
Telomere Length and Hearing Loss: A Two-Sample Mendelian Randomization

Abstract
:1. Introduction
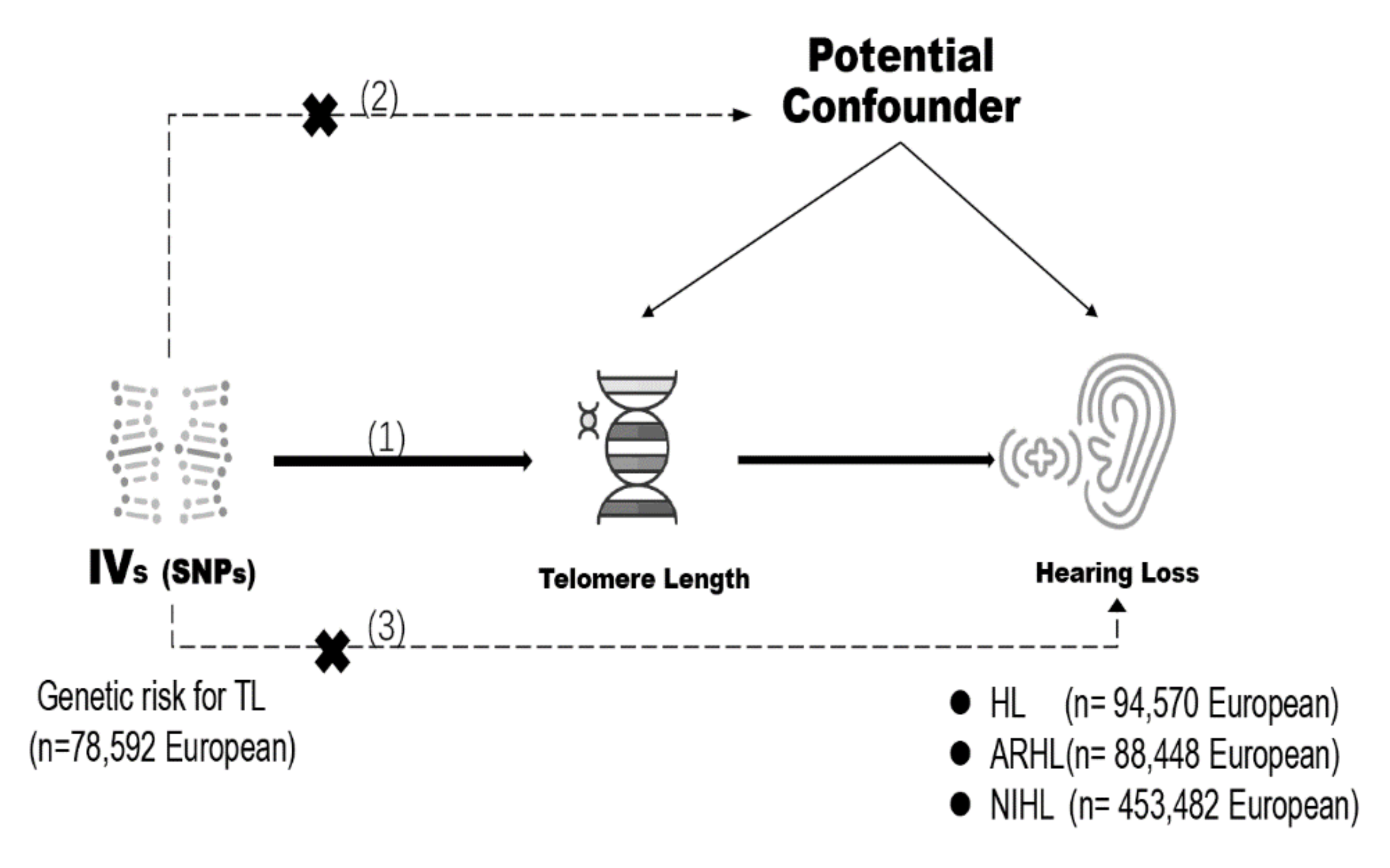
2. Materials and Methods
2.1. Genetic Instrument Selection
2.2. Genetic Summary Data for Hearing Loss
2.3. Mendelian Randomization Estimates
2.4. Statistical Analysis
3. Results
3.1. Selection of Instrumental Variables
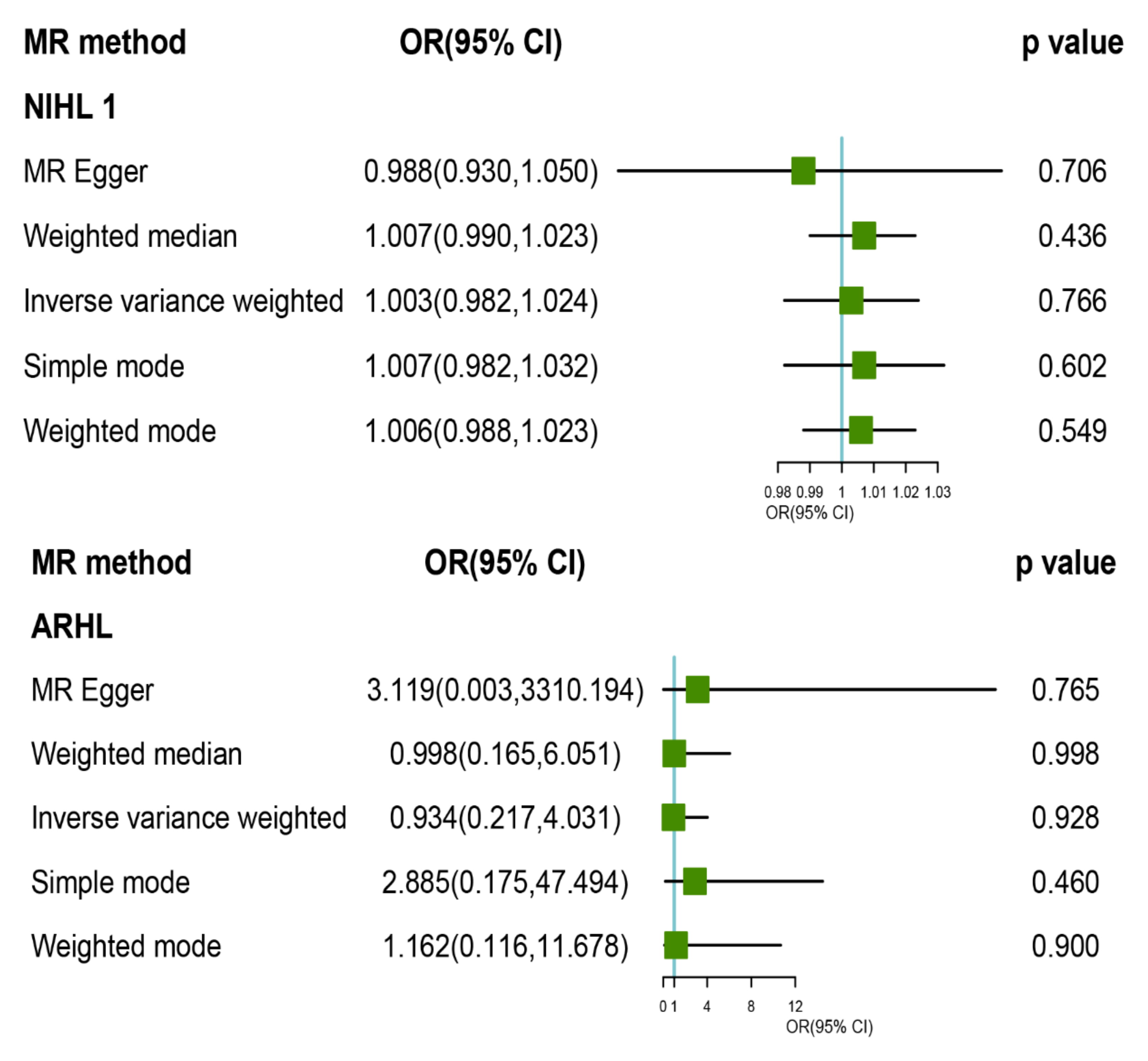
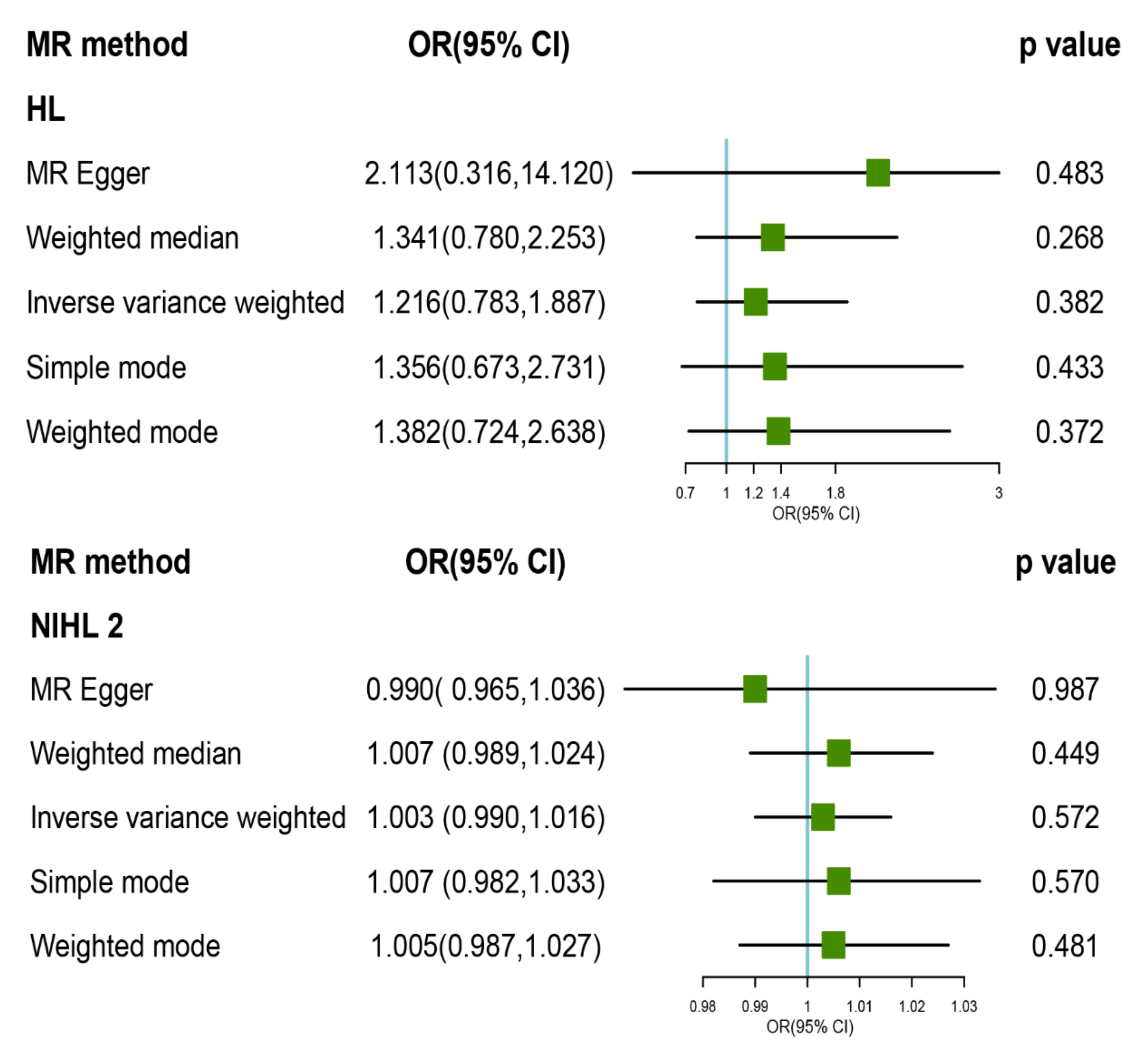
3.2. MR Analysis of TL with Risk of HL and Its Subtypes
3.3. Pleiotropy and Sensitivity Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Eyken, E.; Van Camp, G.; Van Laer, L. The Complexity of Age-Related Hearing Impairment: Contributing Environmental and Genetic Factors. Audiol. Neurotol. 2007, 12, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Przewozny, T.; Gojska-Grymajlo, A.; Kwarciany, M.; Gasecki, D.; Narkiewicz, K. Hypertension and cochlear hearing loss. Blood Press. 2015, 24, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Dhukhwa, A.; Bhatta, P.; Sheth, S.; Korrapati, K.; Tieu, C.; Mamillapalli, C.; Ramkumar, V.; Mukherjea, D. Targeting Inflammatory Processes Mediated by TRPVI and TNF-α for Treating Noise-Induced Hearing Loss. Front. Cell. Neurosci. 2019, 13, 444. [Google Scholar] [CrossRef] [Green Version]
- Park, D.J.; Ha, S.; Choi, J.S.; Lee, S.H.; Park, J.E.; Seo, Y.J. Induced Short-Term Hearing Loss due to Stimulation of Age-Related Factors by Intermittent Hypoxia, High-Fat Diet, and Galactose Injection. Int. J. Mol. Sci. 2020, 21, 7068. [Google Scholar] [CrossRef]
- Griffiths, T.D.; Lad, M.; Kumar, S.; Holmes, E.; McMurray, B.; Maguire, E.A.; Billig, A.J.; Sedley, W. How Can Hearing Loss Cause Dementia? Neuron 2020, 108, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Chadha, S.; Kamenov, K.; Cieza, A. The world report on hearing, 2021. Bull. World Health Organ. 2021, 99, 242. [Google Scholar] [CrossRef] [PubMed]
