) |
| Histologic Diagnosis | |
| PDAC | 10 (52.6) |
| IPMN | 5 (26.3) |
| Chronic Pancreatitis | 2 (10.5) |
| Autoimmune Pancreatitis | 1 (5.3) |
| SCN | 1 (5.3) |
| Biliary Drainage | |
| ERCP | 5 (26.3) |
| With Stenting | 3 (15.8) |
| Preoperative Biopsy | 6 (31.6) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Speth, C.; Bellotti, R.; Schäfer, G.; Rambach, G.; Texler, B.; Thurner, G.C.; Öfner, D.; Lass-Flörl, C.; Maglione, M. Complement and Fungal Dysbiosis as Prognostic Markers and Potential Targets in PDAC Treatment. Curr. Oncol. 2022, 29, 9833-9854. https://doi.org/10.3390/curroncol29120773
Speth C, Bellotti R, Schäfer G, Rambach G, Texler B, Thurner GC, Öfner D, Lass-Flörl C, Maglione M. Complement and Fungal Dysbiosis as Prognostic Markers and Potential Targets in PDAC Treatment. Current Oncology. 2022; 29(12):9833-9854. https://doi.org/10.3390/curroncol29120773
Chicago/Turabian StyleSpeth, Cornelia, Ruben Bellotti, Georg Schäfer, Günter Rambach, Bernhard Texler, Gudrun C. Thurner, Dietmar Öfner, Cornelia Lass-Flörl, and Manuel Maglione. 2022. "Complement and Fungal Dysbiosis as Prognostic Markers and Potential Targets in PDAC Treatment" Current Oncology 29, no. 12: 9833-9854. https://doi.org/10.3390/curroncol29120773