
How to Distinguish Marfan Syndrome from Marfanoid Habitus in a Physical Examination—Comparison of External Features in Patients with Marfan Syndrome and Marfanoid Habitus

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
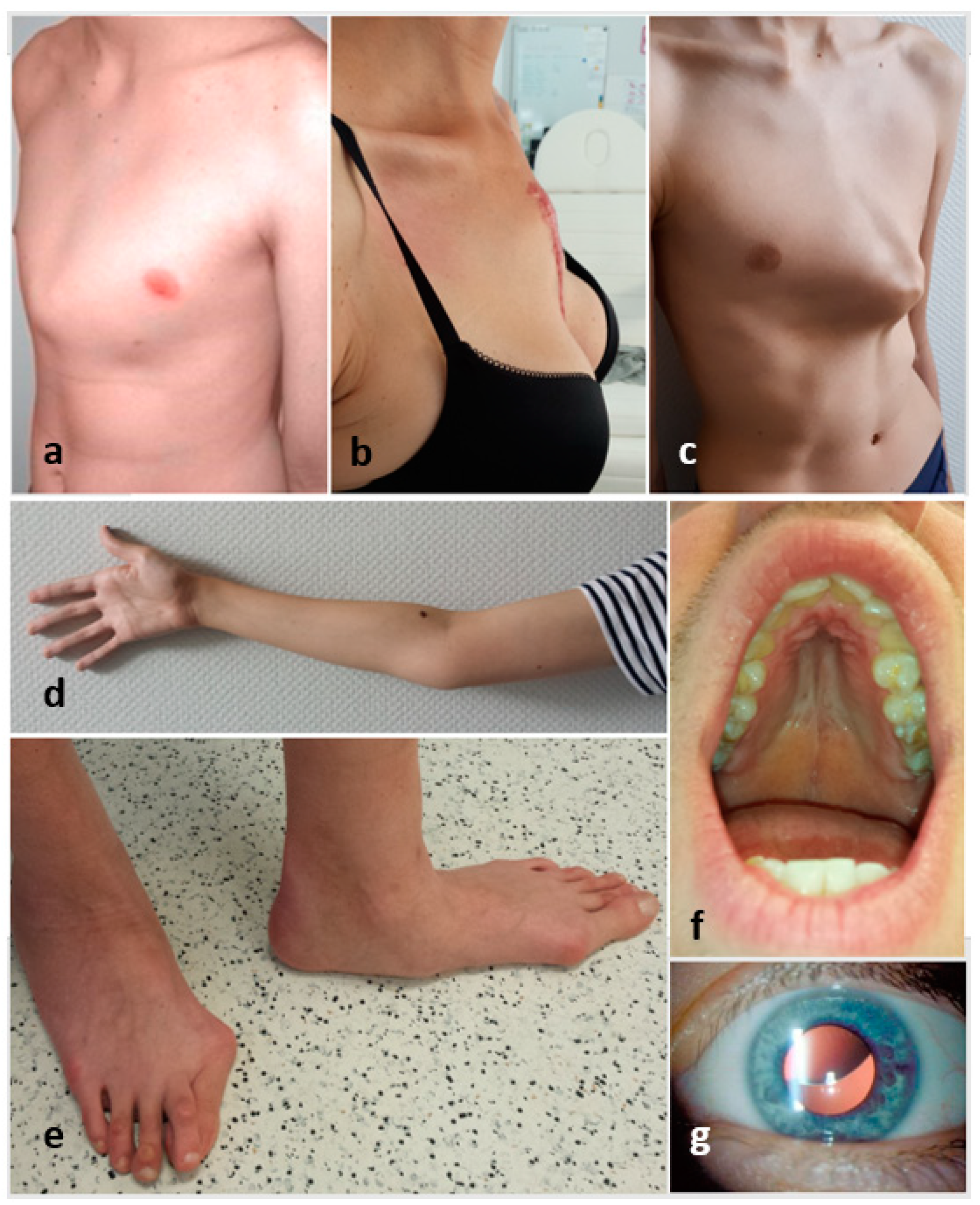
2.1. Detailed Description of the Medical History Taking Process and Physical Examination
2.2. Statistical Analysis
3. Results
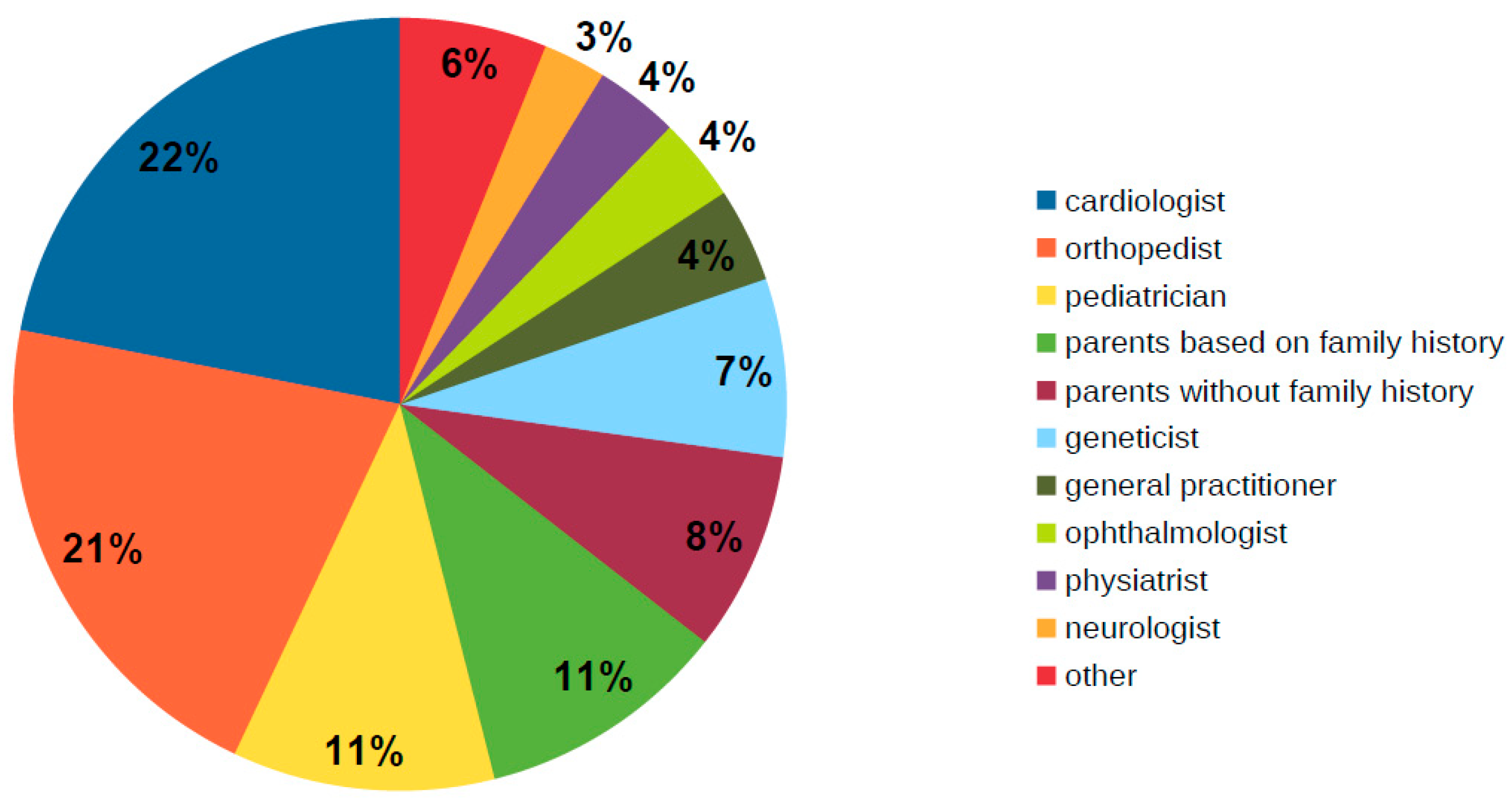
3.1. Patient Characteristics
3.2. Comparative Analysis
3.3. Logistic Regression Study
3.4. Red Flags
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Judge, D.P.; Dietz, H.C. Marfan’s syndrome. Lancet 2005, 366, 1965–1976. [Google Scholar] [CrossRef]
- Rybczynski, M.; Bernhardt, A.M.J.; Rehder, U.; Fuisting, B.; Meiss, L.; Voss, U.; Habermann, C.; Detter, C.; Robinson, P.N.; Arslan-Kirchner, M.; et al. The spectrum of syndromes and manifestations in individuals screened for suspected Marfan syndrome. Am. J. Med. Genet. Part A 2008, 146A, 3157–3166. [Google Scholar] [CrossRef]
- Murdoch, J.L.; Walker, B.A.; Halpern, B.L.; Kuzma, J.W.; McKusick, V.A. Life expectancy and causes of death in the Marfan syndrome. N. Engl. J. Med. 1972, 286, 804–808. [Google Scholar] [CrossRef]
- Erbel, R.; Aboyans, V.; Boileau, C.; Bossone, E.; Bartolomeo, R.D.; Eggebrecht, H.; Evangelista, A.; Falk, V.; Frank, H.; Gaemperli, O.; et al. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2873–2926. [Google Scholar] [CrossRef] [Green Version]
- Mullen, M.; Jin, X.Y.; Child, A.; Stuart, A.G.; Dodd, M.; Aragon-Martin, J.A.; Gaze, D.; Kiotsekoglou, A.; Yuan, L.; Hu, J.; et al. Irbesartan in Marfan syndrome (AIMS): A double-blind, placebo-controlled randomised trial. Lancet 2019, 394, 2263–2270. [Google Scholar] [CrossRef] [Green Version]
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef] [Green Version]
- Trawicka, A.; Bogdanowicz, M.; Woźniak-Mielczarek, L.; Sabiniewicz, R. Families of People with Marfan Syndrome. The Relationship Between the Functioning of the Family System and the Life Quality of the Affected Persons. Psychiatr. Pol. 2020, 55, 815–834. [Google Scholar] [CrossRef]