- Haile, L.M.; Kamenov, K.; Briant, P.S.; Orji, A.U.; Steinmetz, J.D.; Abdoli, A.; Abdollahi, M.; Abu-Gharbieh, E.; Afshin, A.; Ahmed, H.; et al. Hearing loss prevalence and years lived with disability, 1990-2019: Findings from the Global Burden of Disease Study 2019. Lancet 2021, 397, 996–1009. [Google Scholar] [CrossRef]
- Brown, C.S.; Emmett, S.D.; Robler, S.K.; Tucci, D.L. Global Hearing Loss Prevention. Otolaryngol. Clin. N. Am. 2018, 51, 575–592. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Epel, E.S.; Lin, J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 2015, 350, 1193–1198. [Google Scholar] [CrossRef] [Green Version]
- Blasco, M.A. Telomeres and human disease: Ageing, cancer and beyond. Nat. Rev. Genet. 2005, 6, 611–622. [Google Scholar] [CrossRef]
- Opresko, P.L.; Shay, J.W. Telomere-associated aging disorders. Ageing Res. Rev. 2017, 33, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Demanelis, K.; Jasmine, F.; Chen, L.S.; Chernoff, M.; Tong, L.; Delgado, D.; Zhang, C.; Shinkle, J.; Sabarinathan, M.; Lin, H.; et al. Determinants of telomere length across human tissues. Science 2020, 369, eaaz6876. [Google Scholar] [CrossRef] [PubMed]
- Aviv, A. Genetics of leukocyte telomere length and its role in atherosclerosis. Mutat. Res. 2012, 730, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Luo, H.; Yang, T.; Wu, H.; Chen, D. Association of leukocyte telomere length and the risk of age-related hearing impairment in Chinese Hans. Sci. Rep. 2017, 7, 10106. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, D.; Ma, H.; Li, C.; Wang, S.; Wang, Y.; Yang, L.; Xu, L. Association between Leucocyte Telomere Length and Risk of Hearing Loss in the General Population: A Case-Control Study in Zhejiang Province, China. Int. J. Environ. Res. Public Health 2020, 17, 1881. [Google Scholar] [CrossRef] [Green Version]
- Greene, C.C.; McMillan, P.M.; Barker, S.E.; Kurnool, P.; Lomax, M.I.; Burmeister, M.; Lesperance, M.M. DFNA25, a novel locus for dominant nonsyndromic hereditary hearing impairment, maps to 12q21-24. Am. J. Hum. Genet. 2001, 68, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.Y.; Riazuddin, S.; Shahzad, M.; Ahmed, N.; Zafar, A.U.; Rehman, A.U.; Morell, R.J.; Griffith, A.J.; Ahmed, Z.M.; Riazuddin, S.; et al. DFNB79: Reincarnation of a nonsyndromic deafness locus on chromosome 9q34.3. Eur. J. Hum. Genet. EJHG 2010, 18, 125–129. [Google Scholar] [CrossRef]