- Wozniak-Mielczarek, L.; Sabiniewicz, R.; Drezek-Nojowicz, M.; Nowak, R.; Gilis-Malinowska, N.; Mielczarek, M.; Łabuc, A.; Waldoch, A.; Wierzba, J. Differences in Cardiovascular Manifestation of Marfan Syndrome Between Children and Adults. Pediatric Cardiol. 2019, 40, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.N.; Trent, R.J. Aortic complications of Marfan’s syndrome. Lancet 1998, 352, 1722–1723. [Google Scholar] [CrossRef]
- Handisides, J.C.; Hollenbeck-Pringle, D.; Uzark, K.; Trachtenberg, F.L.; Pemberton, V.L.; Atz, T.W.; Bradley, T.J.; Cappella, E.; De Nobele, S.; Groh, G.K.; et al. Health-Related Quality of Life in Children and Young Adults with Marfan Syndrome. J. Pediatr. 2019, 204, 250–255.e1. [Google Scholar] [CrossRef]
- Gritti, A.; Pisano, S.; Catone, G.; Iuliano, R.; Salvati, T.; Gritti, P. Psychiatric and neuropsychological issues in Marfan syndrome:A critical review of the literature. Int. J. Psychiatry Med. 2015, 50, 347–360. [Google Scholar] [CrossRef]
- Stark, V.C.; Arndt, F.; Harring, G.; von Kodolitsch, Y.; Kozlik-Feldmann, R.; Mueller, G.C.; Steiner, K.J.; Mir, T.S. Kid-Short Marfan Score (Kid-SMS) Is a Useful Diagnostic Tool for Stratifying the Pre-Test Probability of Marfan Syndrome in Childhood. Diseases 2015, 3, 24. [Google Scholar] [CrossRef] [Green Version]
- Mueller, G.C.; Stark, V.; Steiner, K.; Weil, J.; von Kodolitsch, Y.; Mir, T.S. The Kid-Short Marfan Score (Kid-SMS)—An easy executable risk score for suspected paediatric patients with Marfan syndrome. Acta Paediatr. 2013, 102, e84–e89. [Google Scholar] [CrossRef]
- Ting, B.L.; Mathur, D.; Loeys, B.L.; Dietz, H.C., 3rd; Sponseller, P.D. The diagnostic value of the facial features of Marfan syndrome. J. Child Orthop. 2010, 4, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Monteil, D.C.; Shikany, A.; Aljeaid, D.; Parrott, A.; Tretter, J.T.; James, J.; Martin, L.J.; Weaver, K.N. Comparison of Evolution of Aortic Root Dilation and Ghent Criteria in Preadolescents and Adolescents with and without Marfan Syndrome. J. Pediatrics 2020, 221, 188–195.e1. [Google Scholar] [CrossRef]
- Faivre, L.; Masurel-Paulet, A.; Collod-Béroud, G.; Callewaert, B.L.; Child, A.H.; Stheneur, C.; Binquet, C.; Gautier, E.; Chevallier, B.; Huet, F.; et al. Clinical and molecular study of 320 children with Marfan syndrome and related type I fibrillinopathies in a series of 1009 probands with pathogenic FBN1 mutations. Pediatrics 2009, 123, 391–398. [Google Scholar] [CrossRef]