- Tayebi, N.; Jamsheer, A.; Flöttmann, R.; Sowinska-Seidler, A.; Doelken, S.C.; Oehl-Jaschkowitz, B.; Hülsemann, W.; Habenicht, R.; Klopocki, E.; Mundlos, S.; et al. Deletions of exons with regulatory activity at the DYNC1I1 locus are associated with split-hand/split-foot malformation: Array CGH screening of 134 unrelated families. Orphanet J. Rare Dis. 2014, 9, 108. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Nguyen, M.T.; Sung, V.; Grobler, A.; Burgner, D.; Saffery, R.; Wake, M. Associations between Telomere Length and Hearing Status in Mid-Childhood and Midlife: Population-Based Cross-Sectional study. Ear Hear. 2019, 40, 1256–1259. [Google Scholar] [CrossRef]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef] [Green Version]
- Burgess, S.; Scott, R.A.; Timpson, N.J.; Davey Smith, G.; Thompson, S.G. Using published data in Mendelian randomization: A blueprint for efficient identification of causal risk factors. Eur. J. Epidemiol. 2015, 30, 543–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, N.M.; Holmes, M.V.; Davey Smith, G. Reading Mendelian randomisation studies: A guide, glossary, and checklist for clinicians. BMJ Clin. Res. Ed. 2018, 362, k601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, E.L.; Nandakumar, S.K.; Liao, X.; Bick, A.G.; Karjalainen, J.; Tabaka, M.; Gan, O.I.; Havulinna, A.S.; Kiiskinen, T.T.J.; Lareau, C.A.; et al. Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature 2020, 586, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.L.; Ahsan, H.; VanderWeele, T.J. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int. J. Epidemiol. 2011, 40, 740–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brion, M.J.A.; Shakhbazov, K.; Visscher, P.M. Calculating statistical power in Mendelian randomization studies. Int. J. Epidemiol. 2013, 42, 1497–1501. [Google Scholar] [CrossRef] [Green Version]