- Stheneur, C.; Tubach, F.; Jouneaux, M.; Roy, C.; Benoist, G.; Chevallier, B.; Boileau, C.; Jondeau, G. Study of phenotype evolution during childhood in Marfan syndrome to improve clinical recognition. Genet. Med. 2014, 16, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Roman, M.J.; Devereux, R.B.; Preiss, L.R.; Asch, F.M.; Eagle, K.A.; Holmes, K.W.; LeMaire, S.A.; Maslen, C.L.; Milewicz, D.M.; Morris, S.A.; et al. Associations of Age and Sex With Marfan Phenotype: The National Heart, Lung, and Blood Institute GenTAC (Genetically Triggered Thoracic Aortic Aneurysms and Cardiovascular Conditions) Registry. Circ. Cardiovasc. Genet. 2017, 10, e001647. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Thumb sign | Positive when after clenching one’s fist, the distal phalanx of the adducted thumb is beyond the ulnar border of the palm (in order to reach maximal effect, assistance by the examiner is acceptable). |
| Wrist sign | Positive when, during the wrapping of the contralateral wrist, the thumb covers the whole nail of the fifth finger. |
| US/LS (upper segment to lower segment ratio) | The lower segment is measured in the standing position with the patient leaning against a wall, from the top of the symphysis pubis to the floor, whereas the upper segment is the difference between the patients’ height and the lower segment. It is considered reduced if it is <0.85. |
| ASHR (arm span to height ratio) | It is considered increased if it is >1.05. |
| Reduced elbow extension | Defined as an angle between the forearm and arm lesser than 170 (degrees). |
| Dolichocephaly | Defined as the ratio between the width and length of the skull, with a value ranging between 0.6–0.76. |
| Stretch marks | It is a clinically important sign when there is no connection between their presence and major weight fluctuations (or pregnancy) and also when they are located at an unusual area, such as the lumbar region, upper arm, mid-back or thigh. |
| Hindfoot deformity | It is a combination of hindfoot valgus with forefoot abduction and the lowering of the midfoot (previously referred to as the medial rotation of the medial malleolus). It should be evaluated from the anterior and posterior view and should be distinguished from the more common “flat foot” without significant hindfoot valgus. |
| Excessive growth (in children) remarkably high stature (in adults) | It is height equal to and above the 97th percentile in children, whilst it is equal to and above 176 cm for women and equal to and above 190 cm for men in the adult population. |
| Deficiency in weight | It is clinically important when BMI is under 18.5 kg/m2 in the adult population whilst in the child population BMI equal and under the 5th percentile. |
| Moderate or severe scoliosis | Scoliosis where the Cobb angle is more than 20 degrees. |
| Moderate or high myopia | It is myopia equal or higher than 3 dioptres |
| Feature | Child and Adult Population (n = 277) | Child Population (n = 178) | Adult Population (n = 99) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Marfan Syndrome (n = 109) | Marfanoid Habitus (n = 168) | p | Marfan Syndrome (n = 46) | Marfanoid Habitus (n = 132) | p | Marfan Syndrome (n = 63) | Marfanoid Habitus (n = 36) | p | |
| deficiency in weight | 27 (24.8) | 42 (25.0) | 0.959 | 13 (28.3) | 36(27.3) | 0.791 | 14 (22.2) | 6 (16.7) | 0.517 |
| excessive growth/remarkably high stature | 54 (49.5) | 46 (27.4) | <0.001 | 19 (41.3) | 33 (25.0) | 0.018 | 35 (55.6) | 13 (36.1) | 0.068 |
| ASHR >1.05 | 37 (35.2) | 18 (10.9) | <0.001 | 19 (43.2) | 13 (10.0) | <0.001 | 18 (29.5) | 5 (14.3) | 0.093 |
| USLS < 0.85 | 95 (87.2) | 155 (92.3) | 0.162 | 40 (87.0) | 122 (92.4) | 0.367 | 55 (87.3) | 33 (91.7) | 0.506 |
| joint laxity | 69 (63.3) | 106 (63.1) | 0.972 | 31 (67.4) | 92 (69.7) | 0.771 | 38 (60.3) | 14 (38.9) | 0.040 |
| wrist sign | 57 (52.3) | 68 (40.5) | 0.053 | 25 (54.3) | 56 (42.4) | 0.162 | 32 (50.8) | 12 (33.3) | 0.093 |
| thumb sign | 75 (68.8) | 97 (57.7) | 0.064 | 34 (73.9) | 85 (64.4) | 0.238 | 41 (65.1) | 12 (33.3) | 0.002 |
| scoliosis | 86 (78.9) | 119 (70.8) | 0.135 | 35 (76.1) | 92 (69.7) | 0.409 | 51 (81.0) | 27 (75.0) | 0.486 |
| moderate or severe scoliosis | 15 (13.8) | 15 (8.9) | 0.206 | 3 (6.5) | 12 (9.1) | 0.763 | 12 (19.0) | 3 (8.3) | 0.153 |
| pectus excavatum | 27 (24.8) | 42 (25.0) | 0.966 | 16 (34.8) | 38 (28.8) | 0.446 | 11 (17.5) | 4 (11.1) | 0.397 |
| pectus carinatum | 44 (40.4) | 22 (13.1) | <0.001 | 12 (26.1) | 17 (12.9) | 0.037 | 32 (50.8) | 5 (13.9) | <0.001 |
| reduced elbow extension | 13 (11.9) | 4 (2.4) | 0.001 | 6 (13.0) | 4 (3.0) | 0.020 | 7 (11.1) | 0 (0.0) | 0.046 |
| flat feet | 64 (58.7) | 91 (54.2) | 0.456 | 31 (67.4) | 76 (57.6) | 0.242 | 33 (52.4) | 15 (41.7) | 0.305 |