- Didelez, V.; Sheehan, N. Mendelian randomization as an instrumental variable approach to causal inference. Stat. Methods Med. Res. 2007, 16, 309–330. [Google Scholar] [CrossRef]
- Sekula, P.; Del Greco, M.F.; Pattaro, C.; Kottgen, A. Mendelian Randomization as an Approach to Assess Causality Using Observational Data. J. Am. Soc. Nephrol. 2016, 27, 3253–3265. [Google Scholar] [CrossRef] [Green Version]
- Nazarzadeh, M.; Pinho-Gomes, A.C.; Bidel, Z.; Dehghan, A.; Canoy, D.; Hassaine, A.; Ayala Solares, J.R.; Salimi-Khorshidi, G.; Smith, G.D.; Otto, C.M.; et al. Plasma lipids and risk of aortic valve stenosis: A Mendelian randomization study. Eur. Hear. J. 2020, 41, 3913–3920. [Google Scholar] [CrossRef] [Green Version]
- Loh, N.Y.; Noordam, R.; Christodoulides, C. Telomere length and metabolic syndrome traits: A Mendelian randomisation study. Aging Cell 2021, 20, e13445. [Google Scholar] [CrossRef]
- Bowden, J.; Smith, G.D.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef] [Green Version]
- Bowden, J.; Smith, G.D.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbanck, M.; Chen, C.Y.; Neale, B.; Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Hemani, G.; Zhengn, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 2018, 7, e34408. [Google Scholar] [CrossRef] [PubMed]
- Haycock, P.C.; Burgess, S.; Nounu, A.; Zheng, J.; Okoli, G.N.; Bowden, J.; Wade, K.H.; Timpson, N.J.; Evans, D.M.; Willeit, P.; et al. Association Between Telomere Length and Risk of Cancer and Non-Neoplastic Diseases: A Mendelian Randomization Study. JAMA Oncol. 2017, 3, 636–651. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Stoma, S.; Lotta, L.A.; Warner, S.; Albrecht, E.; Allione, A.; Arp, P.P.; Broer, L.; Buxton, J.L.; Da Silva Couto Alves, A.; et al. Genome-wide Association Analysis in Humans Links Nucleotide Metabolism to Leukocyte Telomere Length. Am. J. Hum. Genet. 2020, 106, 389–404. [Google Scholar] [CrossRef] [Green Version]