| hindfoot deformity | 42 (38.5) | 36 (21.4) | 0.002 | 25 (54.3) | 35 (26.5) | 0.001 | 17 (27.0) | 1 (2.8) | 0.003 |
| stretch marks | 55 (50.5) | 42 (25.0) | <0.001 | 16 (34.8) | 32 (24.2) | 0.165 | 39 (61.9) | 10 (27.8) | 0.001 |
| dolichocephaly | 45 (41.3) | 68 (40.5) | 0.894 | 26 (56.5) | 59 (44.7) | 0.167 | 19 (30.2) | 9 (25.0) | 0.584 |
| dental crowding | 54 (49.5) | 53 (31.5) | 0.003 | 24 (52.2) | 42 (31.8) | 0.014 | 30 (47.6) | 11 (30.6) | 0.097 |
| gothic palate | 71 (65.1) | 43 (25.6) | <0.001 | 30 (65.2) | 33 (25.0) | <0.001 | 41 (65.1) | 10 (27.8) | <0.001 |
| downslanting palpebral fissures | 52 (47.7) | 42 (25.0) | <0.001 | 24 (52.2) | 35 (26.5) | 0.001 | 28 (44.4) | 7 (19.4) | 0.012 |
| retrognathia | 33 (30.3) | 40 (23.8) | 0.233 | 16 (34.8) | 31 (23.5) | 0.134 | 17 (27.0) | 9 (25.0) | 0.829 |
| micrognation | 55 (50.5) | 64 (38.1) | 0.042 | 24 (52.2) | 44 (33.3) | 0.024 | 31 (49.2) | 20 (55.6) | 0.543 |
| enophthalmia | 71 (65.1) | 95 (56.5) | 0.154 | 28 (60.9) | 78 (59.1) | 0.832 | 43 (68.3) | 17 (47.2) | 0.039 |
| malar hypoplasia | 53 (48.6) | 75 (44.6) | 0.516 | 20 (43.5) | 59 (44.7) | 0.886 | 33 (52.4) | 16 (44.4) | 0.447 |
| age at the time of first suspicion of MFS | 12.9 ± 12.7 | 14.4 ± 7.9 | 0.307 | 5.9 ± 4.1 | 12.0 ± 4.1 | <0.001 | 17.8 ± 14.3 | 23.6 ± 11.6 | 0.035 |
| birth weight | 3514.3 ± 541.0 | 3458.7 ± 575.8 | 0.575 | 3556.9 ± 459.8 | 3477.9 ± 558.2 | 0.456 | - | - | - |
| birth length | 55.5 ± 3.4 | 56.1 ± 3.3 | 0.405 | 55.9 ± 2.9 | 56.0 ± 3.3 | 0.925 | - | - | - |
| joint pain | 57 (52.3) | 68 (40.5) | 0.053 | 24 (52.2) | 47 (35.6) | 0.048 | 33 (52.4) | 21 (58.3) | 0.567 |
| frequent headaches | 44 (40.4) | 53 (31.5) | 0.133 | 13 (28.3) | 32 (24.2) | 0.589 | 31 (49.2) | 21 (58.3) | 0.382 |
| dizziness | 34 (31.2) | 24 (14.3) | 0.001 | 6 (13.0) | 13 (9.8) | 0.582 | 28 (44.4) | 11 (30.6) | 0.174 |
| syncope | 30 (27.5) | 31 (18.5) | 0.075 | 9 (19.6) | 23 (17.4) | 0.745 | 21 (33.3) | 8 (22.2) | 0.243 |
| chest pain | 37 (33.9) | 42 (25.0) | 0.107 | 8 (17.4) | 27 (20.5) | 0.653 | 29 (46.0) | 15 (41.7) | 0.674 |
| palpitations | 27 (24.8) | 24 (14.3) | 0.028 | 5 (10.9) | 14 (10.6) | 1.000 | 22 (34.9) | 10 (27.8) | 0.465 |
| coordination disorders | 38 (34.9) | 59 (35.1) | 1.000 | 18 (39.1) | 49 (37.1) | 0.809 | 20 (31.7) | 10 (27.8) | 0.679 |
| hernias | 26 (23.9) | 16 (9.5) | 0.001 | 9 (19.6) | 12 (9.1) | 0.058 | 17 (27.0) | 4 (11.1) | 0.063 |
| idiopathic pulmonary oedema | 6 (5.5) | 3 (1.8) | 0.161 | 2 (4.3) | 2 (1.5) | 0.275 | 4 (6.3) | 1 (2.8) | 0.650 |
| multiple injuries | 15 (13.8) | 19 (11.3) | 0.543 | 4 (8.7) | 11 (8.3) | 1.000 | 11 (17.5) | 8 (22.2) | 0.563 |
| effort tolerance worse than that of peers | 54 (49.5) | 57 (33.9) | 0.010 | 19 (41.3) | 37 (28.0) | 0.095 | 35 (55.6) | 20 (55.6) | 1.000 |
| lens subluxation | 45 (41.3) | 2 (1.2) | <0.001 | 18 (39.1) | 1 (0.8) | <0.001 | 27 (42.9) | 1 (2.8) | <0.001 |
| myopia ≥ 3 dioptres | 43 (39.4) | 21 (12.5) | <0.001 | 14 (30.4) | 13 (9.8) | 0.001 | 29 (46.0) | 8 (22.2) | 0.018 |
| Feature | Child and Adult Population (n = 277) | Child Population (n = 178) | Adult Population (n = 99) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| p | HR | 95%CI | p | HR | 95%CI | p | HR | 95%CI | |
| ASHR > 1.05 | 0.035 | 3.48 | 1.09–11.11 | 0.003 | 11.48 | 2.29–57.48 | 0.810 | 0.73 | 0.06–9.13 |
| thumb sign | 0.643 | 1.32 | 0.41–4.20 | 0.888 | 1.13 | 0.21–6.03 | 0.269 | 3.78 | 0.36–40.07 |
| wrist sign | 0.939 | 0.96 | 0.32–2.84 | 0.875 | 0.88 | 0.18–4.36 | 0.622 | 0.56 | 0.05–5.74 |
| pectus carinatum | 0.048 | 3.00 | 1.01–8.92 | 0.808 | 0.80 | 0.13–4.94 | 0.131 | 4.75 | 0.63–35.79 |
| scoliosis | 0.557 | 1.40 | 0.45–4.35 | - | - | - | - | - | - |
| assymetry of the chest | 0.410 | 1.58 | 0.53–4.65 | 0.119 | 4.33 | 0.69–27.38 | - | - | - |
| joint laxity | - | - | - | - | - | - | 0.179 | 5.82 | 0.45–76.04 |
| reduced elbow extension | 0.816 | 1.27 | 0.17–9.60 | 0.396 | 3.02 | 0.24–38.59 | 0.999 | 168.49 | 0.76–9605.20 |
| enophthalmia | 0.381 | 1.53 | 0.59–3.93 | - | - | - | 0.040 | 9.60 | 1.11–82.89 |
| micrognation | 0.336 | 0.62 | 0.23–1.64 | 0.545 | 0.58 | 1.00–3.39 | - | - | - |
| retrognathia | - | - | - | 0.919 | 0.91 | 0.15–5.63 | - | - | - |
| downslanting palpebral fissures | 0.167 | 1.94 | 0.76–4.98 | 0.152 | 2.71 | 0.69–10.58 | 0.807 | 0.77 | 1.00–6.23 |
| gothic palate | 0.221 | 1.79 | 0.71–4.53 | 0.534 | 1.59 | 0.37–6.78 | 0.908 | 1.13 | 0.14–9.42 |
| dental crowding | 0.403 | 1.52 | 0.57–4.06 | 0.228 | 2.43 | 0.57–10.26 | 0.361 | 2.60 | 0.34–20.18 |
| stretch marks | 0.159 | 2.04 | 0.76–5.50 | - | - | - | 0.114 | 5.11 | 0.68–38.53 |
| hindfoot deformity | 0.005 | 4.54 | 1.57–13.20 | 0.017 | 6.02 | 1.39–26.15 | 0.119 | 10.61 | 0.55–206.61 |