- Konings, A.; Van Laer, L.; Van Camp, G. Genetic studies on noise-induced hearing loss: A review. Ear Hear. 2009, 30, 151–159. [Google Scholar] [CrossRef]
- Sliwinska-Kowalska, M.; Pawelczyk, M. Contribution of genetic factors to noise-induced hearing loss: A human studies review. Mutat. Res. 2013, 752, 61–65. [Google Scholar] [CrossRef]
- Wu, T.Y.; Lin, J.N.; Luo, Z.Y.; Hsu, C.J.; Wang, J.S.; Wu, H.P. 2,3,4’,5-Tetrahydroxystilbene-2-O-β-D-Glucoside (THSG) Activates the Nrf2 Antioxidant Pathway and Attenuates Oxidative Stress-Induced Cell Death in Mouse Cochlear UB/OC-2 Cells. Biomolecules 2020, 10, 465. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Senescence and immortalization: Role of telomeres and telomerase. Carcinogenesis 2005, 26, 867–874. [Google Scholar] [CrossRef]
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 5, 4172. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Li, X.; Zhang, X.; Zhang, J.; Sun, Q.; Xu, X.; Sun, M.; Tian, Q.; Li, Q.; Wang, H.; et al. No Causal Effect of Telomere Length on Ischemic Stroke and Its Subtypes: A Mendelian Randomization Study. Cells 2019, 8, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasching, C.L. Telomere length measurement as a clinical biomarker of aging and disease. Crit. Rev. Clin. Lab. Sci. 2018, 55, 443–465. [Google Scholar] [CrossRef] [PubMed]
- Chaitidis, N.; Theocharisz, P.; Festas, C.; Aritzi, I. Association of rheumatoid arthritis with hearing loss: A systematic review and meta-analysis. Rheumatol. Int. 2020, 40, 1771–1779. [Google Scholar] [CrossRef]
- Xie, S.B.; Ning, H.T.; She, Y.C.; Jing, Q.C.; Jiang, Q.; Zhang, Y.N.; Mei, L.Y.; Feng, Y.; Wu, X.W. Effect of systemic lupus erythematosus and rheumatoid arthritis on sudden sensorineural hearing loss. Laryngoscope 2020, 130, 2475–2480. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.K.; Loh, P.R.; Finucane, H.K.; Ripke, S.; Yang, J.; Schizophrenia Working Group of the Psychiatric Genomics Consortium; Patterson, N.; Daly, M.J.; Price, A.L.; Neale, B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015, 47, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Morrison, J.; Knoblauch, N.; Marcus, J.H.; Stephens, M.; He, X. Mendelian randomization accounting for correlated and uncorrelated pleiotropic effects using genome-wide summary statistics. Nat. Genet. 2020, 52, 750. [Google Scholar] [CrossRef]
- Wang, J.S.; Zhao, Q.Y.; Bowden, J.; Hemani, G.; Smith, G.D.; Small, D.S.; Zhang, N.R. Causal inference for heritable phenotypic risk factors using heterogeneous genetic instruments. PLoS Genet. 2021, 17, e1009575. [Google Scholar] [CrossRef]
- Gurung, R.L.; Yiamunaa, M.; Moh, A.M.C.; Dorajoo, R.; Liu, S.; Liu, J.J.; Shabbir, A.; So, J.B.Y.; Tan, C.H.; Cheng, A.K.S.; et al. Correlation of Telomere Length in Adipose Tissue and Leukocytes and its Association with Postsurgical Weight Loss. Obesity 2020, 28, 2424–2430. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| SNP | Chr | Nearby Gene | EA | MAF | β | SE | p-Value | F Statistic |
|---|---|---|---|---|---|---|---|---|
| rs10936600 | 3 | LRRC34 | T | 0.243 | −0.086 | 0.006 | 6.00 × 10−51 | 225.300 |
| rs7705526 | 5 | TERT | A | 0.328 | 0.082 | 0.006 | 5.00 × 10−45 | 198.300 |
| rs4691895 | 4 | NAF1 | C | 0.783 | 0.058 | 0.006 | 1.00 × 10−21 | 91.000 |
| rs9419958 | 10 | STN1 | C | 0.862 | −0.064 | 0.007 | 5.00 × 10−19 | 79.500 |
| rs75691080 | 20 | STM3 | T | 0.091 | −0.067 | 0.009 | 6.00 × 10−14 | 56.500 |
| rs59294613 | 7 | POT1 | A | 0.293 | −0.041 | 0.005 | 1.00 × 10−13 | 55.100 |
| rs8105767 | 19 | ZNF257 | G | 0.289 | 0.039 | 0.005 | 5.00 × 10−13 | 52.100 |
| rs3219104 | 1 | PARP1 | C | 0.830 | 0.042 | 0.006 | 9.00 × 10−11 | 42.000 |
| rs2736176 | 6 | AIF1 | C | 0.313 | 0.034 | 0.005 | 3.00 × 10−10 | 39.400 |
| rs3785074 | 16 | TERF2 | G | 0.263 | 0.035 | 0.006 | 4.00 × 10−10 | 38.900 |
| rs7194734 | 16 | MPHOSPH6 | T | 0.782 | −0.037 | 0.006 | 7.00 × 10−10 | 38.100 |
| rs228595 | 11 | ATM | A | 0.417 | −0.028 | 0.005 | 1.00 × 10−8 | 32.200 |
| rs2302588 | 14 | DCAF4 | C | 0.100 | 0.048 | 0.008 | 2.00 × 10−8 | 31.900 |
| rs13137667 | 4 | MOB1B | C | 0.959 | 0.077 | 0.014 | 2.00 × 10−8 | 31.200 |