| myopia ≥ 3 D | 0.224 | 2.00 | 0.65–6.15 | 0.732 | 0.67 | 0.07–6.59 | 0.804 | 1.31 | 0.16–10.82 |
| lens subluxation | <0.001 | 67.12 | 6.51–691.63 | 0.001 | 78.91 | 5.51–1130.44 | 0.998 | 364.67 | 0.78–3698.90 |
| excessive growth/remarkably high stature | 0.001 | 5.46 | 1.99–14.93 | 0.076 | 3.87 | 0.87–17.25 | 0.044 | 14.54 | 1.07–197.83 |
| hernias | 0.004 | 6.45 | 1.83–22.67 | 0.044 | 7.25 | 0.87–17.25 | 0.574 | 2.16 | 0.15–31.86 |
| idiopathic pulmonary oedema | 0.758 | 1.43 | 0.15–14.10 | - | - | - | - | - | - |
| Children | Adults |
|---|---|
| pectus carinatum | |
| reduced elbow extension | |
| hindfoot deformity | |
| gothic palate | |
| downslanting palpebral fissures | |
| lens subluxation | |
| myopia ≥ 3 dioptres | |
| excessive growth/remarkably high stature | |
| ASHR > 1.05 | thumb sign |
| dental crowding | joint laxity |
| micrognation | stretch marks |
| hernias | enophthalmia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wozniak-Mielczarek, L.; Osowicka, M.; Radtke-Lysek, A.; Drezek-Nojowicz, M.; Gilis-Malinowska, N.; Sabiniewicz, A.; Mielczarek, M.; Sabiniewicz, R. How to Distinguish Marfan Syndrome from Marfanoid Habitus in a Physical Examination—Comparison of External Features in Patients with Marfan Syndrome and Marfanoid Habitus. Int. J. Environ. Res. Public Health 2022, 19, 772. https://doi.org/10.3390/ijerph19020772
Wozniak-Mielczarek L, Osowicka M, Radtke-Lysek A, Drezek-Nojowicz M, Gilis-Malinowska N, Sabiniewicz A, Mielczarek M, Sabiniewicz R. How to Distinguish Marfan Syndrome from Marfanoid Habitus in a Physical Examination—Comparison of External Features in Patients with Marfan Syndrome and Marfanoid Habitus. International Journal of Environmental Research and Public Health. 2022; 19(2):772. https://doi.org/10.3390/ijerph19020772
Chicago/Turabian StyleWozniak-Mielczarek, Lidia, Michalina Osowicka, Alicja Radtke-Lysek, Magda Drezek-Nojowicz, Natasza Gilis-Malinowska, Anna Sabiniewicz, Maksymilian Mielczarek, and Robert Sabiniewicz. 2022. "How to Distinguish Marfan Syndrome from Marfanoid Habitus in a Physical Examination—Comparison of External Features in Patients with Marfan Syndrome and Marfanoid Habitus" International Journal of Environmental Research and Public Health 19, no. 2: 772. https://doi.org/10.3390/ijerph19020772