| rs55749605 | 3 | SENP7 | A | 0.579 | −0.037 | 0.007 | 2.00 × 10−8 | 31.200 |
| rs62053580 | 16 | RFWD3 | G | 0.169 | −0.039 | 0.007 | 4.00 × 10−8 | 30.200 |
| HLs | IVW | WM | MR Egger | MR_ PRESSO | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | p | Cochran Q Statistics (df) | I2 | p | OR (95% CI) | p | OR (95% CI) | p | Intercept (se) | p | Cochran Q Statistics (df) | I2 | p | p | |
| HL | 1.216 (0.783, 1.887) | 0.382 | 2.449 (5) | −1.042 | 0.784 | 1.341 (0.780, 2.253) | 0.268 | 2.113 (0.316, 14.120) | 0.483 | −0.027 (0.046) | 0.589 | 2.106 (4) | −1.849 | 0.716 | 0.866 |
| ARHL | 0.934 (0.217, 4.031) | 0.928 | 5.18 (5) | 0.035 | 0.393 | 0.998 (0.151, 6.593) | 0.998 | 3.119 (0.003, 3310.194) | 0.765 | −0.058 (0.167) | 0.745 | 5.037 (4) | −0.191 | 0.284 | 0.520 |
| NIHL | 1.003 (0.982, 1.024) | 0.766 | 41.549 (14) | 0.639 | 0.000 | 1.007 (0.99, 1.023) | 0.436 | 0.988 (0.93, 1.05) | 0.706 | 0.001 (0.002) | 0.609 | 40.687 (13) | 0.631 | 0.0001 | <0.001 |
| NIHLa | 1.003 (0.990, 1.016) | 0.572 | 7.844 (11) | −0.402 | 0.727 | 1.007 (0.989, 1.024) | 0.449 | 0.990 (0.965, 1.036) | 0.987 | 0.000 (0.001) | 0.818 | 7.788 (10) | −0.412 | 0.65 | 0.779 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Liu, S.; Xin, J.; Qian, P.; Guo, S.; Xu, X.; Wang, D.; Yang, L. Telomere Length and Hearing Loss: A Two-Sample Mendelian Randomization. Int. J. Environ. Res. Public Health 2022, 19, 8937. https://doi.org/10.3390/ijerph19158937
Liu Y, Liu S, Xin J, Qian P, Guo S, Xu X, Wang D, Yang L. Telomere Length and Hearing Loss: A Two-Sample Mendelian Randomization. International Journal of Environmental Research and Public Health. 2022; 19(15):8937. https://doi.org/10.3390/ijerph19158937
Chicago/Turabian StyleLiu, Yun, Shuangyan Liu, Jiarui Xin, Peiyi Qian, Shuli Guo, Xiaojun Xu, Dahui Wang, and Lei Yang. 2022. "Telomere Length and Hearing Loss: A Two-Sample Mendelian Randomization" International Journal of Environmental Research and Public Health 19, no. 15: 8937. https://doi.org/10.3390/ijerph19158937